
Membrane Recognition and Dynamics of the RNA Degradosome
Recent discoveries that two ribonucleases with major roles in mRNA degradation, RNase E of Escherichia coli and RNase Y of Bacillus subtilis, are localized to the inner cytoplasmic membrane suggest that spatial separation of transcription and mRNA degradation are general features of the bacterial cell. Here we show that RNase E rapidly diffuses over the entire inner membrane forming short-lived foci. Results of molecular dynamics simulations lead us to suggest that RNase E interacts with the lipid membrane by a novel mechanism permitting a high degree of translational freedom. We show that RNA substrate is necessary for the formation of RNase E foci and that formation of foci correlates with constraints on the diffusion of RNase E. We therefore propose that foci are degradation bodies in which several RNase E molecules engage an RNA substrate. The sequestration of the mRNA degradation machinery to the inner cytoplasmic membrane has important consequences for mRNA turnover. This organization likely favors formation of polyribosomes on nascent transcripts before they are exposed to the degradation machinery. Rapid diffusion of RNase E on the inner cytoplasmic membrane could be part of a scanning mechanism that facilitates recognition of cytoplasmic polyribosomes and cooperative degradation of mRNA.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004961
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004961
Summary
Recent discoveries that two ribonucleases with major roles in mRNA degradation, RNase E of Escherichia coli and RNase Y of Bacillus subtilis, are localized to the inner cytoplasmic membrane suggest that spatial separation of transcription and mRNA degradation are general features of the bacterial cell. Here we show that RNase E rapidly diffuses over the entire inner membrane forming short-lived foci. Results of molecular dynamics simulations lead us to suggest that RNase E interacts with the lipid membrane by a novel mechanism permitting a high degree of translational freedom. We show that RNA substrate is necessary for the formation of RNase E foci and that formation of foci correlates with constraints on the diffusion of RNase E. We therefore propose that foci are degradation bodies in which several RNase E molecules engage an RNA substrate. The sequestration of the mRNA degradation machinery to the inner cytoplasmic membrane has important consequences for mRNA turnover. This organization likely favors formation of polyribosomes on nascent transcripts before they are exposed to the degradation machinery. Rapid diffusion of RNase E on the inner cytoplasmic membrane could be part of a scanning mechanism that facilitates recognition of cytoplasmic polyribosomes and cooperative degradation of mRNA.
Introduction
In Escherichia coli, Salmonella, and many other bacteria, RNase E makes critical contributions to general and regulated mRNA degradation [1, 2]. General mRNA degradation is the default turnover pathway, whereas regulated mRNA degradation is controlled by factors such as sRNA (small RNA) and the RNA binding protein Hfq [3, 4]. RNase E contains a large noncatalytic region that is the scaffold for the assembly of a multienzyme complex known as the RNA degradosome [5]. Recently, RNase E was shown to be localized to the inner cytoplasmic membrane by tagging with fluorescent protein [6, 7], a finding that has been corroborated for the native enzyme as well as other RNA degradosome components by proteomic analyses of the inner membrane [8, 9]. The association of RNase E with the membrane benefits organism fitness as indicated by the slow growth of strains bearing deletions or point mutations that disrupt membrane binding [6], so the interaction is likely to be functionally important. It has been postulated that membrane association physically separates sites of transcription from sites of mRNA degradation and thereby confers a time delay before the onset of decay of a transcript [10]. The general importance of the localization of RNase E has been underscored by the recent finding that RNase Y, a key ribonuclease of mRNA degradation in Bacillus subtilis, is also membrane-localized [11]. What makes this parallel especially striking is that RNase E and RNase Y share no common evolutionary ancestor and their functional analogy therefore arose through convergent evolution.
The basis for the interaction of RNase E with a phospholipid bilayer was established by the identification of a 15-residue MTS in the noncatalytic region that is necessary and sufficient for membrane localization [6]. The MTS, with the propensity to form an amphipathic α-helix, bears signature features conserved in RNase E homologs throughout the γ-Proteobacteria including the clustering of bulky aromatic residues on one face of the helix, small hydrophilic residues on the opposite face and the presence of basic residues flanking the hydrophobic core. Mutation of signature residues of the MTS of E. coli RNase E confirmed their importance for membrane localization in vivo and for interaction with protein-free phospholipid vesicles in vitro [6]. To elucidate the structural basis for RNase E recognition of the cytoplasmic membrane, we have undertaken molecular dynamics simulations with a realistic model of the E. coli inner membrane and the MTS peptide, and we have performed binding studies using a fluorescein-labelled derivative of this peptide. These analyses shed light on the geometry, energetics and dynamics of the interaction of the MTS with the lipid bilayer.
Recent reports suggest that RNase E and RhlB, which is a DEAD-box RNA helicase component of the RNA degradosome, form a membrane-associated cytoskeletal-like structure, and that RhlB localizes to the cytoskeleton independently of RNase E [7, 12, 13]. The question therefore arises how RNase E and RhlB interact in vivo, and to what degree RNase E and RhlB are free to diffuse on the inner cytoplasmic membrane. To address this question, we have conducted microscopy studies in live cells in which RNase E and RhlB were tagged with fluorescent protein. Under live cell conditions, the association of RhlB with the membrane depends on its interaction with RNase E. In addition, we observe that RNase E rapidly diffuses on the inner cytoplasmic membrane forming transient foci. The likely impact of the membrane dynamics of RNase E on its access to RNA substrates and the coordinated activities of the degradosome will be discussed.
Results
Recruitment of RhlB to the inner cytoplasmic membrane
Although there is evidence that the DEAD-box RNA helicase RhlB associates with RNase E through a direct protein-to-protein interaction [5, 14, 15], recent reports have suggested that RhlB by itself can form a membrane-associated cytoskeletal-like structure [12, 13]. We therefore explored the structural requirement for the localization of RhlB to the inner cytoplasmic membrane by constructing strains in which RNase E and RhlB were tagged at their C-terminal ends with mCherry and CFP (Cyan Fluorescence Protein), respectively. These constructs are functional single copy chromosome replacements in the NCM3416 background, which is a wild type E. coli K12 strain [16]. Additional constructs contain variants of RNase E-mCherry in which the MTS, protein Scaffold (Sca) or HBS (Helicase Binding Site) were deleted based on previous work mapping these sites [17].
Fig. 1 presents a gallery of micrographs showing images of strains expressing RhlB-CFP and RNase E-mCherry. In Fig. 1, a few cells were chosen from a large field (S1 Fig.). Cultures were grown to mid logarithmic phase in MOPS-glycerol-amino acids media at 30°C. Similar results were obtained in LB media and at 37°C. In the wild type strain (top panel), RNase E and RhlB are enriched in foci at the periphery of the cell. RNase E and RhlB do not co-localize in these images, which were made with a 4 s exposure time due to the weak RhlB-CFP fluorescence signal. The apparent lack of co-localization is likely due to rapid movement of RNase E under the live cell conditions used in these experiments (S2 Fig. and results below). In the second panel, RNase E ∆MTS is a variant in which the MTS has been deleted. Both RNase E and RhlB are delocalized from the periphery and the signal is cytoplasmic and diffuse. These results demonstrate that the membrane localization of RhlB depends on the MTS of RNase E. In the third panel, RNase E ∆Sca is a variant with a deletion of the scaffold, which interacts with RhlB, enolase and PNPase. Like the wild type protein, RNase E ∆Sca is localized in foci at the periphery of the cell, whereas RhlB is cytoplasmic and diffuse. These results demonstrate that the membrane localization of RhlB requires an interaction with the scaffold region of RNase E, either directly or indirectly via an interaction with another component of the RNA degradosome. In the fourth panel, RNase E ∆HBS is a variant of RNase E in which the binding site for RhlB has been deleted. RhlB is localized to the cell interior as in the RNase E ∆Sca construct. These results demonstrate that the localization of RhlB to the membrane depends on a direct protein-to-protein interaction with RNase E. Due to the limit of resolution of light microscopy, we cannot exclude the possibility that some molecules of RhlB-CFP remain membrane associated in the RNase E ∆Sca and ∆HBS constructs. We nevertheless conclude that most if not all of the membrane localization of RhlB-CFP depends on a protein interaction with RNase E and that RhlB by itself is not a membrane binding protein. Throughout the construction and imaging of these strains, as well as imaging in other strain backgrounds [6], we have not observed cytoskeletal-like structures that were reported in other studies [7, 12, 13].

Molecular dynamics simulations of the interaction of the MTS with a phospholipid bilayer
To better understand the molecular basis for the interaction of the MTS with the phospholipid bilayer, and to determine what constraints, if any, this type of membrane association has on the diffusion of RNase E, we performed coarse-grain molecular dynamics simulations. We used a membrane model with realistic E. coli lipid composition, namely cardiolipin (CARD), dipalmitoylphosphatidylglycerol (PG) and dipalmitoylphosphatidylethanolamine (PE) in a ratio of approximately 1:2:6, respectively [18, 19]. We used a 21-residue sequence corresponding to the residues 565–585 of RNase E, which includes the 15-residue core of the MTS (Fig. 2A). This peptide was used in previous biophysical measurements of the interaction of the MTS with a phospholipid bilayer made with purified E. coli lipids [6]. In the coarse-grain simulation, the peptide starts in bulk solution in a helical conformation, and with time randomly encounters the phospholipid bilayer, whereupon it adheres to the membrane surface and then penetrates into the acyl interior (Fig. 2B). A molecular graphics movie of the simulation is provided (S1 Video). The simulation shows that the MTS inserts in the membrane with the helical axis oriented parallel to the bilayer plane and hydrophobic side chains buried deep into the acyl core of the lipids. The peptide interacts with approximately 50 phospholipids (S3 Fig.), in reasonable agreement with previous calorimetry measurements in which the binding isotherm was fitted to an interaction with approximately 40 phospholipids [6]. The helical conformation of the MTS was retained throughout the coarse-grain simulation. Atomistic simulations of the MTS without any restraints on the secondary structure revealed that the MTS is a stable helix in the environment of the phospholipid bilayer. This result is consistent with previous circular dichroism measurements showing that the peptide has greater propensity to form an α-helical conformation upon interaction with a phospholipid bilayer composed of E. coli lipids [6].

Molecular dynamics simulations were also undertaken with MTS variants that were previously studied experimentally [6]. These are a double substitution of phenylalanine with alanine (F574A/F575A); a substitution of phenylalanine with glutamic acid (F575E); and a proline insertion between residues F574 and F575. In the wild type peptide, the hydrophobic amino acid side chains penetrate into the acyl interior of the lipid bilayer, the small hydrophilic and basic amino acid side chains interact at the level of the glycerol moieties (Fig. 2C). The 3-residue N- and C-terminal extensions flanking the MTS interact at the level of the phosphate and ethanolamine moieties. Estimates of the energy of interaction (S4 Fig.) correlate with the depth of insertion of the hydrophobic amino acids into the membrane. In the F574A/F575A and F575E variants, the hydrophobic side chains protrude into the aqueous layer and the small hydrophilic and basic residues interact at the level of the ethanolamine moieties (Fig. 2C). In previous calorimetry experiments employing the corresponding peptides, no heat of interaction with lipid vesicles was detected with peptides corresponding to the F574A/F575A and F575E variants [6] suggesting that interaction of the hydrophobic side chains with the acyl core of the lipid bilayer drives the binding reaction. Microscopy of fluorescein-labelled peptides that correspond to the sequences in the molecular dynamics simulations confirms binding in vitro to vesicles made with purified E. coli lipids (Fig. 2D). The degree of fluorescence at the membrane diminishes with the F574A/F575A variant, in accord with the findings from the molecular dynamics simulations and the diminished membrane association of the corresponding RNase E variant in vivo [6]. The simulation with the proline insertion variant shows that it has a more favorable energy of interaction than wild-type (S4 Fig.), probably due to additional contacts with the inserted proline, which is buried deep in the acyl interior of the lipid bilayer (Fig. 2C). The results of the molecular dynamics calculations are in agreement with experimental work showing that full-length RNase E with a proline insertion at this position interacts with phospholipid vesicles in vitro and localizes to the cytoplasmic membrane in vivo [6]. The stability of the helix in the membrane and the estimated free energy explain why the proline insertion does not disrupt interaction with the phospholipid bilayer. The congruence between the previous biophysical measurements and the properties predicted by the molecular dynamics simulation validate the coarse-grain approach used here.
Since membrane composition and curvature can affect protein binding [20, 21], we asked whether these parameters are predicted to affect the interaction of the MTS with the phospholipid bilayer. The molecular dynamics simulations indicate that the MTS exhibits preferential interaction with anionic lipids (CARD and PG) in comparison to Zwitterionic lipids (PE) (S5 Fig.). This result suggests that the basic residues flanking the hydrophobic core of the amphipathic α-helix, which is a conserved feature of the MTS [6], form favorable electrostatic contacts with anionic lipids that help to stabilize the α-helix and/or increase the energy of interaction. Membrane composition could therefore affect the interaction of the MTS with the phospholipid bilayer. The molecular dynamics simulations presented here are based on planar lipid bilayers, but simulations of small lipid vesicles with curved surfaces composed of phosphatidlycholine (PC) or a realistic mixture of E. coli lipids gave similar results suggesting that the interaction of the MTS with the phospholipid bilayer is not sensitive to membrane curvature.
Diffusion can be important for the function of a membrane protein since it affects interactions with substrate and other proteins. In the coarse grain simulation using a 10 µs period, the rate of diffusion for the MTS on the membrane was predicted to be in the order of 103 µm2/s. We know of no other study of a bacterial peripheral membrane protein that can be used for comparison. The rate of diffusion of the membrane anchor of GRP1, which is a eukaryotic peripheral membrane protein, is 330-fold slower than the predicted rate of diffusion of the MTS [22]. The membrane anchor of GRP1 makes a specific high affinity interaction with phosphatidylinositol-3,4,5-trisphosphate (PIP3). Experimental work including molecular dynamics simulations suggests that the rate of diffusion of GRP1 on the membrane is limited by the frictional coefficient of PIP3. The much faster predicted rate of diffusion of the MTS of RNase E suggests a different mechanism of translocation. We propose that the MTS ‘glides’ in the phospholipid bilayer by making a large number of weak contacts that are rapidly broken and reformed. Although RNase E in the cell would likely have a slower rate of diffusion, our results suggest that it would have a high degree of translational freedom in the absence of interactions with other components such as membrane proteins or polyribosomes.
Distribution and movement of RNase E
During the cell imaging work shown in Fig. 1 and previous work with the KSL2000/pVK207 strain, which expresses RNase E-YFP (Yellow Fluorescent Protein) [6], we noticed that the RNase E fluorescence signal in live cells rapidly fluctuated, giving the impression that foci of RNase E circulate on the periphery of the cell. Fixation of the cells with formaldehyde arrests this motion. To better define the distribution of RNase E in the cell, we first examined formaldehyde fixed cells (KSL2000/pVK207) by confocal microscopy (Fig. 3A). Deconvolution of this image shows that RNase E localizes to the periphery of the cell. We then used TIRFm to selectively excite RNase E-YFP in a thin layer adjacent to the coverslip. Fig. 3B shows wide field and TIRF images of two fields of live cells. The diagram in the lower right hand corner of each image indicates the plain of focus. In these images, an exposure time of 100 ms was used to minimize motion during the acquisition, which can result in apparent but artifactual polymer-like structures. No elongated polymeric or helical structures were observed under these high-speed imaging conditions. Rather, randomly distributed clusters of RNase E were observed. S2 Fig. shows two high-speed TIRFm images of live cells taken 4 s apart. An overlay of these images artificially colored red and green shows massive redistribution of RNase E in the 4 s time interval. Using an approach that has been previously used to track MreB movement [23–25], the distribution and directionality of RNase E movement was analyzed in the kymograms shown in Fig. 3C. A single cell was scanned along its long and short axes. Heat maps derived from the intensity of the fluorescent signal were accumulated to construct the kymograms. Inspection of the fixed cell shows that the distribution of RNase E on the membrane is unchanged over a period of 3 seconds, whereas the distribution rapidly fluctuates in live cells. The lack of any clear repeat pattern in the kymograms of the live cell suggests that the movement of RNase E is random; that is, strongly correlated movement should appear as tracks or waves in the kymograms.
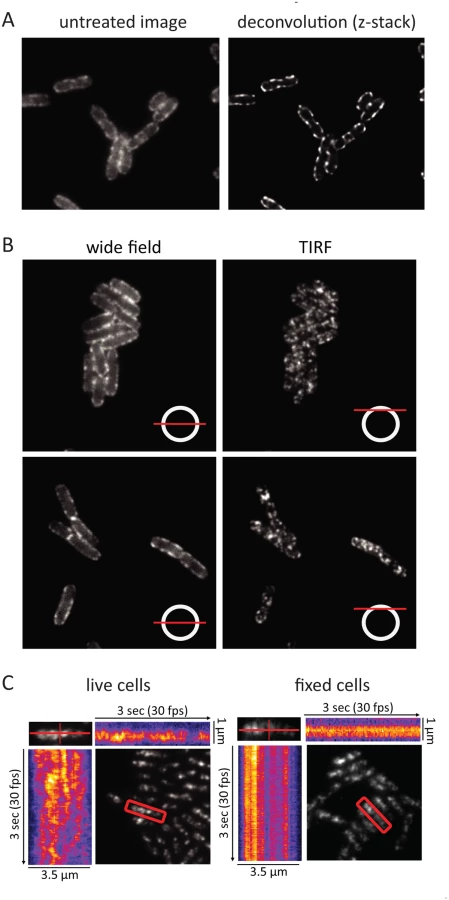
In the KSL2000/pVK207 strain analyzed in Fig. 3, RNase E-YFP was expressed from a low copy number plasmid that complemented a deletion of the gene encoding RNase E on the chromosome [6]. Comparable results were obtained using the Kti164 strain in which RNase E-GFP was expressed from a single copy construct on the chromosome, but the GFP signal is less intense than the YFP signal. Experimental work suggests that this difference is due to the intrinsic relative brightness of the YFP and GFP constructs since the level of RNase E-YFP and RNase E-GFP, as determined by SDS-PAGE and Western blotting is comparable to wild-type RNase E levels ([6] and S6 Fig.). We interpret the results of epifluorescence microscopy, confocal microscopy and TIRFm as evidence for the rapid diffusion of RNase E over the entire inner membrane and the formation of short-lived foci containing multiple molecules of RNase E.
Forces acting on RNase E
We wanted to know if the localization or diffusion of RNase E on the membrane depends on an energy source or forces generated by transcription or translation. We therefore analyzed the distribution of RNase on the membrane after treatment of the cells with carbonyl cyanide m-chlorophenyl hydrazone (CCCP), kanamycin or rifampicin. CCCP collapses the transmembrane proton gradient, while kanamycin and rifampicin are inhibitors of translation and transcription, respectively. Treatment of the cells with CCCP or kanamycin had no discernable effect on RNase E localization making it unlikely that the electrochemical gradient, ATP generation or translation influences the cellular distribution of RNase E (S7 Fig.). In contrast, after treatment with rifampicin the appearance of RNase E on the membrane is different as evidenced by the loss of foci and smooth distribution along the perimeter of the cell (Fig. 4A). The intensity of fluorescence along the membrane was measured by the line scans shown in Fig. 4B providing a method to quantify changes in the distribution of RNase E on the membrane. This analysis was applied to a field of cells to generate plots of average pixel intensity and variance (Fig. 4C). Average pixel intensity is a measure of the level of RNase E in the cell. The plot in Fig. 4C shows that the distribution of average pixel intensity is not affected by rifampicin treatment. This is expected since RNase E is a stable protein and rifampicin treatment is not predicted to change its level. In contrast, the average variance is clearly lower after treatment with rifampicin showing that the smooth distribution of RNase E along the perimeter of the cell (Fig. 4A) is a general property of a population of cells.

To further examine the effect of rifampicin, we used the intrinsic photobleaching that occurs in TIRFm to measure the diffusion of RNase E on the membrane. Briefly, since only a portion of the membrane is excited in TIRFm, the rate of photobleaching is related to the rate of diffusion of the fluorescent protein. If a fluorescent protein diffuses rapidly relative to the intrinsic rate of photobleaching, then the pool of bleachable molecules will be slowly depleted since individual molecules only spend a short time on the surface that is excited. If, on the other hand, diffusion is slow, then the pool of bleachable molecules will be depleted faster since individual molecules will spend a longer time on the surface that is excited. With the appropriate controls, it is possible to estimate a relative rate of diffusion by this technique [21]. Fig. 5A shows snapshots of RNase E distribution by TIRFm before and after treatment with rifampicin, Fig. 5B is a photobleaching time course and Fig. 5C is the quantification of the photobleaching experiment. Rifampicin treatment results in a diffuse distribution of RNase E with few if any intense foci as compared to the untreated control. Remarkably, rifampicin decreases the rate of photobleaching, indicating an increase in the rate of RNase E diffusion. To test if rifampicin treatment has a general effect on the diffusion of membrane proteins, we measured the photobleaching rate of the F1Fo ATP synthase before and after treatment with rifampicin (S8 Fig.). As the rate of diffusion of the F1Fo ATP synthase is not affected, we conclude that rifampicin specifically affects RNase E diffusion.

Requirement of RNA substrate to form RNase E foci
To explore whether RNA substrate is required to form RNase E foci, we exploited the ENS134 strain encoding bacteriophage T7 RNA polymerase [26, 27], which is insensitive to inhibition by rifampicin. In this strain, it is possible to inhibit E. coli RNA polymerase under conditions in which T7 RNA polymerase actively transcribes genes with a bacteriophage T7 promoter. The ENS134 strain has a chromosomal copy of the gene for T7 RNA polymerase under the control of an inducible lac promoter, and a chromosomal copy of the gene encoding lacZ under the control of a bacteriophage T7 promoter (Fig. 6A). The lacZ gene is fused to a tRNA gene followed by a transcription terminator. Transcription by T7 RNA polymerase results in high level synthesis of a lacZ-tRNA transcript. RNase E is necessary for degradation of the lacZ mRNA [27]. Maturation of the tRNA is predicted to require RNase E and RNase P considering the established pathway in E. coli [28]. In order to visualize RNase E in the ENS134 strain, we introduced pVK207, which is the low copy number plasmid encoding the RNase E-YFP fusion used in the work shown in Figs. 3, 4 and 5. Fig. 6B shows epifluorescence images of the ENS134/pVK207 strain. The control panel (-rifampicin, -IPTG) shows that RNase E-YFP distribution is comparable to what we observed in the KLS2000/pVK207 strain. Autoregulation of RNase E expression, which is predicted to down regulate expression of plasmid rne-yfp and chromosomal rne genes, results in a level of total RNase E (RNase E + RNase E-YFP) comparable to the normal RNase E level [6, 29, 30]. Since there are approximately 5 plasmid copies of rne-yfp for each chromosomal copy of rne, RNase E-YFP is the predominant form of RNase E in these cells.

Images in Fig. 6B show that cells treated first with IPTG and then rifampicin have a pattern of peripheral RNase E foci similar to cells that have not been treated with rifampicin (+IPTG, +rifampicin vs. +IPTG, ‑rifampicin). This result suggests that synthesis of the lacZ-tRNA transcript in the presence of rifampicin results in an interaction that stimulates the formation of RNase E foci. The effect of rifampicin on a field of cells was quantified (Fig. 6C). This result shows that changes in distribution of RNase E along the perimeter of the cell is a general property of the population of cells treated with rifampicin. The small reduction in variance after treatment with rifampicin (+IPTG, +rifampicin vs. +IPTG, ‑rifampicin) is likely due to inhibition of E. coli RNA polymerase. From these results we conclude that the effect of rifampicin on formation of RNase E foci is not a secondary consequence of the shut off of E. coli transcription. We next analyzed RNase E-YFP photobleaching. Fig. 6D shows that there is no effect on the rate of diffusion of RNase E if IPTG is added before rifampicin. This result suggests that synthesis of the lacZ-tRNA transcript in the presence of rifampicin results in an interaction that constrains the diffusion of RNase E. Taken together, these results show that there is a direct correlation between formation of foci and constraints on the diffusion of RNase E. Considering this experimental work, we propose that the formation of RNase E foci requires interaction with RNA substrate, that foci formation constrains the diffusion of RNase E, and that rifampicin acts on the foci indirectly by depleting the pool of RNA substrate.
Discussion
Live cell microscopy shows that RNase E is located on the cytoplasmic membrane and that the DEAD-box RNA helicase RhlB is associated with RNase E, but neither protein forms cytoskeletal-like structures as reported earlier [12, 13]. This is the first report in which RhlB has been visualized directly in live cells as previous work employed indirect immunofluorescence. The YFP, GFP and CFP tags used here are A206K variants that have been reported to minimize dimerization [31]. The fusion proteins are present at levels comparable to wild type. We have used agarose pads that do not disturb the physiology of the cell, whereas cells immobilized on glass slides were imaged in the work showing cytoskeletal-like structures. Under the conditions used here, RNase E is highly mobile over the entire surface of the membrane. The movement is spontaneous since it does not appear to be driven by an energy dependent process suggesting that the motion of degradosome assembly is due to continuous buffeting by other macromolecules in the densely packed milieu of the cell including the membrane-associated cell wall synthesis machinery [21]. Recent biophysical studies of live bacterial cells suggest that particle size has a disproportionate influence on diffusion. Small particles including free ribosomes and multienzyme complexes can be treated as components in a liquid-like state whereas larger particles such as polyribosomes are apparently constrained in a solid-like state that requires metabolic activity for ‘mixing’ [32, 33].
Molecular dynamics simulations provide a conceptual framework for visualizing how the MTS anchors RNase E to the inner cytoplasmic membrane by a novel mechanism permitting a high degree of translational freedom. Although the interaction of an individual MTS with the membrane represents a weak force compared to integral membrane proteins with multiple trans-membrane segments, the cumulative effect of having four MTS elements spatially co-localized as a consequence of RNase E tetramerization should have a strong binding effect through chelate cooperativity [34], which is consistent with biochemical work showing that the solubilization and purification of RNase E requires high concentrations of nonionic detergent [35]. Considering previous measurements of the interaction of a peptide corresponding to the MTS with a phospholipid bilayer composed of E. coli lipids (Kd = 1.3 × 10-6 M) [6] and the effect of chelate cooperativity, we believe that RNase E should be fully membrane-bound, which is consistent with the confocal microscope image presented here. Membrane association could aid the organization of the RNase E tetramer and bring the noncatalytic C-terminal region, which interacts with the other components of the RNA degradosome, within a restricted hemisphere in which they may cooperate with the catalytic core of RNase E. The interaction of the MTS with the membrane is predicted to affect the local concentration of lipid species. The spatial co-localization of the four MTS elements in the RNase E tetramer could accentuate a preference for anionic lipids at the site of membrane docking. Cooperativity in the interaction between anionic lipids with basic residues flanking the hydrophobic core of four MTS elements could further increase avidity of RNase E for the membrane.
We have described a highly dynamic distribution on the membrane in which RNase E forms short-lived foci. Analysis of the dynamics of RNase E motion by TIRFm suggests that diffusion on the membrane is random with no indication of correlation with the long or short axis of the cell. This result excludes models in which RNase E moves along ‘tracks’ in the membrane or is constrained by the machinery involved in cell wall synthesis as has recently been described for MreB [23, 24]. Depletion of RNA substrates by inhibition of transcription results in disappearance of foci and an increase in the rate of diffusion of RNase E. Our results suggest that foci are sites of degradation in which several molecules of RNase E as well as other components of the RNA degradosome interact with an RNA substrate. RNase E foci could therefore have a function analogous to eukaryotic P-bodies and stress granules, which are ribonucleoprotein particles containing factors involved in translation inhibition and mRNA degradation [36, 37]. RNase E foci are nevertheless much smaller and much more short-lived than P-bodies or stress granules.
An important activity of RNase E is general and regulated mRNA degradation. An example of regulated mRNA degradation is the rapid response associated with quorum sensing in Vibrio cholera, which involves sRNA, Hfq and RNase E [38]. A caveat to such regulation is that the target mRNA finds its way to RNase E and is actively degraded. Recent work suggests that RNase E can interact with polyribosomes and sRNA/Hfq complexes [39, 40]. It can therefore be envisaged that RNase E associated with an sRNA/Hfq complex could interrogate a polyribosome as mRNA spools through the translational machinery providing a window of opportunity for degradation if the sRNA can match a recognition region in the transcript. General mRNA degradation, which is initiated in the absence of a regulatory factor, could also involve polyribosome interrogation with direct competition between translation re-initiation and cleavage by RNase E. In either case, a productive interaction would lead to the formation of ribosome-free mRNA facilitating the recruitment of additional RNase E molecules. Sequestration of the RNA degradosome to the two dimensional surface of the inner cytoplasmic membrane and rapid diffusion could increase cooperativity in general and regulated mRNA degradation as well as other processes involving RNase E such as tRNA maturation. It is also possible that this system has a quality control function in the degradation of defective transcripts that fail to form polyribosomes or other ribonucleoprotein complexes. Characterization of RNA substrates localized to RNase E clusters could help to identify cellular processes that are facilitated by the formation of cooperative degradation bodies.
Materials and Methods
Atomistic and coarse grain molecular dynamics simulations
Atomistic simulations were performed using the GROMACS simulation package, version 4.5.1 [41] with the GROMOS53a6 force-field [42, 43] and the SPC water model [44]. Simulations were performed in the NPT ensemble, with the Nosé Hoover thermostat [45, 46] with a time constant of 0.5 ps and the Parrinello—Rahman barostat [47] with a time constant of 5.0 ps used to maintain a temperature of 310 K and a pressure of 1 bar. Long-range electrostatic interactions were treated using the smooth particle mesh Ewald method and a long-range dispersion correction was applied to the energy and pressure beyond the cut-off. The neighbor list was updated every 5 steps during the simulations. All bonds were constrained using the LINCS algorithm [48] allowing a 2 fs timestep to be applied in all simulations. The protonation states of all titratable residues of the peptides were assigned using pH 7. Repeats of all of the simulations were performed using different randomly assigned starting velocities. Peptides were manually positioned in the bulk solvent just above the lipid headgroups of one leaflet of the pre-equilibrated DPPC lipid bilayers.
Within the GROMACS simulation package, the MARTINI force-field was used for the lipids and a variant of this was used to represent the interactions between lipid and protein [49]. The peptide secondary structure was retained using weak restraints between backbone particles to represent hydrogen-bonds, as has been shown to work well for other peptides [50]. The different bilayers (E. coli membrane models contained 14 cardiolipin, 88 DPPE, and 26 DPPG lipid molecules) were setup by substituting the appropriate lipids into pre-equilibrated DPPC bilayers. The non-bonded neighbor list was updated every 10 steps. The integration time step was 40 ps. All simulations were performed at constant temperature (310 K) and pressure (1 bar), using the Berendsen thermostat and barostat [51]. Lennard-Jones interactions were shifted to zero between 9 Å and 12 Å, and electrostatics were shifted to zero between 0 Å to 12 Å, with a relative dielectric constant of 15.
Strains construction
Strains and plasmids are listed in Table 1. The Kti series of strains was constructed in the NCM3416 background using the λ Red recombination system [52, 53]. PCR products (S1 Table) were transformed into NCM3416/pKD20. Cmr (Chloramphenicol resistant) transformants were selected at 30°C and then streaked at 42°C to eliminate pKD20. The constructs were purified by bacteriophage P1 transduction of the Cmr marker into NMC3416. The strains were then transformed with pCP20 and ampicillin resistant colonies selected at 30°C. To eliminate pCD20 and the Cmr cassette, transformants were streaked at 43°C on ampicillin and then tested for loss of the Cmr marker. The coding sequence of the fusion protein and flanking regions were then sequenced. All constructs are C-terminal fusions and the FRT scar is located downstream of the translation stop codon. For construction of the strains expressing RNase E-mCherry, RNase E-GFP and RhlB-CFP, DNA fragments generated by crossover PCR were transformed into NCM3416/pKD20. For construction of strains expressing RNase E(∆MTS)-mCherry, RNase E(∆Sca)-mCherry and RNase E(∆HBS)-mCherry, plasmids containing the mutant rne allele fused to mCherry and the Cmr cassette were constructed (S1 Table). These plasmids were then used as templates to produce PCR products to transform NCM3416/pKD20.
Deletions in RNase E variants are as follows (wild type RNase E coordinates): ∆MTS, 567–582; ∆scaffold, 702–1061; ∆HBS, 705–737. SDS-PAGE showed that the fusion proteins are stable and their levels are comparable to the wild type protein. For Western blotting, the transfer of RNase E-GFP and RNase E-mCherry is inefficient and not sufficiently reproducible to be quantified. We therefore visualized the fusion proteins directly by SDS-PAGE and Coomassie staining (S6A–S6B Fig.), which is possible because the fusions are amongst the largest proteins in the cell and they migrate as distinct bands. As it is difficult to raise specific high-titer antibodies against RhlB, we examined RhlB and RhlB-CFP using affinity purified polyclonal rabbit antibody as described [54] or antibody against CFP (S6C–S6D Fig.). These results show that RhlB-CFP is stable and that its level in the cell is comparable to wild type RhlB. Strains expressing defective RNase E variants grow slower than an isogenic wild type control and the defective variant is expressed at a higher level due to autoregulation [6, 29, 30]. Since the growth rate and RNase E levels of the strains expressing RNase E-mCherry, RNase E-GFP and RNase E-YFP are normal (S6 Fig. and [6]), we conclude that these fusion proteins are fully active. RhlB is not essential, but its activity can be tested in vivo since ∆rhlB in a pcnB- background results in the accumulation of mRNA degradation intermediates [55]. RhlB is part of a 3′ exonucleolytic mRNA degradation pathway involving RhlB and PNPase as components of the RNA degradosome. S9 Fig. shows a Northern blot probed for an mRNA degradation intermediate known to accumulate in the ∆rhlB, pcnB- background. A probe for 5S ribosomal RNA was included as a loading control. In the pcnB- background, the mRNA degradation intermediate accumulates in the rhlB-cfp strain, but the level is much higher in the ∆rhlB strain. We therefore conclude that the RhlB-CFP fusion is active in vivo albeit at a lower level than wild type RhlB.
Cell growth and microscopy
Liquid cultures were grown at 30°C with vigorous aeration in LB or MOPS medium [56] supplemented with glycerol (0.5%) and amino acids (50 µg/ml each l-amino acid except tryptophane, tyrosine and phenylalanine). Microscope setups are listed in S2 Table. Bacteria were prepared for microscopy as described [57]. Aliquots (0.5–1.0 µl) from cultures grown to mid logarithmic phase were spotted on microscope slides covered with a thin layer of agarose (1.2% in water). After a few minutes to allow absorption of the cells, the agarose pad was covered with a slip and the slide immediately mounted on the microscope to take images. Preparation of the microscope slide and imaging was performed at room temperature. To chemically fix cells, aliquots from cultures grown to mid logarithmic phase were treated with formaldehyde (1%) for 10 min at 30°C with agitation and then quenched with glycine (100 mM). Images were analyzed using ImageJ [58, 59]. TIRFm photobleaching and quantitative analyses to determine relative diffusion rates were performed as described [21].
For the analysis of peptide binding, FITC-labelled PAQPGLLSRFFGALKALFSGGK (wild type) or PAQPGLLSRAAGALKALFSGGK (F574A/F574A variant) were mixed with liposomes and visualized by spinning disk confocal fluorescence microscopy. Liposomes were prepared from E. coli lipid extracts (Avanti Polar Lipids) using Octyl-Glucoside detergent dialysis method as described [57]. Liposomes were diluted to 1 mg/ml in 5 mM Tris pH 7.5, 150 mM KCl, and peptides were added at concentration of 100 ug/ml. 2 mg/ml BSA was included as unspecific protein. Incubation (5 min) and microscopy were performed at 30°C.
Ethics statement
Not applicable. This study did not involve human participants, specimens or tissue samples, or vertebrate animals, embryos or tissues.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Gorna MW, Carpousis AJ, Luisi BF (2012) From conformational chaos to robust regulation: the structure and function of the multi-enzyme RNA degradosome. Q Rev Biophys 45: 105–145. doi: 10.1017/S003358351100014X 22169164
2. Bandyra KJ, Bouvier M, Carpousis AJ, Luisi BF (2013) The social fabric of the RNA degradosome. Biochim Biophys Acta 1829: 514–522. doi: 10.1016/j.bbagrm.2013.02.011 23459248
3. Waters LS, Storz G (2009) Regulatory RNAs in bacteria. Cell 136: 615–628. doi: 10.1016/j.cell.2009.01.043 19239884
4. Beisel CL, Storz G (2010) Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol Rev 34: 866–882. doi: 10.1111/j.1574-6976.2010.00241.x 20662934
5. Carpousis AJ (2007) The RNA degradosome of Escherichia coli: an mRNA-degrading machine assembled on RNase E. Annu Rev Microbiol 61: 71–87. doi: 10.1146/annurev.micro.61.080706.093440 17447862
6. Khemici V, Poljak L, Luisi BF, Carpousis AJ (2008) The RNase E of Escherichia coli is a membrane-binding protein. Mol Microbiol 70: 799–813. 18976283
7. Taghbalout A, Rothfield L (2007) RNaseE and the other constituents of the RNA degradosome are components of the bacterial cytoskeleton. Proc Natl Acad Sci U S A 104: 1667–1672. doi: 10.1073/pnas.0610491104 17242352
8. Papanastasiou M, Orfanoudaki G, Koukaki M, Kountourakis N, Sardis MF, et al. (2013) The Escherichia coli peripheral inner membrane proteome. Mol Cell Proteomics 12: 599–610. doi: 10.1074/mcp.M112.024711 23230279
9. Lopez-Campistrous A, Semchuk P, Burke L, Palmer-Stone T, Brokx SJ, et al. (2005) Localization, annotation, and comparison of the Escherichia coli K-12 proteome under two states of growth. Mol Cell Proteomics 4: 1205–1209. doi: 10.1074/mcp.D500006-MCP200 15911532
10. Mackie GA (2013) RNase E: at the interface of bacterial RNA processing and decay. Nat Rev Microbiol 11: 45–57. doi: 10.1038/nrmicro2930 23241849
11. Shahbabian K, Jamalli A, Zig L, Putzer H (2009) RNase Y, a novel endoribonuclease, initiates riboswitch turnover in Bacillus subtilis. EMBO J 28: 3523–3533. doi: 10.1038/emboj.2009.283 19779461
12. Taghbalout A, Rothfield L (2008) RNaseE and RNA helicase B play central roles in the cytoskeletal organization of the RNA degradosome. J Biol Chem 283: 13850–13855. doi: 10.1074/jbc.M709118200 18337249
13. Taghbalout A, Yang Q, Arluison V (2014) The Escherichia coli RNA processing and degradation machinery is compartmentalized within an organized cellular network. Biochem J 458: 11–22. doi: 10.1042/BJ20131287 24266791
14. Worrall JA, Howe FS, McKay AR, Robinson CV, Luisi BF (2008) Allosteric activation of the ATPase activity of the Escherichia coli RhlB RNA helicase. J Biol Chem 283: 5567–5576. doi: 10.1074/jbc.M708620200 18165229
15. Ait-Bara S, Carpousis AJ (2010) Characterization of the RNA degradosome of Pseudoalteromonas haloplanktis: conservation of the RNase E-RhlB interaction in the {gamma}-Proteobacteria. J Bacteriol. doi: 10.1128/JB.00592-10 20729366
16. Soupene E, van Heeswijk WC, Plumbridge J, Stewart V, Bertenthal D, et al. (2003) Physiological studies of Escherichia coli strain MG1655: growth defects and apparent cross-regulation of gene expression. J Bacteriol 185: 5611–5626. doi: 10.1128/JB.185.18.5611-5626.2003 12949114
17. Bouvier M, Carpousis AJ (2011) A tale of two mRNA degradation pathways mediated by RNase E. Mol Microbiol 82: 1305–1310. doi: 10.1111/j.1365-2958.2011.07894.x 22074454
18. Sweetman G, Trinei M, Modha J, Kusel J, Freestone P, et al. (1996) Electrospray ionization mass spectrometric analysis of phospholipids of Escherichia coli. Mol Microbiol 20: 233–238. doi: 10.1111/j.1365-2958.1996.tb02504.x 8861220
19. Oursel D, Loutelier-Bourhis C, Orange N, Chevalier S, Norris V, et al. (2007) Lipid composition of membranes of Escherichia coli by liquid chromatography/tandem mass spectrometry using negative electrospray ionization. Rapid Commun Mass Spectrom 21: 1721–1728. doi: 10.1002/rcm.3013 17477452
20. Strahl H, Hamoen LW (2012) Finding the corners in a cell. Curr Opin Microbiol 15: 731–736. doi: 10.1016/j.mib.2012.10.006 23182676
21. Strahl H, Burmann F, Hamoen LW (2014) The actin homologue MreB organizes the bacterial cell membrane. Nat Commun 5: 3442. doi: 10.1038/ncomms4442 24603761
22. Knight JD, Lerner MG, Marcano-Velazquez JG, Pastor RW, Falke JJ (2010) Single molecule diffusion of membrane-bound proteins: window into lipid contacts and bilayer dynamics. Biophys J 99: 2879–2887. doi: 10.1016/j.bpj.2010.08.046 21044585
23. Garner EC, Bernard R, Wang W, Zhuang X, Rudner DZ, et al. (2011) Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science 333: 222–225. doi: 10.1126/science.1203285
24. Dominguez-Escobar J, Chastanet A, Crevenna AH, Fromion V, Wedlich-Soldner R, et al. (2011) Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science 333: 225–228. doi: 10.1126/science.1203466 21636744
25. van Teeffelen S, Wang S, Furchtgott L, Huang KC, Wingreen NS, et al. (2011) The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc Natl Acad Sci U S A 108: 15822–15827. doi: 10.1073/pnas.1108999108 21903929
26. Lopez PJ, Iost I, Dreyfus M (1994) The use of a tRNA as a transcriptional reporter: the T7 late promoter is extremely efficient in Escherichia coli but its transcripts are poorly expressed. Nucleic Acids Res 22: 2434. doi: 10.1093/nar/22.7.1186 8036178
27. Iost I, Dreyfus M (1995) The stability of Escherichia coli lacZ mRNA depends upon the simultaneity of its synthesis and translation. EMBO J 14: 3252–3261. 7542588
28. Ow MC, Kushner SR (2002) Initiation of tRNA maturation by RNase E is essential for cell viability in E. coli. Genes Dev 16: 1102–1115. doi: 10.1101/gad.983502
29. Jain C, Belasco JG (1995) RNase E autoregulates its synthesis by controlling the degradation rate of its own mRNA in Escherichia coli: unusual sensitivity of the rne transcript to RNase E activity. Genes Dev 9: 84–96. doi: 10.1101/gad.9.1.84 7530223
30. Leroy A, Vanzo NF, Sousa S, Dreyfus M, Carpousis AJ (2002) Function in Escherichia coli of the non-catalytic part of RNase E: role in the degradation of ribosome-free mRNA. Mol Microbiol 45: 1231–1243. doi: 10.1046/j.1365-2958.2002.03104.x 12207692
31. Shaner NC, Steinbach PA, Tsien RY (2005) A guide to choosing fluorescent proteins. Nat Methods 2: 905–909. doi: 10.1038/nmeth819 16299475
32. Bakshi S, Siryaporn A, Goulian M, Weisshaar JC (2012) Superresolution imaging of ribosomes and RNA polymerase in live Escherichia coli cells. Mol Microbiol 85: 21–38. doi: 10.1111/j.1365-2958.2012.08081.x 22624875
33. Parry BR, Surovtsev IV, Cabeen MT, O’Hern CS, Dufresne ER, et al. (2014) The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 156: 183–194. doi: 10.1016/j.cell.2013.11.028 24361104
34. Hunter CA, Anderson HL (2009) What is cooperativity? Angew Chem Int Ed Engl 48: 7488–7499. doi: 10.1002/anie.200902490 19746372
35. Carpousis AJ, Leroy A, Vanzo N, Khemici V (2001) Escherichia coli RNA degradosome. Methods Enzymol 342: 333–345. doi: 10.1016/S0076-6879(01)42556-0 11586906
36. Decker CJ, Parker R (2012) P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol 4: a012286. doi: 10.1101/cshperspect.a012286 22763747
37. Stoecklin G, Kedersha N (2013) Relationship of GW/P-bodies with stress granules. Adv Exp Med Biol 768: 197–211. doi: 10.1007/978-1-4614-5107-5_12 23224972
38. Shao Y, Feng L, Rutherford ST, Papenfort K, Bassler BL (2013) Functional determinants of the quorum-sensing non-coding RNAs and their roles in target regulation. EMBO J 32: 2158–2171. doi: 10.1038/emboj.2013.155 23838640
39. Tsai YC, Du D, Dominguez-Malfavon L, Dimastrogiovanni D, Cross J, et al. (2012) Recognition of the 70S ribosome and polysome by the RNA degradosome in Escherichia coli. Nucleic Acids Res 40: 10417–10431. doi: 10.1093/nar/gks739 22923520
40. Morita T, Maki K, Aiba H (2005) RNase E-based ribonucleoprotein complexes: mechanical basis of mRNA destabilization mediated by bacterial noncoding RNAs. Genes Dev 19: 2176–2186. doi: 10.1101/gad.1330405 16166379
41. Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, et al. (2005) GROMACS: fast, flexible, and free. J Comput Chem 26: 1701–1718. doi: 10.1002/jcc.20291 16211538
42. Poger D, Mark AE (2009) On the Validation of Molecular Dynamics Simulations of Saturated and cis-Monounsaturated Phosphatidylcholine Lipid Bilayers: A Comparison with Experiment. J Chem Theory Comput 6: 325–336. doi: 10.1021/ct900487a
43. Poger D, Van Gunsteren WF, Mark AE (2010) A new force field for simulating phosphatidylcholine bilayers. J Comput Chem 31: 1117–1125. doi: 10.1002/jcc.21396 19827145
44. Berendsen H, Postma J, van Gunsteren W, Hermans J (1981) Interaction Models for Water in Relation to Protein Hydration. In: Pullman B, editor. Intermolecular Forces. Dordrecht, The Netherlands: Reidel Publishing Company.
45. Nose S (1984) A molecular dynamics method for simulations in the canonical ensemble. Mol Phys 52: 255–268. doi: 10.1080/00268978400101201
46. Hoover WG (1985) Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A 31: 1695–1697. doi: 10.1103/PhysRevA.31.1695 9895674
47. Parrinello M, Rahman AJ (1981) J Appl Phys 52: 7182–7190. doi: 10.1063/1.328693
48. Hess B, Bekker H, Berendsen HJC, Fraaije J (1997) LINCS: a linear constraint solver for molecular simulations. J Comput Chem 18: 1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12%3C1463::AID-JCC4%3E3.3.CO;2-L
49. Bond PJ, Sansom MS (2006) Insertion and assembly of membrane proteins via simulation. J Am Chem Soc 128: 2697–2704. doi: 10.1021/ja0569104 16492056
50. Bond PJ, Holyoake J, Ivetac A, Khalid S, Sansom MS (2007) Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J Struct Biol 157: 593–605. doi: 10.1016/j.jsb.2006.10.004 17116404
51. Berendsen HJC, Postma JMP, van Gunsteren WF, DiNola A, Haak JR (1984) Molecular dynamics with coupling to an external bath. J Chem Phys 81: 3684–3690. doi: 10.1063/1.448118
52. Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97: 6640–6645. doi: 10.1073/pnas.120163297 10829079
53. Uzzau S, Figueroa-Bossi N, Rubino S, Bossi L (2001) Epitope tagging of chromosomal genes in Salmonella. Proc Natl Acad Sci U S A 98: 15264–15269. doi: 10.1073/pnas.261348198 11742086
54. Carpousis AJ, Khemici V, Ait-Bara S, Poljak L (2008) Co-immunopurification of multiprotein complexes containing RNA-degrading enzymes. Methods Enzymol 447: 65–82. doi: 10.1016/S0076-6879(08)02204-0 19161838
55. Khemici V, Carpousis AJ (2004) The RNA degradosome and poly(A) polymerase of Escherichia coli are required in vivo for the degradation of small mRNA decay intermediates containing REP-stabilizers. Mol Microbiol 51: 777–790. doi: 10.1046/j.1365-2958.2003.03862.x 14731278
56. Neidhardt FC, Bloch PL, Smith DF (1974) Culture medium for enterobacteria. J Bacteriol 119: 736–747. 4604283
57. Strahl H, Hamoen LW (2010) Membrane potential is important for bacterial cell division. Proc Natl Acad Sci U S A 107: 12281–12286. doi: 10.1073/pnas.1005485107 20566861
58. Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675. doi: 10.1038/nmeth.2089 22930834
59. Collins TJ (2007) ImageJ for microscopy. Biotechniques 43: 25–30. doi: 10.2144/000112517 17936939
60. Vanzo NF, Li YS, Py B, Blum E, Higgins CF, et al. (1998) Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome. Genes Dev 12: 2770–2781. doi: 10.1101/gad.12.17.2770 9732274
61. Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, et al. (2004) Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22: 1567–1572. doi: 10.1038/nbt1037
62. Ah-Seng Y, Rech J, Lane D, Bouet JY (2013) Defining the role of ATP hydrolysis in mitotic segregation of bacterial plasmids. PLoS Genet 9: e1003956. doi: 10.1371/journal.pgen.1003956 24367270
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
- Souvislost haplotypu M2 genu pro annexin A5 s opakovanými reprodukčními ztrátami
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Příjem alkoholu a menstruační cyklus
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates