
The SUMO Isopeptidase Ulp2p Is Required to Prevent Recombination-Induced Chromosome Segregation Lethality following DNA Replication Stress
SUMO conjugation is a key regulator of the cellular response to DNA replication stress, acting in part to control recombination at stalled DNA replication forks. Here we examine recombination-related phenotypes in yeast mutants defective for the SUMO de-conjugating/chain-editing enzyme Ulp2p. We find that spontaneous recombination is elevated in ulp2 strains and that recombination DNA repair is essential for ulp2 survival. In contrast to other SUMO pathway mutants, however, the frequency of spontaneous chromosome rearrangements is markedly reduced in ulp2 strains, and some types of rearrangements arising through recombination can apparently not be tolerated. In investigating the basis for this, we find DNA repair foci do not disassemble in ulp2 cells during recovery from the replication fork-blocking drug methyl methanesulfonate (MMS), corresponding with an accumulation of X-shaped recombination intermediates. ulp2 cells satisfy the DNA damage checkpoint during MMS recovery and commit to chromosome segregation with similar kinetics to wild-type cells. However, sister chromatids fail to disjoin, resulting in abortive chromosome segregation and cell lethality. This chromosome segregation defect can be rescued by overproducing the anti-recombinase Srs2p, indicating that recombination plays an underlying causal role in blocking chromatid separation. Overall, our results are consistent with a role for Ulp2p in preventing the formation of DNA lesions that must be repaired through recombination. At the same time, Ulp2p is also required to either suppress or resolve recombination-induced attachments between sister chromatids. These opposing defects may synergize to greatly increase the toxicity of DNA replication stress.
Published in the journal:
. PLoS Genet 7(3): e32767. doi:10.1371/journal.pgen.1001355
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001355
Summary
SUMO conjugation is a key regulator of the cellular response to DNA replication stress, acting in part to control recombination at stalled DNA replication forks. Here we examine recombination-related phenotypes in yeast mutants defective for the SUMO de-conjugating/chain-editing enzyme Ulp2p. We find that spontaneous recombination is elevated in ulp2 strains and that recombination DNA repair is essential for ulp2 survival. In contrast to other SUMO pathway mutants, however, the frequency of spontaneous chromosome rearrangements is markedly reduced in ulp2 strains, and some types of rearrangements arising through recombination can apparently not be tolerated. In investigating the basis for this, we find DNA repair foci do not disassemble in ulp2 cells during recovery from the replication fork-blocking drug methyl methanesulfonate (MMS), corresponding with an accumulation of X-shaped recombination intermediates. ulp2 cells satisfy the DNA damage checkpoint during MMS recovery and commit to chromosome segregation with similar kinetics to wild-type cells. However, sister chromatids fail to disjoin, resulting in abortive chromosome segregation and cell lethality. This chromosome segregation defect can be rescued by overproducing the anti-recombinase Srs2p, indicating that recombination plays an underlying causal role in blocking chromatid separation. Overall, our results are consistent with a role for Ulp2p in preventing the formation of DNA lesions that must be repaired through recombination. At the same time, Ulp2p is also required to either suppress or resolve recombination-induced attachments between sister chromatids. These opposing defects may synergize to greatly increase the toxicity of DNA replication stress.
Introduction
As part of the DNA damage response, homologous recombination (HR), particularly template switch recombination through the post-replication DNA repair pathway (PRR), provides an important mechanism for restarting stalled replication forks and filling in un-replicated gaps in DNA (reviewed in [1], [2]). These recombination events must be managed carefully, however. DNA strand exchange during HR, followed by re-initiating replication using the nascent sister chromatid as a template, can result in the formation of DNA linkages between daughter chromosomes. Failure to resolve these linkages, called sister chromatid junctions (SCJs), leads to chromosome breakage or aneuploidy, and may contribute to genome instability in many forms of cancer (reviewed in [3]).
A variety of studies implicate SUMO post-translational modification as an important regulator of HR in response to replication stress. Following activation of the SUMO precursor protein, SUMO modification is catalyzed by the E2 conjugating enzyme Ubc9p, which typically acts through one of several E3 ligases to covalently join SUMO moieties to lysine residues on substrate proteins (reviewed in [4]). One SUMO substrate that plays an especially prominent role in controlling HR at replication forks is Pol30p/PCNA, which is modified to recruit different activities to the replisome. During S phase, Ubc9p works through the E3 ligase Siz1p to sumoylate PCNA on K164 and K127 [5]. SUMO modified PCNA recruits the Srs2p helicase [6], [7], which suppresses unscheduled HR by disassembling Rad51p nucleoprotein filaments [8]-[10]. Following replication fork stalling at MMS-induced DNA lesions, however, PRR proteins catalyze either mono- or poly-ubiquitinylation of PCNA K164 [5]. These modifications recruit trans-lesion bypass polymerases or induce template switching HR, respectively, providing alternative mechanisms to bypass the lesion and restart replication [5], . The existence of additional SUMO substrates that control HR is suggested by the observations that mutations affecting both Ubc9p and the E3 ligase Mms21p, which is not required for PCNA sumoylation, confer sensitivity to the replication impeding drugs hydroxyurea (HU) and methyl methansulfonate (MMS) [5], [14]-[19]. Mms21p exists in a complex with two members of the structural maintenance of chromosomes family of proteins, Smc5p and Smc6p, which are also required for HU and MMS-resistance [15], [16], [20]-[22]. Notably, in response to MMS, ubc9, mms21, smc5 and smc6 mutants show an accumulation of X-shaped DNA structures that are thought to represent either regressed forks-a possible intermediate in fork restart-or hemi-catenate SCJs [17], [19], [23]. In this sense, they resemble mutants defective for the Sgs1p/Top3p/Rmi1p complex, which, through concerted helicase/topoisomerase activities, catalyzes the dissolution of hemi-catenates and other DNA linkages [24]-[27]. These findings suggest complex roles for sumoylation in either preventing excessive/improper HR at stalled replication forks and/or mediating the active dissolution of SCJs.
As with the forward SUMO pathway, SUMO de-conjugation is also required to tolerate replication stress. Budding yeast contains two members of the SENP/Ulp family of SUMO isopeptidases, Ulp1p and Ulp2p, which catalyze removal of SUMO [28]-[30]. Ulp1p is an essential enzyme that is preferentially localized to the nuclear pore [28]-[32], whereas Ulp2p is distributed throughout the nucleus [29], [30], [33]. Ulp2p (first identified as Smt4p; [34]) is not essential, but ulp2 mutants grow poorly and exhibit a complex assortment of phenotypes, including chromosome segregation and cell division defects. [29], [30], [35]-[42]. Ulp1p and Ulp2p also mediate functions that promote SUMO modification. Ulp1p is required to cleave the SUMO precursor to expose a glycine residue necessary for conjugation [28], while Ulp2p possesses a chain editing activity that prevents formation of aberrantly poly-sumoylated substrates [43]. Poly-sumoylation has the potential to interfere with the functional role of SUMO addition. Moreover, recent evidence has revealed that some poly-sumoylated substrates are targeted for degradation by the SUMO-targeted Slx5p-Slx8p ubiquitin ligase [44]-[46].
Although Ulp1p and Ulp2p play different roles in the SUMO pathway, one trait shared by ulp1 and ulp2 strains is that both exhibit sensitivity to HU and MMS [29], [30], [43]. Previously, a ulp1-I615N mutant was shown to accumulate single-stranded gaps during DNA replication, to exhibit increased spontaneous recombination, and to become dependent on Srs2p and HR for viability, suggesting a role for Ulp1p in suppressing replication errors that induce HR [47]. Insight into the replication stress sensitivity of ulp2 mutants has come from the important finding that Ulp2p is required for cells to complete mitosis following DNA damage checkpoint arrest [41]. From this, de-sumoylation of Ulp2p substrates may be necessary to restart the chromosome segregation machinery once the checkpoint block to mitosis has been relieved [41], [48]. But whether Ulp2p, like other components of the SUMO pathway, is also involved in controlling HR during DNA damage or replication stress has not yet been examined. In this study, we find that, following replication fork stalling by MMS, ulp2 mutants accumulate persistent recombination intermediates that are likely to correspond to SCJs. This mis-regulation is accompanied by a severe, recombination-dependent, block to chromosome segregation, revealing a critical role for Ulp2p in allowing sister chromatids to disjoin following HR DNA repair.
Results
Recombination is elevated and essential in ulp2 mutants
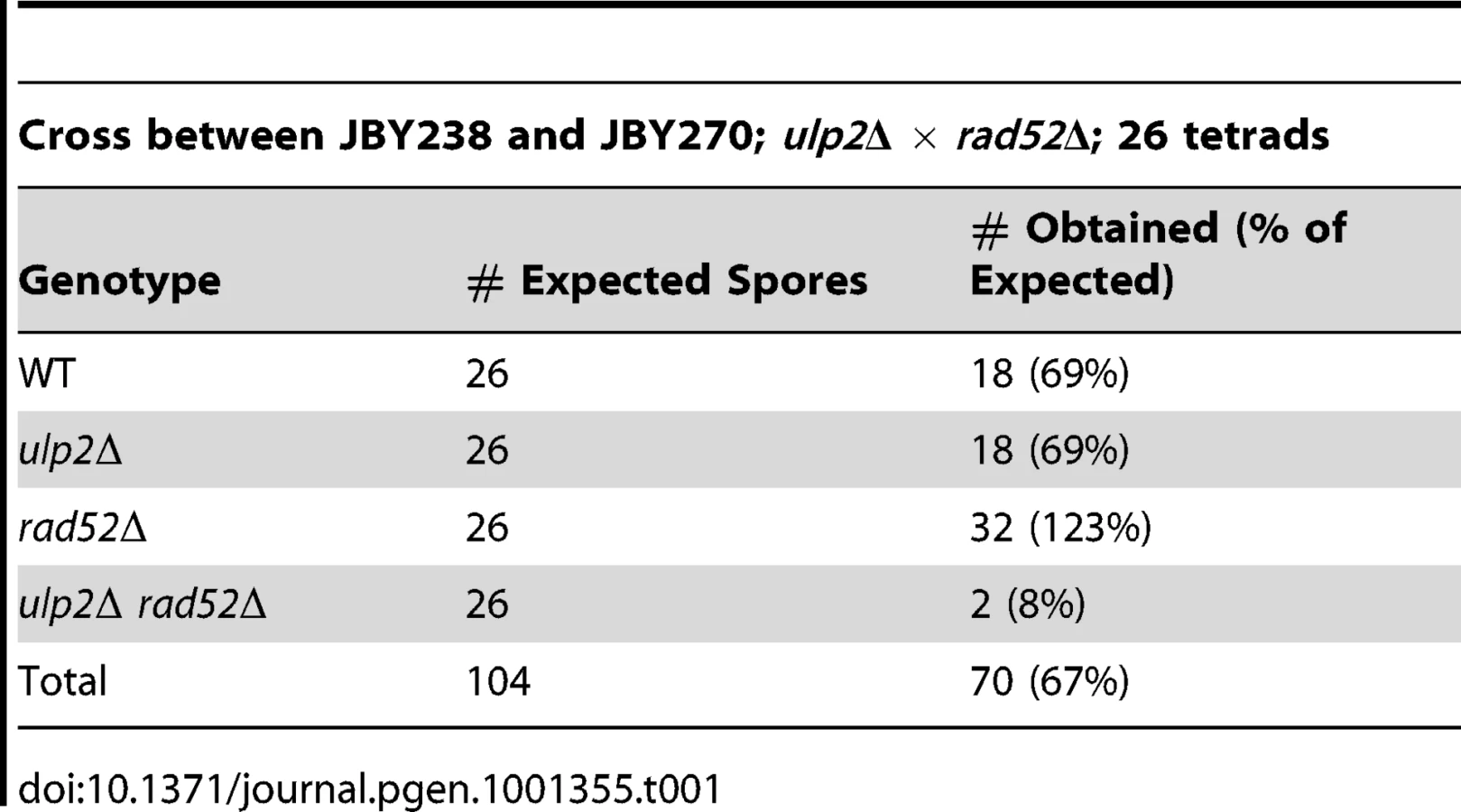
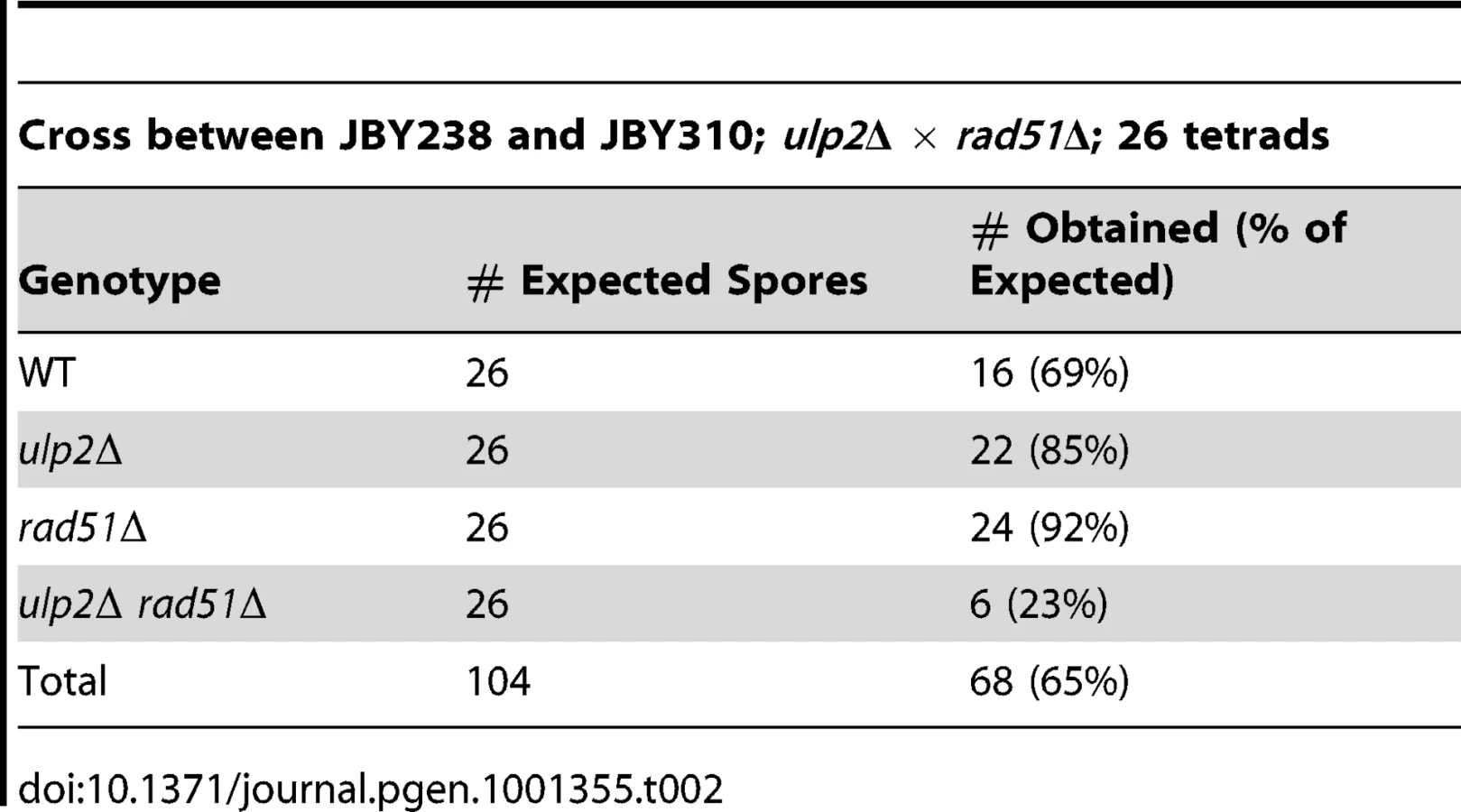
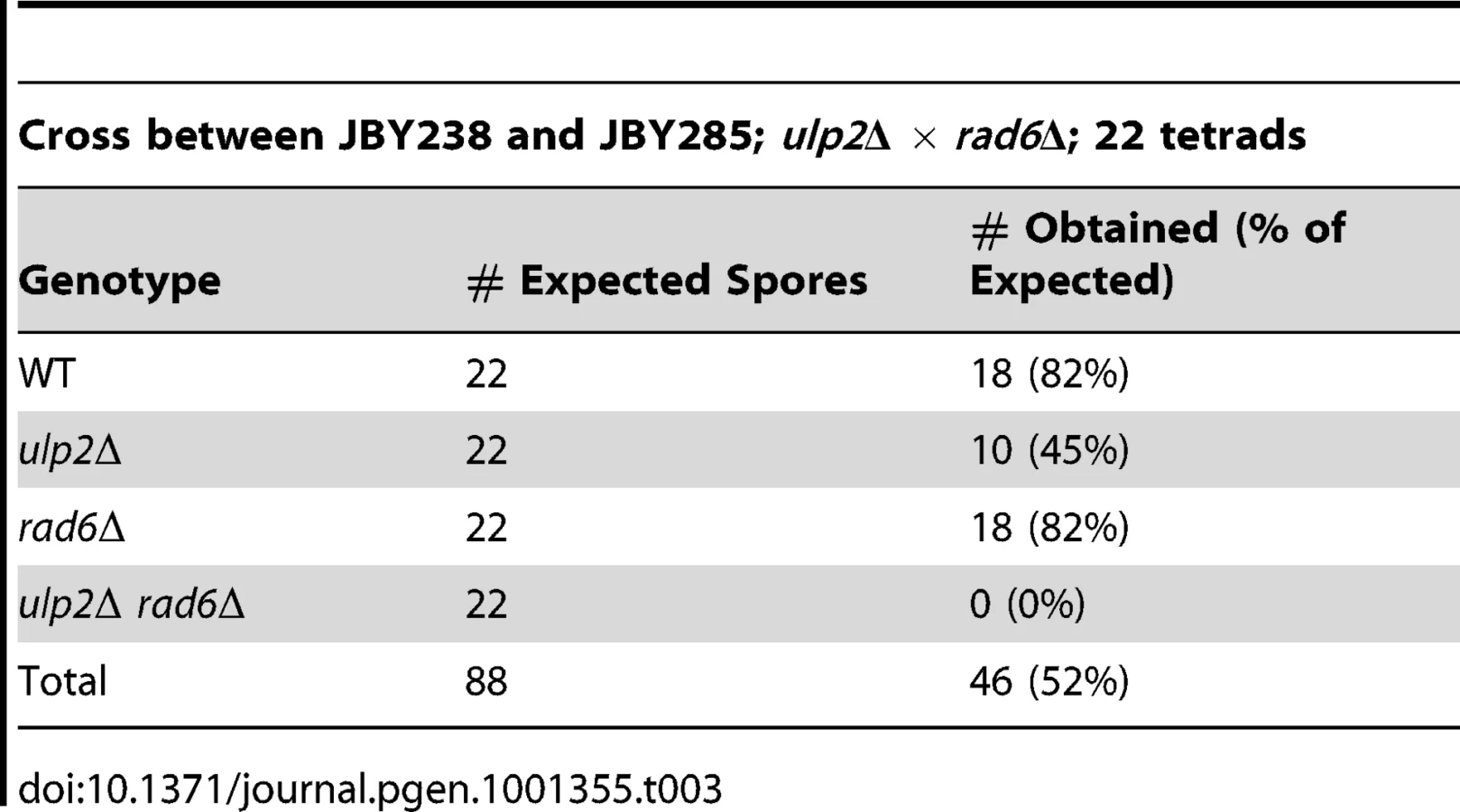
We initially set out to determine if ulp2 mutants displayed a similar dependency on recombination as ulp1-I615N strains [47]. A ulp2 deletion mutant (ulp2Δ) was mated to rad52Δ, rad51Δ and rad6Δ strains. Rad51p and Rad52p are required for most forms of HR [2], while Rad6p controls trans-lesion synthesis and template switching PRR [1]. ulp2Δ rad52Δ, ulp2Δ rad51Δ and ulp2Δ rad6Δ double mutants were either not obtained or were obtained at lower than expected frequencies from these crosses (Table 1, Table 2, Table 3). For rad52Δ, we examined this apparent synthetic lethality further by isolating ulp2Δ rad52Δ segregants harboring a wild type (WT) copy of RAD52 on a URA3 minichromosome (pRAD52). ulp2Δ rad52Δ/pRAD52 mutants grew weakly, if at all, on media containing 5-FOA, a drug that only allows growth if cells are capable of losing pRAD52 (Figure 1A). Thus, Rad52p is essential for proliferation of ulp2Δ cells.
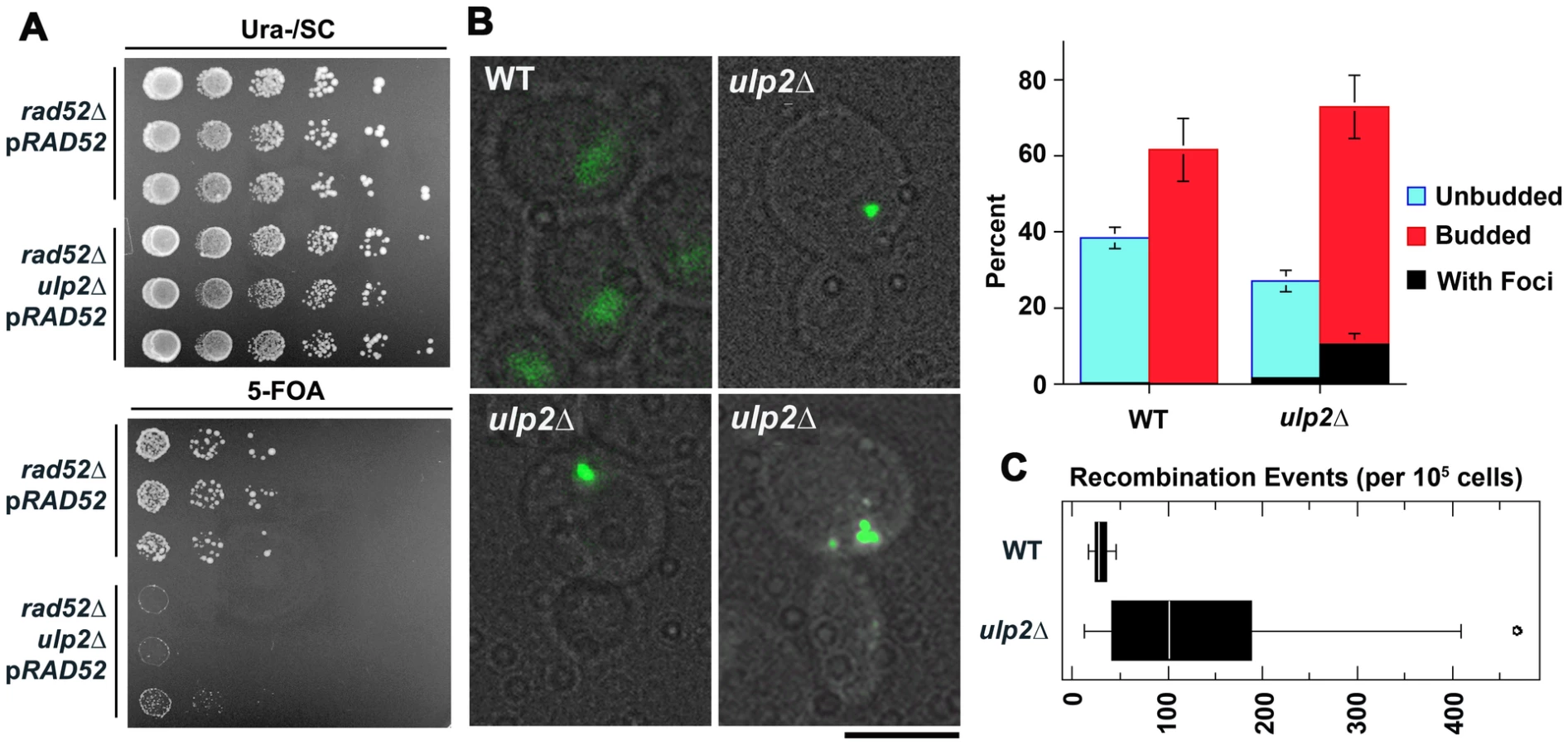
The essential role of Rad52p prompted us to examine whether HR was elevated in the absence of Ulp2p. Yeast cells exhibit a uniform nuclear distribution of fluorescent Rad52p-GFP in the absence of DNA damage (Figure 1B, [49]), but Rad52p-GFP rapidly assembles into intra-nuclear foci during HR DNA repair [49]. We found that an average of 17% of ulp2Δ cells in mid-logarithmic phase cultures displayed Rad52p-GFP foci, a significant increase (p = 0.0074) compared to less than 1% in WT cells. (Figure 1B). As a second assay, we utilized a reporter in which recombination events between direct repeats on chromosome XV can be selected because they restore an intact HIS3 gene [50]. ulp2Δ cells exhibited a 4.7-fold increase in the median frequency of this form of recombination (Figure 1C; p = 0.044), indicating spontaneous HR at this genomic locus is significantly increased in ulp2Δ mutants.
Ulp2 mutants display a reduced frequency of chromosome rearrangements
DNA replication errors are potent inducers of HR and can initiate chromosome rearrangements [51], [52]. Based on this, we used a yeast artificial chromosome (YAC) assay to examine the frequency of spontaneous gross chromosomal rearrangements (GCRs) in ulp2Δ cells ([53]; Figure 2A). For comparison, we also measured GCR frequencies in ulp1-333, smt3-331 and ubc9-1 SUMO pathway mutants (SMT3 encodes the single SUMO isoform in budding yeast). Using the YAC system, we obtained median GCR frequencies of 252×10−7 for WT cells, 5490×10−7 for smt3-331 cells, 6109×10−7 for ubc9-1 cells, and 2617×10−7 for ulp1-333 cells (Figure 2B), representing 22-, 24-, and 10-fold increases, respectively, compared to WT controls. In contrast, and counter to initial expectations, it proved difficult to recover spontaneous GCRs in ulp2Δ mutants, with a median GCR frequency of 56×10−7 (Figure 2C). This represents a significant (p = 0.025) 4.5-fold decrease compared to WT.
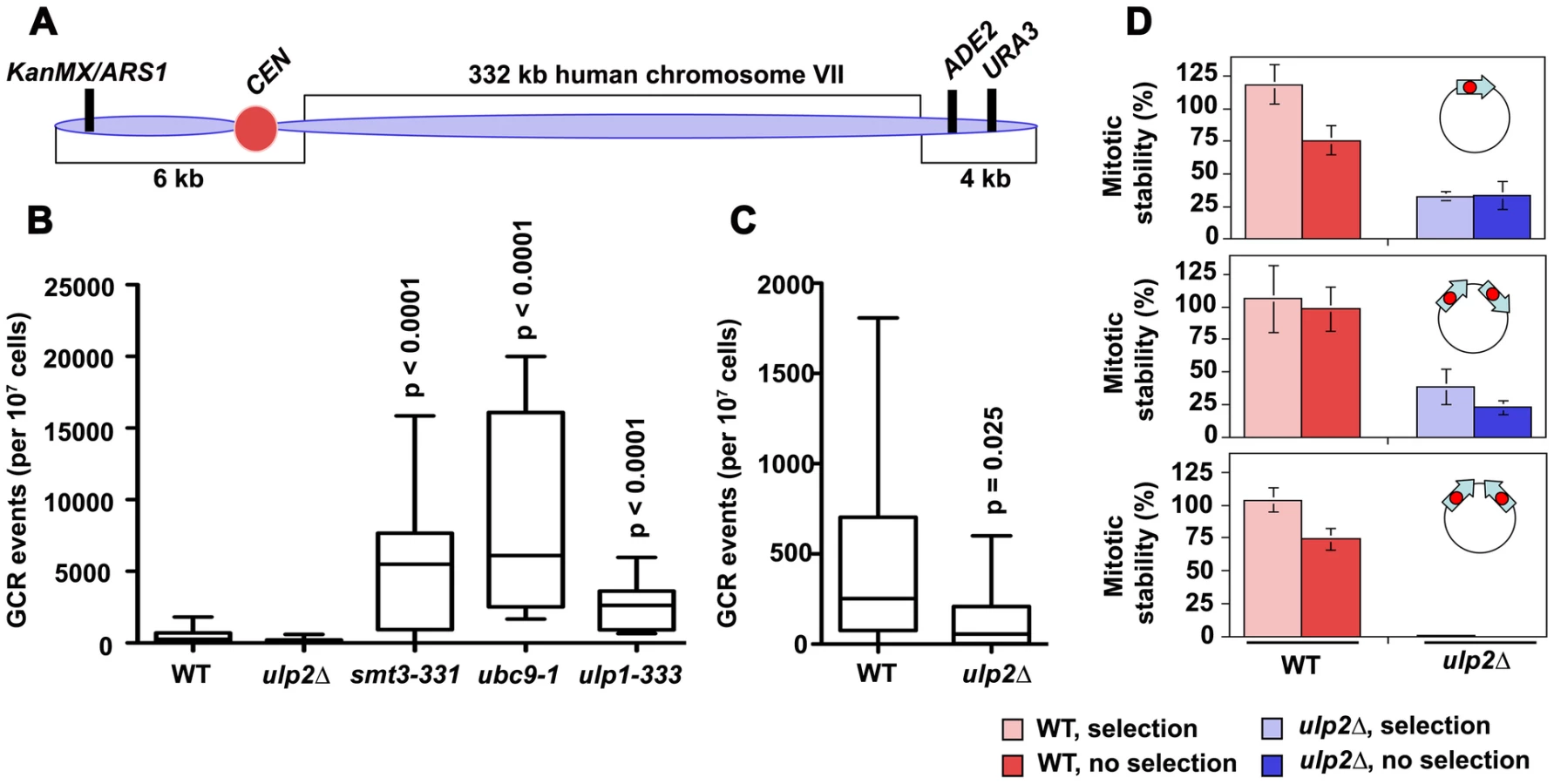
To further monitor chromosome rearrangements we examined two circular dicentric minichromosomes. In one (p2XCENdirect), two copies of a CEN sequence were oriented in a direct repeat configuration. In the other (p2XCENinvert), the same CEN duplication was oriented as inverted repeats. Previous studies have shown that both direct and inverted repeat dicentrics can be efficiently transformed into yeast, and are initially retained through a combination of co-orientation of the two CENs on the spindle and non-disjunction following dicentric bridging [54], [55]. During outgrowth, however, rearranged minichromosomes that have deleted one of the CENs accumulate. For direct CEN repeats these deletions tend to arise through loop out events, whereas inverted CEN repeats are resolved through more complex re-arrangements. Consistent with this characterization, in WT transformants p2XCENdirect and p2XCENinvert exhibited similar mitotic stabilities to p1XCEN controls (Figure 2D). Analysis of minichromosomes rescued from these cells revealed precise CEN1 excision for p2XCENdirect and a diversity of plasmid species for p2XCENinvert (not shown). In ulp2Δ mutants, p1XCEN was only retained in ∼30% of the cells; this result is in keeping with previous studies showing reduced minichromosome stability in the absence of Ulp2p [29]. p2XCENdirect demonstrated a similar stability to p1XCEN (Figure 2D), and underwent the same precise CEN deletions observed in WT (not shown). In contrast, p2XCENinvert proved extremely unstable, with less than 1% of ulp2Δ cells maintaining the mini-chromosome. These results suggest that some chromosome re-arrangements either fail to occur or cannot be tolerated in ulp2 mutants.
MMS–induced HR intermediates accumulate in ulp2Δ mutants
In order to more directly examine the consequences of HR in ulp2Δ mutants, we used MMS to induce recombination. As an initial experiment, we examined chromosome integrity following exposure to MMS by pulse-field gel electrophoresis. WT and ulp2Δ cells were arrested in G1, released into media containing 0.01% MMS for 2 hr, and then allowed to recover in MMS-free media. Following MMS treatment a lower molecular weight DNA smear was observed in both WT and ulp2Δ strains (Figure 3A), reflecting MMS-induced chromosome breakage [17]. For both strains, a one hr recovery largely restored the normal chromosome banding pattern. This suggests Ulp2p is not obviously required for healing MMS-induced DNA breaks.
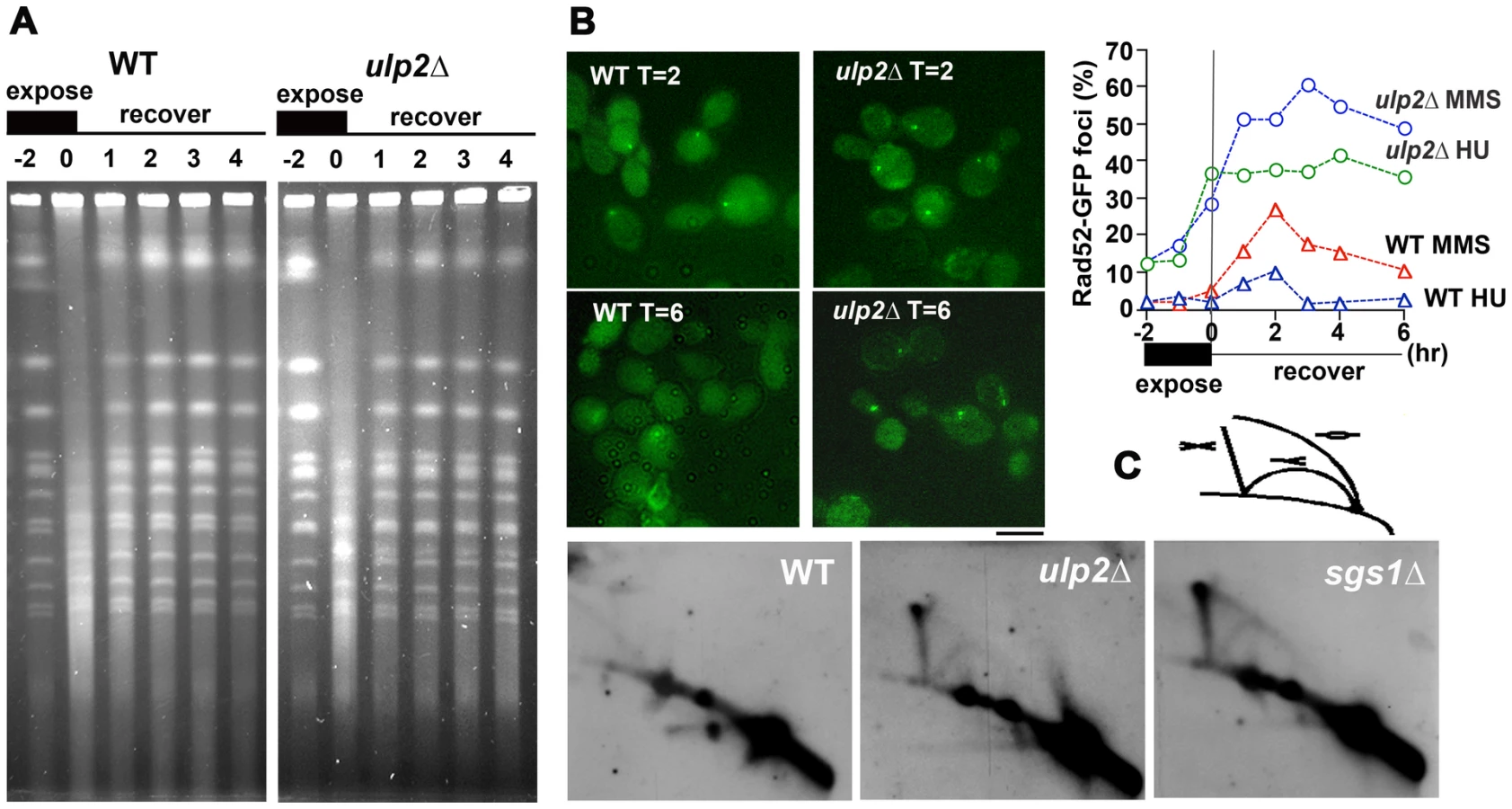
We next examined processing of MMS-induced DNA lesions. In the experiment shown in Figure 3B, WT cells and ulp2Δ mutants expressing RAD52-GFP were treated with 0.01% MMS and allowed to recover. After a 2 hr recovery, ∼30% of WT cells accumulated Rad52p-GFP foci (Figure 3B). By 6 hr, however, the percentage of cells with Rad52p-GPF foci had substantially declined and many cells were proceeding with the next round of cell division. In contrast, ulp2Δ mutants showed a much stronger accumulation of Rad52p-GFP foci, reaching a maximum of ∼60% (Figure 3B), and these foci tended to persist for the duration of the recovery period. We also examined Rad52p-GFP foci in ulp2Δ cells treated with 200 mM HU. HU does not normally induce Rad52p foci because the integrity of the replisome is maintained by the S phase checkpoint (Figure 3B, [56]). HU treated ulp2Δ cells, however, exhibited a strong induction of Rad52p-GFP foci.
In response to MMS, proper regulation of HR is required to prevent X-shaped recombination intermediates from accumulating in the vicinity of origins of replication [17], [19], [23]. On two-dimensional gels these structures migrate as a “X-spike” that is distinct from replication forks and bubbles [57], [58]. To determine whether ulp2Δ mutants accumulated this type of HR intermediate, ulp2Δ cells, along with WT and sgs1Δ controls, were released from a G2/M nocodazole block and treated with 0.033% MMS for 3 hr as previously described [17]. Genomic DNAs were fractionated on two-dimensional gels, and probed with a DNA fragment corresponding to ARS305. A prominent X-spike signal was observed in sgs1Δ and ulp2Δ samples (Figure 3D). Thus, Ulp2p deconjugating and/or chain editing activities are required to prevent accumulation of MMS-induced HR intermediates.
Interactions between Ulp2p and Sgs1p
Based on current evidence, Sgs1p is one SUMO target that could be connected to Ulp2p's role in HR. In particular, a recent study has shown that a single prominent SUMO species of Sgs1p accumulates after MMS exposure, and K621 has been identified as the acceptor lysine that is responsible for this modification [59]. We were able to confirm that treatment with 0.3% MMS resulted in a substantial fraction of Sgs1p-myc shifting into a reduced mobility species (Figure 4A and Figure S1), and that a decreased amount of this form was observed following treatment with a lower MMS concentration (0.033%; Figure 4B, 4C). The appearance of this form was abolished in ubc9-1 strains (Figure 4B) and a sgs1-K621R mutant (Figure S1), indicating it is likely to correspond to the previously reported K621 conjugate. In ulp2Δ strains, however, a marked increase in this putative Sgs1p SUMO species was observed (Figure 4B, 4C), which persisted for at least 3 hr after removal of MMS (Figure 4C). In sum, these results suggest that sumoylation of Sgs1p is likely to be regulated by Ulp2p.
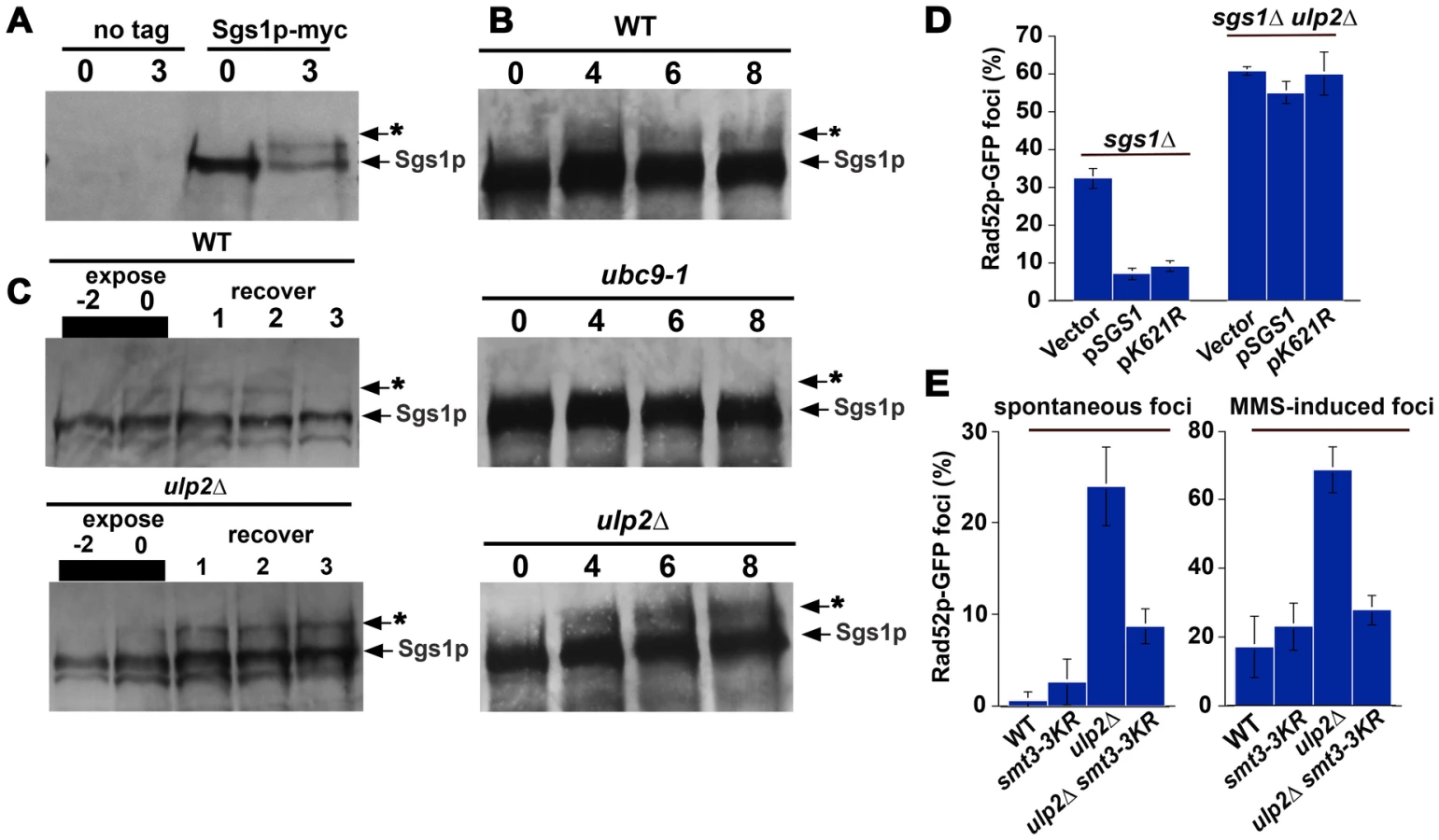
If failure to properly control Sgs1p sumoylation was responsible for ulp2Δ HR defects, SUMO-resistant Sgs1p might ameliorate these phenotypes. We therefore examined whether a plasmid-born copy of the sgs1-K621R allele could prevent Rad52p foci accumulation. Following a two hr treatment with 0.010% MMS, however, no significant reduction in ulp2Δ sgs1-K621R cells displaying Rad52p-GFP foci was observed (Figure 4D). Previous studies have shown that a form of Smt3 (smt3-3KR) that cannot form polymeric SUMO chains can rescue the HU and MMS sensitivity of ulp2 mutants [43], leading us to test whether smt3-3KR could prevent Rad52p foci accumulation. This proved to be the case, as smt3-3KR ulp2Δ double mutants did in fact show a substantial reduction in the accumulation of both spontaneous and MMS-induced Rad52p foci (Figure 4F). Thus, proper SUMO chain editing through Ulp2p is likely to be important in controlling HR.
ulp2Δ mutants fail in chromosome segregation after exposure to MMS
In our experiments, it was apparent that ulp2Δ cells frequently remained blocked in the cell cycle during recovery from MMS, similar to previous results examining ulp2 recovery following HU treatment and in response to an irreparable DNA double strand break [41]. We took four experimental approaches to investigate the basis for the apparent MMS recovery defect of ulp2Δ cells. First, phospho-activation of the Rad53p checkpoint kinase during the DNA damage response results in a series of slower migrating gel mobility variants [60], and collapse of these forms provides a means to assess silencing of the checkpoint. In WT cells, Rad53p phospho-variants almost completely disappeared during a 2–4 hr recovery after treatment with 0.01% MMS (Figure 5A). A similar pattern was observed in ulp2Δ strains, although the accumulation and disappearance of shifted Rad53p appeared to be slightly delayed.
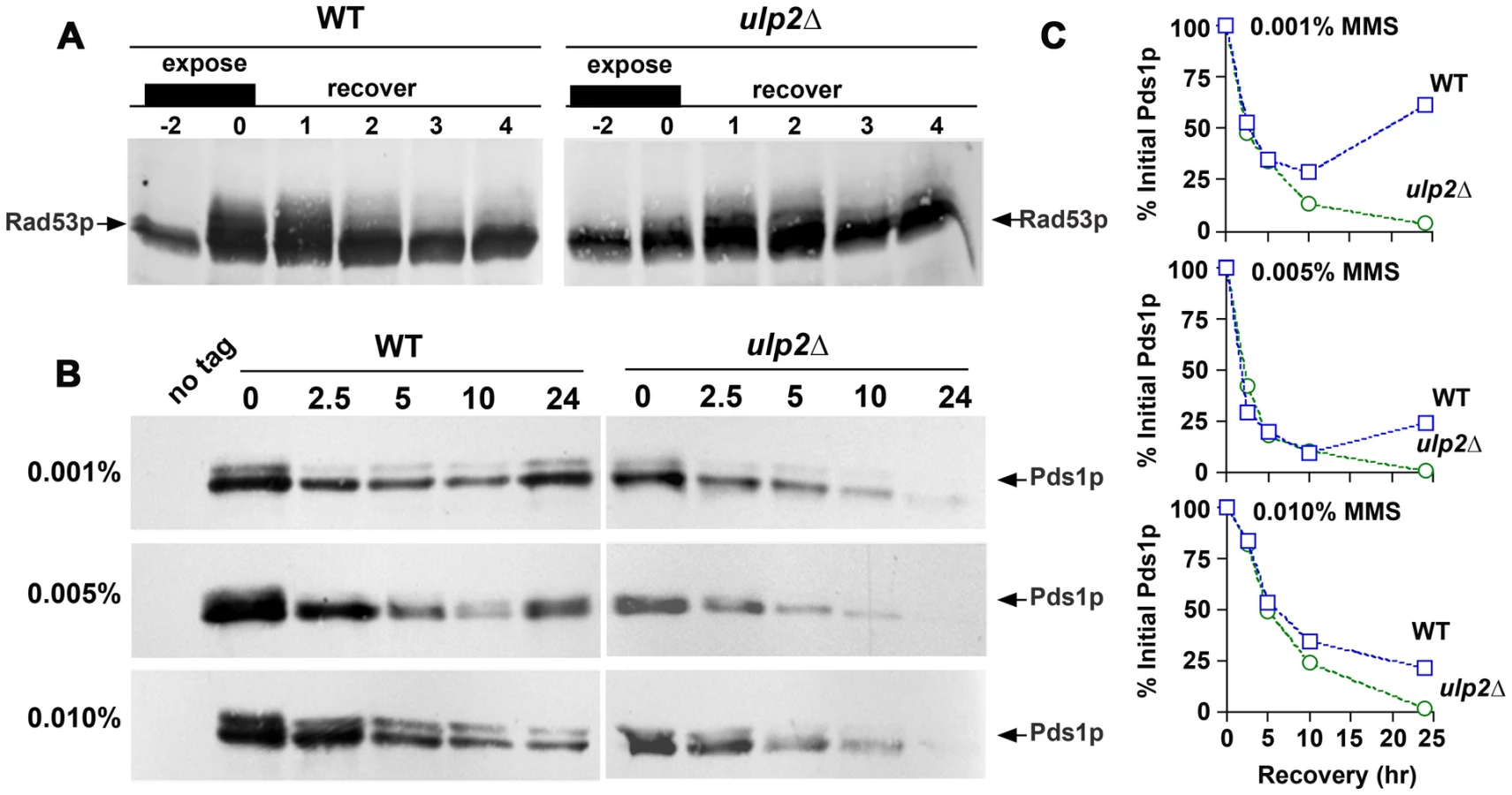
Second, we examined degradation of Pds1p/securin. Pds1p is a downstream target of the DNA damage checkpoint that is stabilized to block cohesin proteolysis and anaphase entry [61], [62]. The kinetics of Pds1p degradation therefore provides a read-out of commitment to anaphase. In these experiments, we used the cdc14-1 allele to block Pds1p re-synthesis once cells recovered from the checkpoint. cdc14-1 PDS1-myc and cdc14-1 ulp2Δ PDS1-myc cells were treated with 0.001%, 0.005% and 0.01% MMS for 2 hr, allowed to recover at a cdc14-1 non-permissive temperature, and Pds1p-myc abundance was monitored over a 24 hr period. In cdc14-1 cells, Pds1p-myc degradation proceeded in a dose-dependent manner until 10 hr post-treatment (Figure 5B, 5C). At this point, Pds1p started to increase in the 0.001% and 0.005% MMS cultures, probably reflecting leakage through the cdc14-1 arrest. These degradation kinetics were virtually indistinguishable in cdc14-1 ulp2Δ cells, although re-synthesis of Pds1p was not observed (Figure 5B, 5C). These results suggest that MMS treated ulp2Δ cells can terminate checkpoint signaling and commit to anaphase.
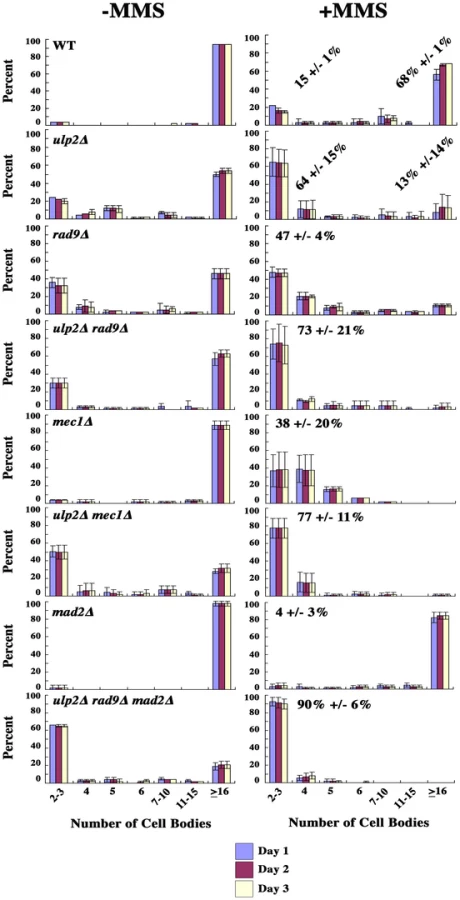
Third, we used micro-colony analysis to determine whether getting rid of the checkpoint relieved the restraint on cell division. Cells from MMS treated and untreated cultures were positioned on agar plates, and the appearance of cell bodies was examined over time. A budded yeast cell arrested at the DNA damage checkpoint consists of two cell bodies. If this cell completes mitosis and one of progeny cells sends forth a bud, the microcolony now contains three cell bodies, and the number of cell bodies increases exponentially with continued division. We found that an average of 68% of WT cells were able to form microcolonies containing ≥ 16 cell bodies within a 3 day period after transient exposure to MMS, indicating the majority recovered efficiently (Figure 6). In comparison, even in the absence of MMS, 20% of ulp2Δ cells remained blocked at the 2–3 cell body stage. This lethality was strongly exacerbated by MMS treatment, with 64% of ulp2Δ cells failing to proliferate beyond 2–3 cell bodies. Inactivating the DNA damage checkpoint in rad9Δ ulp2Δ mutants, or both the DNA damage and S phase checkpoints in mec1Δ ulp2Δ mutants, did not relieve the ulp2Δ block to cell division (Figure 6). ulp2 cells fail to maintain chromatid cohesion at centromeric regions during DNA damage checkpoint arrest [36], [42], which could potentially activate the spindle assembly checkpoint (SAC). We therefore tested whether abolishing the SAC could restore ulp2Δ division. However, ∼60% of MMS treated ulp2Δ mad2Δ mutants still remained blocked with 2–3 cell bodies (Figure S2). We further generated a ulp2Δ rad9Δ mad2Δ triple mutant to abolish both DNA damage and SAC checkpoint responses. This triple mutant grew extremely poorly, and, following exposure to MMS, ∼90% of the cells failed to recover (Figure 6). Thus, MMS treated ulp2Δ mutants experience a terminal block to cell division even in the absence of pre-anaphase checkpoint controls.
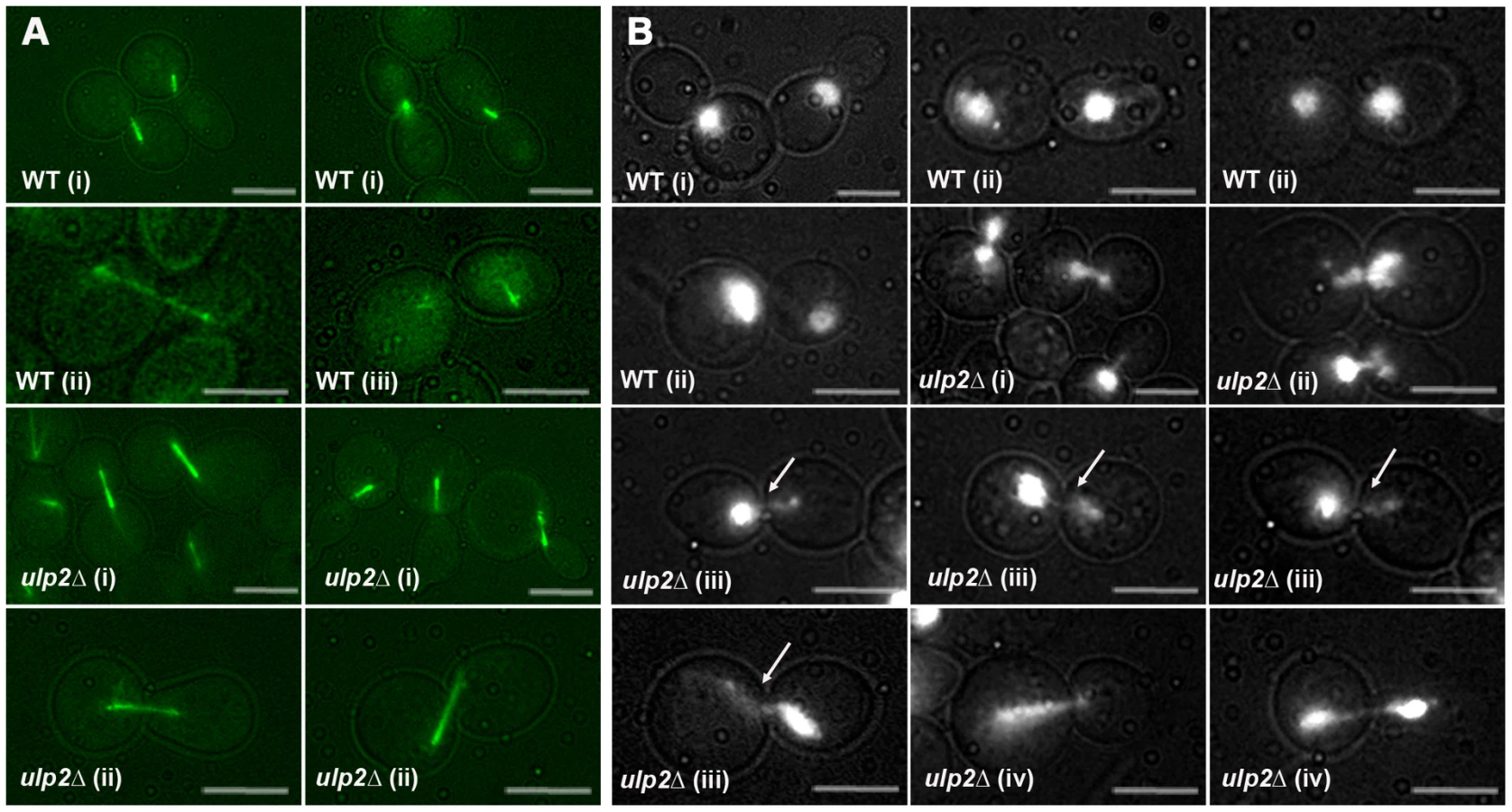
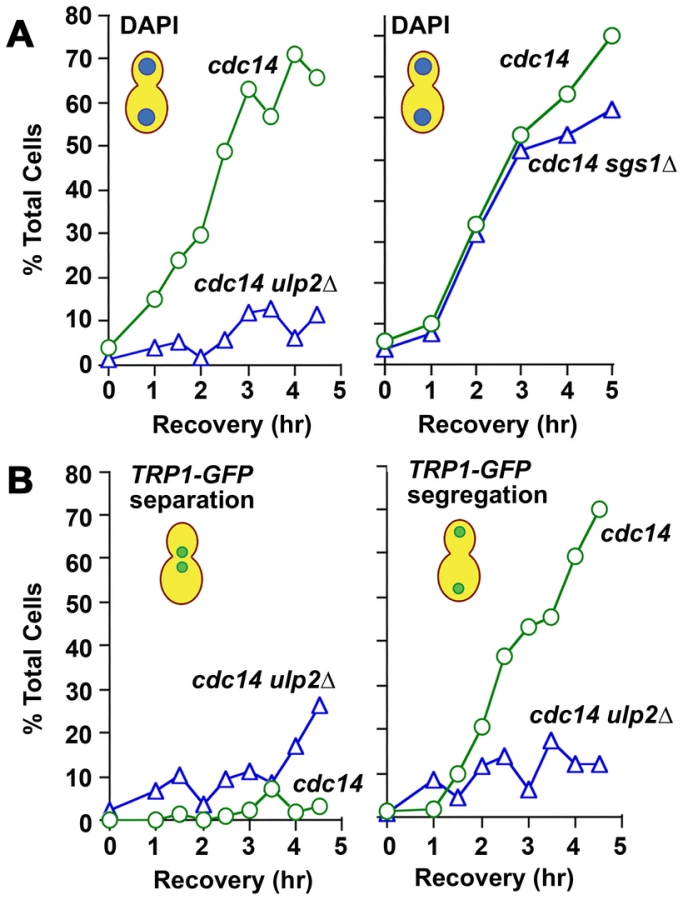
Fourth, we examined mitotic progression in ulp2Δ cells by cytology and flow cytometry. Following a 2 hr treatment with 0.01% MMS, WT cells arrested at the DNA damage checkpoint typically showed short pre-anaphase spindles and an undivided mass of chromatin (Figure 7A, 7B). Completion of mitosis during recovery was characterized by normal spindle extension and chromosome transmission. As monitored by DAPI staining and a Lac operator-GFP chromosome tag (TRP1-GFP), ∼70% of cdc14-1 cells underwent chromosome separation and segregation during recovery (Figure 8A, 8B), and FACS analysis indicated that many cells proceeded with additional rounds of cell division (Figure S3). In contrast, many MMS treated ulp2Δ cells showed partial, incomplete spindle extension during recovery, accompanied by an apparent block to nuclear division (Figure 7A, 7B). In some cells it was possible to visualize chromatin fibers that appeared to be pulled away from an undivided mass of chromatin (Figure 7B iii; arrows). In others, chromosome separation appeared more complete, but chromatin was stretched to varying degrees along the spindle (Figure 7B iv). DAPI staining indicated less than 20% of cdc14-1 ulp2Δ cells successfully completed chromosome segregation (Figure 8A). ∼30% of cdc14-1 ulp2Δ cells underwent TRP1-GFP separation during recovery, but the separated foci largely failed to segregate (Figure 8B). FACS analysis suggested that MMS treated ulp2Δ cells potentially tried to proceed with a second round of DNA replication following this block chromosome segregation, although the FACS profiles were quite heterogeneous and did not clearly resolve into a peak of cells with a 4N content of DNA (Figure S3).
Since sgs1Δ and ulp2Δ mutants both accumulate HR intermediates that might be expected to link sister chromatids (Figure 3C), we additionally examined chromosome segregation during MMS recovery in cdc14-1 sgs1Δ cells. Compared to the ulp2Δ defect, the fraction of MMS treated cdc14-1 sgs1Δ cells that could segregate their chromosomes to an extent necessary to form two distinct nuclear masses was only slightly reduced compared to cdc14-1 controls (Figure 8A; see Figure S4 for a more complete description). Taken as a whole, these results allow us to conclude that, although they commit to anaphase, ulp2Δ mutants are unable to separate their chromosomes efficiently following MMS treatment. Furthermore, this non-disjunction defect appears more severe than that observed in a sgs1Δ strain.
Blocking HR restores chromosome segregation in MMS–treated ulp2 mutants
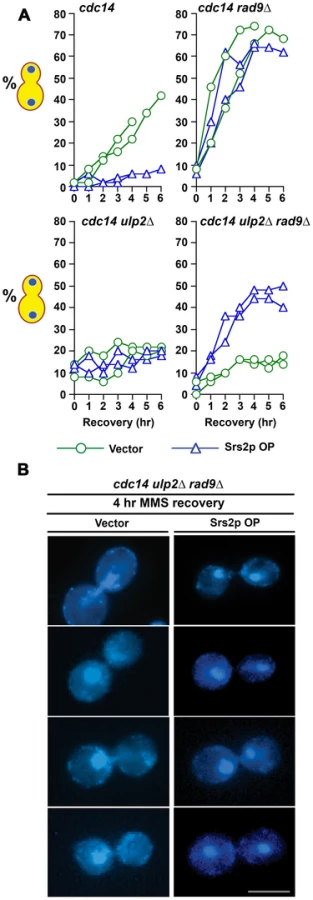
If defective HR in MMS treated ulp2Δ cells is causally linked to the chromosome separation defect that we observed in our experiments, blocking recombination should restore chromosome segregation. Given that HR is essential in ulp2Δ mutants (Figure 1) our approach to test this was to overproduce (OP) the Srs2p helicase. In addition to antagonizing nucleoprotein filament assembly [8]-[10], Srs2p also appears to exert anti-recobinogenic activity by unwinding D-loop intermediates [63], [64]. Srs2p OP should therefore be an effective way to short circuit early stages of HR. cdc14-1, cdc14-1 rad9Δ, cdc14-1 ulp2Δ and cdc14-1 rad9Δ ulp2Δ strains were transformed with a vector control or a high copy plasmid in which SRS2 was expressed under control of its endogenous promoter (pSRS2). The transformants were then treated with 0.01% MMS for 2 hr and allowed to recover at a cdc14-1 non-permissive temperature. Compared to vector controls, cdc14-1/pSRS2 cells remained blocked in a pre-anaphase configuration for the duration of the recovery period (Figure 9A). This delay was abolished in cdc14-1 rad9Δ/pSRS2 transformants, suggesting Srs2p OP was able to prolong DNA damage checkpoint arrest. In the absence of Ulp2p, however, inactivating the checkpoint in the cdc14-1 rad9Δ ulp2Δ/vector strain was insufficient to allow cells to proceed with chromosome segregation (Figure 9A, 9B). Significantly, Srs2p OP demonstrated a remarkable ability to allow ulp2Δ strains to escape this mitotic block, with ∼50% of cdc14-1 rad9Δ ulp2Δ/pSRS2 cells now segregating their chromosomes in a seemingly normal anaphase (Figure 9A, 9B). Thus, Srs2p OP substantially relieves the block to chromosome separation in MMS treated ulp2Δ cells.
Discussion
HR and genome stability in ulp2 mutants
One principal finding of this study is that, even in the absence of exogenous DNA replication stress, spontaneous recombination is increased in ulp2Δ cells. This conclusion is based on two observations. First, by genetic criteria, spontaneous recombination at a genomic location on chromosome XV is elevated in ulp2Δ strains. Second, ulp2Δ mutants also display an increase in the frequency of spontaneous Rad52p DNA repair foci. A similar increase in Rad52p foci has been observed in a number of other SUMO pathway mutants, and has been shown to be largely attributable to a requirement for sumoylation in preventing inappropriate recombination events involving the 2 µm circle, an endogenous plasmid found in most S. cerevisiae strains [65]. Since we have not directly examined the effect of the 2 µm circle on recombination in ulp2 mutants, destabilization of this extrachromosomal element may well contribute to the ulp2Δ increase in Rad52p foci. However, as the 2 µm circle is not required for S. cerevisiae growth, our finding that HR DNA repair becomes essential in ulp2Δ strains strongly suggests that Ulp2p acts to suppress the formation of genomic DNA lesions that must be repaired through recombination. Previous analyses of the SUMO pathway support this possibility. For example, SUMO conjugation-defective ubc9-1 mutants exhibit synthetic growth defects in the absence of HR and, at the non-permissive temperature, accumulate DNA structures that activate Rad53p [17]. Furthermore, as described in the Introduction, ulp1-I615N mutants also show increased HR and require HR for viability; in this case, the requirement for HR was shown to correspond with single-stranded DNA gaps arising during S phase [47]. It is striking that perturbations to Ulp1p and Ulp2p, which appear to target largely distinct sets of SUMO substrates [29], impose such seemingly similar dependencies on HR. Another observation that lends credence to the idea that Ulp2p suppresses recombinogenic DNA lesions is that ulp2Δ mutants greatly induce the formation of Rad52p foci following HU treatment. Such foci are not observed in HU treated WT cells [56], consistent with an underlying replication problem in ulp2Δ mutants that is exacerbated by slowed fork progression.
In analyzing genome stability in ulp2Δ strains, we observed two interesting differences compared to other SUMO pathway mutants. First, whereas our data indicate that Rad6p-dependent PRR is essential in ulp2 mutants, mis-regulation of SUMO conjugation in ulp1-I165N rad18 [47], ubc9-1 rad18 [19], siz1 rad18 [11], pol30-K164R rad18 and pol30-K164R rad6 [5] mutants can actually compensate for defective PRR. One scenario that might account for this difference is if poly-sumoylation of a Ulp2p substrate(s) caused a distinct perturbation to DNA replication that was repaired through PRR-mediated HR. In keeping with this interpretation, we find that blocking poly-SUMO chain formation reduces the accumulation of both spontaneous and MMS-induced HR foci in ulp2Δ mutants.
A second apparent difference concerns the formation of GCRs. In contrast to smt3-331, ubc9-1 and ulp1-333 strains, where spontaneous GCRs are increased, ulp2Δ mutants show reduced GCRs. Formally, Ulp2p could promote GCR formation by stimulating error prone DNA repair. There is precedence for this, as a previous study found that, in the absence of template switch PRR, Siz1p-mediated sumoylation of PCNA was required to form certain types of GCRs [66]. Alternatively, Ulp2p could be required for cells that would give rise to GCRs to recover and propagate efficiently. Our observations with dicentric minichromosomes are consistent with the idea that repair events leading to some GCRs may not be tolerated in ulp2Δ strains. We were able to recover re-arranged dicentrics from ulp2Δ mutants when duplicated CEN sequences were present in a direct repeat configuration. Such deletions can occur through single-strand annealing, an intra-chromosomal form of recombination [67]. In contrast, CEN deletion GCRs were not recovered when the two CENs were oriented as inverted repeats. Recent studies have shown that faulty template switch PRR is frequently involved in initiating deletions between inverted repeats [68], [69]. As discussed below, one possibility is that such recombination events are accompanied by formation of SCJs or other types of chromatid attachments that fail to be resolved in ulp2 cells.
Ulp2p prevents accumulation of HR intermediates
Our results led us to suspect that HR DNA repair, while required for viability, might at the same time be toxic to ulp2 cells, prompting us to examine processing of MMS-induced recombination events. From this analysis, one conclusion is that, similar to Ubc9p, Mms21p, Smc5p/Smc6p, and Sgs1p/Top3p [17], [19], Ulp2p is required to prevent X-shaped DNA structures from accumulating at sites of replication fork stalling/collapse. We also find that, whereas Rad52p foci disappear during MMS recovery in WT cells, the incidence of these foci remains elevated in ulp2Δ strains, suggesting a possible role for Ulp2p in terminating recombination. Determining the molecular basis for how Ulp2p prevents accumulation of HR intermediates, and whether this function is related to or separate from Ulp2p's role in Rad52p foci disassembly, are important future questions.
Based on current information, Ulp2p could be connected to HR through a number of different SUMO substrates. First, Mms21p-mediated sumoylation of unknown substrates, probably in conjunction with Smc5p/Smc6p [22], [70], has been proposed to prevent excessive template switch recombination through PRR [19]. Alternatively, more recent evidence suggests Smc5p/Smc6p may instead act downstream of PRR to facilitate the dissolution of HR intermediates [71]. Second, Sgs1p is sumoylated under conditions when it is active in SCJ dissolution [17], [59], although apparently through an Mms21-independent pathway [17]. Third, Ubc9p/Siz1p-controlled sumoylation of PCNA and recruitment of Srs2p may suppress PRR-independent recombination at replication forks [6], [7], [19]. Fourth, Srs2p has also been shown to be sumoylated, with poly-sumoylation being proposed to trigger Srs2p degradation through the Slx5p/Slx8p pathway [72]. Fifth, a fraction of Rad52p [73]-[75], and other HR proteins [76], are sumoylated in response to MMS, which may be involved in fine-tuning processing of broken DNA. Finally, a growing number of protein-protein interactions within HR foci have been found to be controlled by sumoylation (reviewed in [77]). As part of completion of repair, Ulp2p may catalyze the disassembly of these networks.
As a first step in placing Ulp2p in these pathways, we tested whether mis-regulation of Sgs1p sumoylation was connected to ulp2Δ HR defects. Overproduction of Ulp2p was recently shown to block Sgs1p sumoylation on K621 following MMS treatment [59], and, as we report here, MMS-induced sumoylation of Sgs1p is elevated in the absence of Ulp2p. It is therefore likely that Ulp2p acts as the SUMO deconjugating enzyme for Sgs1p. Despite this, short-circuiting Sgs1p sumoylation using the sgs1-K621R mutation did not reduce Rad52p foci accumulation in ulp2Δ cells, indicating mis-regulation of other Ulp2p substrates is likely to be involved in modulating HR.
HR and the ulp2 recovery defect
The failure of ulp2 mutants to resume cell division following DNA damage is one of the most intriguing aspects of the ulp2 phenotype. The first study to document this phenomenon showed that, following adaptation to a persistent DNA break, only a fraction of ulp2 cells were able to proceed with nuclear division, frequently accompanied by abnormally extended or broken mitotic spindles [41]. Inactivating the DNA damage checkpoint rescued this defect, suggesting a critical role for Ulp2p in re-initiating chromosome segregation following completion of the checkpoint response [41], [48].
While our results are largely in accord with this study, we observed a potentially informative difference in the role of the checkpoint in manifesting the ulp2Δ recovery defect. During MMS recovery, ulp2Δ cells dephosphorylated Rad53p and degraded Pds1p on schedule, suggesting they were competent to silence the checkpoint and initiate anaphase. Despite this, sister chromatids failed to disjoin, resulting in a dramatic failure in chromosome segregation. OP of Srs2p, which antagonizes HR [8]-[10], [63], [64], was able to largely restore chromosome segregation. In addition to modulating nucleo-protein filament assembly, Srs2p has also been shown to be required for full activation of the DNA damage checkpoint and for recovery from DNA damage checkpoint arrest [78], [79]. In our experiments, we observed that Srs2p OP greatly extended DNA damage checkpoint arrest in MMS treated WT cells. Based on the above considerations, this extended arrest could presumably reflect either mis-regulation of the checkpoint pathway, or, by interfering with HR DNA repair, elevated Srs2p could simply prolong normal checkpoint signaling. While the effects of Srs2 OP on checkpoint signaling and HR may be multi-faceted, the key point we wish to emphasize here is that abolishing the DNA damage checkpoint (or the SAC) did not allow ulp2Δ cells to divide more times during recovery from MMS treatment. Furthermore, preventing DNA damage checkpoint arrest in MMS treated ulp2Δ rad9Δ cells was insufficient to relieve the block to chromosome separation; OP of Srs2 was also necessary. In sum, these findings strongly suggest that, following replication fork stalling by MMS, downstream events initiated through HR, rather than checkpoint arrest per se, appear to play a causal role in interfering with chromosome segregation.
A key question concerns how HR could have this effect. Perhaps the simplest idea is that unresolved SCJs block chromatid disjunction. Whether this is a sufficient explanation, however, is unclear. First, in the experiments examining ulp2 adaptation to a persistent, endonuclease-targeted DNA break, both chromatids would be expected to be cut, preventing HR strand exchange [41]. Thus, the only way in which DNA linkages could form between chromosomes in these cells would be if extensive resection during prolonged checkpoint arrest triggered illegitimate recombination events. Second, we show that MMS treated sgs1Δ mutants, which are clearly defective in the dissolution of SCJs [17], [25], [27], do not show as severe a block to chromosome separation as Ulp2p-deficient cells. This is consistent with a recent study that showed, from among a collection of helicase-, nuclease-, and topoisomerase-deficient mutants, only smc5, smc6 and mms21 strains showed chromosome segregation defects after a pulse of MMS delivered in G1 [71]. This suggests a role for Mms21p-mediated sumoylation and the Smc5p/Smc6p complex in resolving SCJs or other types of chromatid linkages outside the Sgs1p/Top3p pathway [71]. Along these lines, it is notable that Ulp2p has been implicated in multiple facets of chromatid separation, including controlling sumoylation of cohesin regulatory proteins [37], [42], condensin [35], [38], and DNA topoisomerase II [36], [40]. Speculatively, following induction of HR, there may be an increased requirement for Ulp2p in the vicinity of DNA lesions, not only to prevent accumulation of HR intermediates, but also to complete replication, to disentangle DNA or to release protein-based forms of cohesion. Given the dramatic way in which the absence of Ulp2p potentiates the ability of replication toxins to block cell proliferation, a further understanding of the ulp2 recovery defect could lead to insights that are relevant to cancer treatment.
Materials and Methods
Yeast strains and culture
All S. cerevisiae strains used in this study were derived from the W303-related CRY1 strain and are listed in Table S1. A description of how different genetic elements were introduced into the CRY1 background can be found in Text S1. For all experiments, cells were cultured in standard formulations of yeast extract/peptone/dextrose (YPD) and synthetic complete minimal (SC) media. For G1 synchronization, alpha factor (Bio-Synthesis Corp.) was used at 10 µg/ml. For arresting cells in G2/M, nocodazole (Sigma-Aldrich) was used at 15 µg/ml in YPD. MMS and HU were purchased from Sigma-Aldrich. 5-FOA was purchased from Biovectra/Fisher and used at 1 mg/ml. G418 was purchased from Mediatech/Fisher and used at 200 µg/ml in YPD.
Recombination frequency
pLAY202 ([50]; provided by A. Bailis, City of Hope National Medical Center, Duarte, CA) was linearized with BstXI and targeted to the HIS3 locus, placing a URA3 marker between partially duplicated HIS3 sequences. pLAY202 integrants were propagated in Ura−/SC media, and, following overnight incubation, cell density was quantified using a hemacytometer. Viable cell counts were determined by plating a defined number of cells onto YPD and counting the resulting colonies. Recombination events were selected by plating a larger number of cells onto His−/SC media, and replica plating colonies that arose onto 5-FOA. Colonies that reverted to a His+, Ura− phenotype were scored as recombinants.
GCR frequency
YAC yWss1572-1 ([53]; provided by D. Koshland, Univ. of California at Berkeley, Berkeley, CA) was modified so that the TRP1 marker on the left arm of the YAC was replaced with kanMX. This was performed by PCR amplifying a trp1Δ::kanMX disruption cassette using the following primers
5′-GCATATAAAAATAGTTCAGGCACTCCGAAATACTTGGTTGGCGTGTTTC
GTCAGCTGAAGCTTCGTACGC (CO354)
5′-TCTGGCGTCAGTCCACCAGCTAACATAAAATGTAAGCTTTCGGGGCGCAT
AGGCCACTAGTGGATCTG (CO355)
and pFA6a/kanMX2 [80] as template. G418Res, Trp− transformants were analyzed by PCR to verify correct targeting. The resulting YAC, named yWss1572Δtrp1, was subsequently transferred between strains using cytotransduction [81] or standard genetic crosses. To isolate GCRs, strains containing yWss1572Δtrp1 were grow in Ura−/SC media at 30°C for WT, ulp2Δ, ubc9-1 and smt3-331 strains, and 34°C for ulp1-333 mutants; these represent semi-permissive temperatures for the ubc9-1, smt3-331 and ulp1-333 alleles. Cell densities were quantified using a hemacytometer, and dilutions of the cultures were plated onto YPD to monitor plating efficiency. Aliquots of 105, 106, 107 and 108 cells were plated on 5-FOA to select for loss of the URA3 marker on the YAC. Colonies arising on 5-FOA were replica plated to YPD/G418 and Ade−/SC media. Clones growing on 5-FOA and YPD/G418, but not on Ade−/SC (G418Res, 5-FOASen, Ade−) were considered to arise from GCRs deleting the right arm of the YAC. In contrast, clones that were able to grow on 5-FOA, but could not grow on YPD/G418 or Ade−/SC (G418Sen, 5-FOASen, Ade−) were considered to arise through YAC mis-segregation events. For each culture, the total number of GCR clones arising on all the assay plates was used to calculate GCR frequency.
Minichromosome loss
To monitor the mitotic stability of dicentric minichromosomes, p2XCENdirect (pJBN152; a YRp14-derived minichromosome containing two copies of a 1.7 kb CEN1 DNA fragment in a direct repeat configuration, see Text S1) and p2XCENinvert (pJBN151; similar to pJBN152 but with the CEN1 duplication oriented as an inverted repeat) were transformed into WT and ulp2Δ strains and compared to p1XCEN (YRp14/CEN1) controls. Transformants were inoculated into parallel YPD and Ura−/SC cultures and incubated at 30°C. After ∼15 hr of outgrowth, appropriate dilutions were plated onto YPD and Ura−/SC media. Mitotic stability was calculated by dividing the number of Ura+ colonies by the total number of colonies obtained on YPD.
Microscopy and flow cytometry
Cultures for microscopy were supplemented with 50 µg/ml adenine to quench auto-fluorescence. To visualize Rad52p-GFP and TRP1-GFP, cells were fixed in 1% formaldehyde for 1.5 min and washed into PBS. DAPI staining was performed using Vecta-Shield (Vector Laboratories) containing 10 µg/ml DAPI. TUB1-GFP and HHF2-YFP strains were visualized as live mounts. HHF2-YFP is typically propagated as a heterogyzous diploid (HHF2-YFP-HIS3/+) to minimize selective pressure for rearranged variants that lose the fluorescent marker. However, in order to compare the response of HHF2-YFP strains to MMS concentrations similar to those used in our other recovery experiments, we chose to examine HHF2-YFP haploid segregants that were generated on an experiment-by-experiment basis. This proved to allow propagation of haploid strains with robust Hhf2-YFP fluorescence. In all cases, cells were visualized on Nikon E-800 or Nikon Eclipse 80i microscopes equipped with florescence optics and 100X (1.4 NA) or 60X (1.4 NA) objectives. Rad52p-GFP foci were typically scored using a number 4 neutral density filter to minimize photobleaching. A Zeiss Axioskop 40 microscope equipped with a 25 µm diameter optical fiber dissection needle was used to micromanipulate yeast cells for microcolony analysis. FACS analysis was performed by staining ethanol fixed yeast cells with propidium iodide as previously described [82].
Pulse field gel electrophoresis
10 ml aliquots of OD600 0.8 cultures were harvested by centrifugation and concentrated into 400 µl cell suspension buffer (10 mM Tris, 20 mM NaCl, 50 mM EDTA, pH 7.2). The cell suspension was warmed to 55°C and mixed with 400 µl 2% low melting temperature agarose (SeaKem) dissolved in TBE gel electrophoresis buffer (kept molten at 55°C) containing lyticase (Sigma L4025; final concentration 1 mg/ml). The cell suspension was transferred into molds and allowed to solidify to form plugs (4°C, 15 min). Plugs were pushed out into 50 ml conical tubes and incubated with 5 ml 1 mg/ml lyticase dissolved in 10 mM Tris, 50 mM EDTA, pH 7.2 for one hr at 37°C, followed by treatment 1 mg/ml Proteinase K (Sigma) dissolved in 100 mM EDTA, 0.2% Na Deoxycholate, 1% Na lauryl sarcosine, pH 8.0 at 50°C overnight. Plugs were washed (20 mM Tris, 50 mM EDTA, pH 8.0) 4 times one hour each and stored in wash buffer. Prior to electrophoresis, plugs were placed on a glass plate and trimmed to fit electrophoresis wells. Samples were then fractionated on 1% agarose gels in TBE using a Bio-Rad CHEF-DR II pulsed field electrophoresis system at 6V/cm for 22 hrs with a switch ramp time ramped from 50 to 90 sec at 14°C. Gels were stained with ethidium bromide (0.5 µg/ml, 15 min) prior to photography.
Two-dimensional gel analysis
Genomic DNA preparations and two-dimensional gel electrophoresis were performed according to detailed online methods available from the Brewer-Raghuraman laboratory:
(http://fangman-brewer.genetics.washington.edu/DNA_prep.html)
(http://fangman-brewer.genetics.washington.edu/2Dgel.html)
In brief, cells were grown in 500 ml YPD until the cultures reached an OD600 of 0.6. The cultures were synchronized in nocodazole for 2 hr, washed, and released into fresh YPD containing 0.033% MMS. After a 3 hr treatment, cells were harvested by centrifugation and stored in 5 ml of NIB buffer (17% glycerol, 50 mM MPOS free acid, 150 mM potassium acetate, 2 mM magnesium chloride, 150 µM spermine and 500 µM spermidine, pH 7.2). Cells were lysed by bead beating in NIB buffer, and genomic DNA was purified on cesium chloride density gradients. The resulting DNA samples were digested with HindIII and EcoRV. For first dimension separation, ∼30 µg of digested DNA was loaded onto 0.35% agarose gels and fractionated at 22 volts for 42–48 hr at room temperature. Gel slices containing DNA in the 3–10 kb range were excised and positioned onto a 0.95% agarose gel. Electrophoresis in the second dimension was performed at 4°C at 80 volts for 17 hr at room temperature and 130 volts for another 1.5 hr. Following transfer to nylon membranes (Hybond-XL, GE Healthcare), samples were hybridized with a 280 bp ARS305 DNA fragment PCR amplified from genomic DNA using the following primers:
5′-CTCCGTTTTTAGCCCCCCGTG-
5′-GATTGAGGCCACAGCAAGACCG
The PCR product was radio-labeled (Megaprime DNA labeling system, GE Healthcare) and hybridized using Southern blot procedures as previously described [83].
Protein techniques
Protein extracts were prepared by mechanical beakage of cells in 20% TCA as previously described [36]. 6% SDS-PAGE gels were used to fractionate samples for analysis of Sgs1p-myc and Pds1p-myc, while 12% SDS-PAGE gels (acrylamide: bis = 30:0.39) were used to analyze phosphorylated species of Rad53p. α-myc (9E10, 1:1000, Covance), α-Rad53p (SC-6749, 1∶2000, Santa Cruz), and HRP conjugated secondary (1∶25,000; Jackson ImmunoResearch) antibodies were used for immunoblotting.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BroomfieldS
HryciwT
XiaoW
2001 DNA postreplication repair and mutagenesis in Saccharomyces cerevisiae. Mutat Res 486 167 184
2. San FilippoJ
SungP
KleinH
2008 Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77 229 257
3. BranzeiD
FoianiM
2010 Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11 208 219
4. JohnsonES
2004 Protein modification by SUMO. Annu Rev Biochem 73 355 382
5. HoegeC
PfanderB
MoldovanGL
PyrowolakisG
JentschS
2002 RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419 135 141
6. PfanderB
MoldovanGL
SacherM
HoegeC
JentschS
2005 SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436 428 433
7. PapouliE
ChenS
DaviesAA
HuttnerD
KrejciL
2005 Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19 123 133
8. KrejciL
Van KomenS
LiY
VillemainJ
ReddyMS
2003 DNA helicase Srs2 disrupts the Rad51 presynaptic filament. Nature 423 305 309
9. VeauteX
JeussetJ
SoustelleC
KowalczykowskiSC
Le CamE
2003 The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423 309 312
10. AntonyE
TomkoEJ
XiaoQ
KrejciL
LohmanTM
2009 Srs2 disassembles Rad51 filaments by a protein-protein interaction triggering ATP turnover and dissociation of Rad51 from DNA. Mol Cell 35 105 115
11. StelterP
UlrichHD
2003 Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425 188 191
12. HaracskaL
Torres-RamosCA
JohnsonRE
PrakashS
PrakashL
2004 Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol Cell Biol 24 4267 4274
13. KannouchePL
WingJ
LehmannAR
2004 Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14 491 500
14. MaedaD
SekiM
OnodaF
BranzeiD
KawabeY
2004 Ubc9 is required for damage-tolerance and damage-induced interchromosomal homologous recombination in S. cerevisiae. DNA Repair (Amst) 3 335 341
15. AndrewsEA
PalecekJ
SergeantJ
TaylorE
LehmannAR
2005 Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol Cell Biol 25 185 196
16. ZhaoX
BlobelG
2005 A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci U S A 102 4777 4782
17. BranzeiD
SollierJ
LiberiG
ZhaoX
MaedaD
2006 Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 127 509 522
18. van WaardenburgRC
DudaDM
LancasterCS
SchulmanBA
BjornstiMA
2006 Distinct functional domains of Ubc9 dictate cell survival and resistance to genotoxic stress. Mol Cell Biol 26 4958 4969
19. BranzeiD
VanoliF
FoianiM
2008 SUMOylation regulates Rad18-mediated template switch. Nature 456 915 920
20. OnodaF
TakedaM
SekiM
MaedaD
TajimaJ
2004 SMC6 is required for MMS-induced interchromosomal and sister chromatid recombinations in Saccharomyces cerevisiae. DNA Repair (Amst) 3 429 439
21. Torres-RosellJ
MachinF
FarmerS
JarmuzA
EydmannT
2005 SMC5 and SMC6 genes are required for the segregation of repetitive chromosome regions. Nat Cell Biol 7 412 419
22. AmpatzidouE
IrmischA
O'ConnellMJ
MurrayJM
2006 Smc5/6 is required for repair at collapsed replication forks. Mol Cell Biol 26 9387 9401
23. SollierJ
DriscollR
CastellucciF
FoianiM
JacksonSP
2009 The Saccharomyces cerevisiae Esc2 and Smc5-6 proteins promote sister chromatid junction-mediated intra-S repair. Mol Biol Cell 20 1671 1682
24. KarowJK
ConstantinouA
LiJL
WestSC
HicksonID
2000 The Bloom's syndrome gene product promotes branch migration of holliday junctions. Proc Natl Acad Sci U S A 97 6504 6508
25. LiberiG
MaffiolettiG
LuccaC
ChioloI
BaryshnikovaA
2005 Rad51-dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19 339 350
26. PlankJL
WuJ
HsiehTS
2006 Topoisomerase IIIalpha and Bloom's helicase can resolve a mobile double Holliday junction substrate through convergent branch migration. Proc Natl Acad Sci U S A 103 11118 11123
27. MankouriHW
NgoHP
HicksonID
2007 Shu proteins promote the formation of homologous recombination intermediates that are processed by Sgs1-Rmi1-Top3. Mol Biol Cell 18 4062 4073
28. LiSJ
HochstrasserM
1999 A new protease required for cell-cycle progression in yeast. Nature 398 246 251
29. LiSJ
HochstrasserM
2000 The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin-like Smt3 protein. Mol Cell Biol 20 2367 2377
30. SchwienhorstI
JohnsonES
DohmenRJ
2000 SUMO conjugation and deconjugation. Mol Gen Genet 263 771 786
31. LiSJ
HochstrasserM
2003 The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J Cell Biol 160 1069 1081
32. PanseVG
KusterB
GerstbergerT
HurtE
2003 Unconventional tethering of Ulp1 to the transport channel of the nuclear pore complex by karyopherins. Nat Cell Biol 5 21 27
33. KroetzMB
SuD
HochstrasserM
2009 Essential role of nuclear localization for yeast Ulp2 SUMO protease function. Mol Biol Cell 20 2196 2206
34. MeluhPB
KoshlandD
1995 Evidence that the MIF2 gene of Saccharomyces cerevisiae encodes a centromere protein with homology to the mammalian centromere protein CENP-C. Mol Biol Cell 6 793 807
35. StrunnikovAV
AravindL
KooninEV
2001 Saccharomyces cerevisiae SMT4 encodes an evolutionarily conserved protease with a role in chromosome condensation regulation. Genetics 158 95 107
36. BachantJ
AlcasabasA
BlatY
KlecknerN
ElledgeSJ
2002 The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol Cell 9 1169 1182
37. SteadK
AguilarC
HartmanT
DrexelM
MeluhP
2003 Pds5p regulates the maintenance of sister chromatid cohesion and is sumoylated to promote the dissolution of cohesion. J Cell Biol 163 729 741
38. D'AmoursD
StegmeierF
AmonA
2004 Cdc14 and condensin control the dissolution of cohesin-independent chromosome linkages at repeated DNA. Cell 117 455 469
39. BachantJ
JessenSR
KavanaughSE
FieldingCS
2005 The yeast S phase checkpoint enables replicating chromosomes to bi-orient and restrain spindle extension during S phase distress. J Cell Biol 168 999 1012
40. TakahashiY
Yong-GonzalezV
KikuchiY
StrunnikovA
2006 SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II. Genetics 172 783 794
41. SchwartzDC
FelberbaumR
HochstrasserM
2007 The Ulp2 SUMO protease is required for cell division following termination of the DNA damage checkpoint. Mol Cell Biol 27 6948 6961
42. BaldwinML
JuliusJA
TangX
WangY
BachantJ
2009 The yeast SUMO isopeptidase Smt4/Ulp2 and the polo kinase Cdc5 act in an opposing fashion to regulate sumoylation in mitosis and cohesion at centromeres. Cell Cycle 8 3406 3419
43. BylebylGR
BelichenkoI
JohnsonES
2003 The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J Biol Chem 278 44113 44120
44. UzunovaK
GottscheK
MitevaM
WeisshaarSR
GlanemannC
2007 Ubiquitin-dependent proteolytic control of SUMO conjugates. J Biol Chem 282 34167 34175
45. TathamMH
GeoffroyMC
ShenL
PlechanovovaA
HattersleyN
2008 RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat Cell Biol 10 538 546
46. MullenJR
BrillSJ
2008 Activation of the Slx5-Slx8 ubiquitin ligase by poly-small ubiquitin-like modifier conjugates. J Biol Chem 283 19912 19921
47. SoustelleC
VernisL
FreonK
Reynaud-AngelinA
ChanetR
2004 A new Saccharomyces cerevisiae strain with a mutant Smt3-deconjugating Ulp1 protein is affected in DNA replication and requires Srs2 and homologous recombination for its viability. Mol Cell Biol 24 5130 5143
48. FelberbaumR
HochstrasserM
2008 Ulp2 and the DNA damage response: desumoylation enables safe passage through mitosis. Cell Cycle 7 52 56
49. LisbyM
RothsteinR
MortensenUH
2001 Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci U S A 98 8276 8282
50. MainesS
NegrittoMC
WuX
MantheyGM
BailisAM
1998 Novel mutations in the RAD3 and SSL1 genes perturb genome stability by stimulating recombination between short repeats in Saccharomyces cerevisiae. Genetics 150 963 976
51. LambertS
WatsonA
SheedyDM
MartinB
CarrAM
2005 Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell 121 689 702
52. LemoineFJ
DegtyarevaNP
KokoskaRJ
PetesTD
2008 Reduced levels of DNA polymerase delta induce chromosome fragile site instability in yeast. Mol Cell Biol 28 5359 5368
53. HuangD
KoshlandD
2003 Chromosome integrity in Saccharomyces cerevisiae: the interplay of DNA replication initiation factors, elongation factors, and origins. Genes Dev 17 1741 1754
54. MannC
DavisRW
1983 Instability of dicentric plasmids in yeast. Proc Natl Acad Sci U S A 80 228 232
55. KoshlandD
RutledgeL
Fitzgerald-HayesM
HartwellLH
1987 A genetic analysis of dicentric minichromosomes in Saccharomyces cerevisiae. Cell 48 801 812
56. BarlowJH
RothsteinR
2009 Rad52 recruitment is DNA replication independent and regulated by Cdc28 and the Mec1 kinase. Embo J 28 1121 1130
57. ZouH
RothsteinR
1997 Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell 90 87 96
58. LopesM
Cotta-RamusinoC
LiberiG
FoianiM
2003 Branch migrating sister chromatid junctions form at replication origins through Rad51/Rad52-independent mechanisms. Mol Cell 12 1499 1510
59. LuCY
TsaiCH
BrillSJ
TengSC
2010 Sumoylation of the BLM ortholog, Sgs1, promotes telomere-telomere recombination in budding yeast. Nucleic Acids Res 38 488 498
60. SanchezY
DesanyBA
JonesWJ
LiuQ
WangB
1996 Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271 357 360
61. Cohen-FixO
PetersJM
KirschnerMW
KoshlandD
1996 Anaphase initiation in Saccharomyces cerevisiae is controlled by the APC-dependent degradation of the anaphase inhibitor Pds1p. Genes Dev 10 3081 3093
62. Cohen-FixO
KoshlandD
1997 The anaphase inhibitor of Saccharomyces cerevisiae Pds1p is a target of the DNA damage checkpoint pathway. Proc Natl Acad Sci U S A 94 14361 14366
63. DupaigneP
Le BretonC
FabreF
GangloffS
Le CamE
2008 The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol Cell 29 243 254
64. PrakashR
SatoryD
DrayE
PapushaA
SchellerJ
2009 Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23 67 79
65. XiongL
ChenXL
SilverHR
AhmedNT
JohnsonES
2009 Deficient SUMO attachment to Flp recombinase leads to homologous recombination-dependent hyperamplification of the yeast 2 microm circle plasmid. Mol Biol Cell 20 1241 1251
66. MotegiA
KuntzK
MajeedA
SmithS
MyungK
2006 Regulation of gross chromosomal rearrangements by ubiquitin and SUMO ligases in Saccharomyces cerevisiae. Mol Cell Biol 26 1424 1433
67. Fishman-LobellJ
RudinN
HaberJE
1992 Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12 1292 1303
68. PaekAL
KaocharS
JonesH
ElezabyA
ShanksL
2009 Fusion of nearby inverted repeats by a replication-based mechanism leads to formation of dicentric and acentric chromosomes that cause genome instability in budding yeast. Genes Dev 23 2861 2875
69. MizunoK
LambertS
BaldacciG
MurrayJM
CarrAM
2009 Nearby inverted repeats fuse to generate acentric and dicentric palindromic chromosomes by a replication template exchange mechanism. Genes Dev 23 2876 2886
70. IrmischA
AmpatzidouE
MizunoK
O'ConnellMJ
MurrayJM
2009 Smc5/6 maintains stalled replication forks in a recombination-competent conformation. Embo J 28 144 155
71. Bermudez-LopezM
CeschiaA
de PiccoliG
ColominaN
PaseroP
2010 The Smc5/6 complex is required for dissolution of DNA-mediated sister chromatid linkages. Nucleic Acids Res
72. SaponaroM
CallahanD
ZhengX
KrejciL
HaberJE
2010 Cdk1 targets Srs2 to complete synthesis-dependent strand annealing and to promote recombinational repair. PLoS Genet 6 e1000858 doi:10.1371/journal.pgen.1000858
73. SacherM
PfanderB
HoegeC
JentschS
2006 Control of Rad52 recombination activity by double-strand break-induced SUMO modification. Nat Cell Biol 8 1284 1290
74. Torres-RosellJ
SunjevaricI
De PiccoliG
SacherM
Eckert-BouletN
2007 The Smc5-Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9 923 931
75. OhuchiT
SekiM
BranzeiD
MaedaD
UiA
2008 Rad52 sumoylation and its involvement in the efficient induction of homologous recombination. DNA Repair (Amst) 7 879 889
76. BurgessRC
RahmanS
LisbyM
RothsteinR
ZhaoX
2007 The Slx5-Slx8 complex affects sumoylation of DNA repair proteins and negatively regulates recombination. Mol Cell Biol 27 6153 6162
77. BerginkS
JentschS
2009 Principles of ubiquitin and SUMO modifications in DNA repair. Nature 458 461 467
78. LiberiG
ChioloI
PellicioliA
LopesM
PlevaniP
2000 Srs2 DNA helicase is involved in checkpoint response and its regulation requires a functional Mec1-dependent pathway and Cdk1 activity. Embo J 19 5027 5038
79. VazeMB
PellicioliA
LeeSE
IraG
LiberiG
2002 Recovery from checkpoint-mediated arrest after repair of a double-strand break requires Srs2 helicase. Mol Cell 10 373 385
80. WachA
BrachatA
PohlmannR
PhilippsenP
1994 New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 10 1793 1808
81. SpencerF
HugeratY
SimchenG
HurkoO
ConnellyC
1994 Yeast kar1 mutants provide an effective method for YAC transfer to new hosts. Genomics 22 118 126
82. Schober-DitmoreW
BachantJ
2000 Yeast DNA flow cytometry.
DiamondR
DeMaggioS
In Living Color: Protocols in Flow Cytometry and Cell Sorting New York Springer Lab Manuals 455 460
83. WarsiTH
NavarroMS
BachantJ
2008 DNA topoisomerase II is a determinant of the tensile properties of yeast centromeric chromatin and the tension checkpoint. Mol Biol Cell 19 4421 4433
84. ChristiansonTW
SikorskiRS
DanteM
SheroJH
HieterP
1992 Multifunctional yeast high-copy-number shuttle vectors. Gene 110 119 122
85. MankouriHW
CraigTJ
MorganA
2002 SGS1 is a multicopy suppressor of srs2: functional overlap between DNA helicases. Nucleic Acids Res 30 1103 1113
86. DesanyBA
AlcasabasAA
BachantJB
ElledgeSJ
1998 Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev 12 2956 2970
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 3
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- Whole-Exome Re-Sequencing in a Family Quartet Identifies Mutations As the Cause of a Novel Skeletal Dysplasia
- Origin-Dependent Inverted-Repeat Amplification: A Replication-Based Model for Generating Palindromic Amplicons
- FUS Transgenic Rats Develop the Phenotypes of Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration
- Limited dCTP Availability Accounts for Mitochondrial DNA Depletion in Mitochondrial Neurogastrointestinal Encephalomyopathy (MNGIE)