
GC-Content Evolution in Bacterial Genomes: The Biased Gene Conversion Hypothesis Expands
Classical population genetics models indicate that the efficiency of selection, and hence adaptation, depends on a number of non-selective factors, such as the size of a population or the intensity of recombination. In the last 10 years, evidence has accumulated that another mechanism called GC-Biased Gene Conversion (gBGC) can interfere with selection and even mimic its effects. This phenomenon, which arises from a particularity of the recombination machinery, was first thought to be restricted to sexual eukaryotic organisms. Here, we show that this mechanism probably exists in Bacteria and has a strong impact on their genome evolution. This discovery not only explains many previously unconnected features of bacterial genome evolution, but also highlights the importance of non-adaptive evolutionary processes in Bacteria.
Published in the journal:
. PLoS Genet 11(2): e32767. doi:10.1371/journal.pgen.1004941
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004941
Summary
Classical population genetics models indicate that the efficiency of selection, and hence adaptation, depends on a number of non-selective factors, such as the size of a population or the intensity of recombination. In the last 10 years, evidence has accumulated that another mechanism called GC-Biased Gene Conversion (gBGC) can interfere with selection and even mimic its effects. This phenomenon, which arises from a particularity of the recombination machinery, was first thought to be restricted to sexual eukaryotic organisms. Here, we show that this mechanism probably exists in Bacteria and has a strong impact on their genome evolution. This discovery not only explains many previously unconnected features of bacterial genome evolution, but also highlights the importance of non-adaptive evolutionary processes in Bacteria.
Introduction
Comparative genomics is a fundamental key to the inner workings of genomes. The identification of genes and other functional elements such as regulatory regions, as well as the understanding of their influence on the fitness of organisms rely essentially on the detection of signatures of natural selection within genomes [1]. In that respect, devising a model of sequence evolution in the absence of selective constraints (a neutral model) is critical for the detection of functional sequences. Indeed, to explain the features of a given genomic segment, comparing the fit of a neutral model to that of a model that also invokes selection (either purifying or positive) is the operational way to infer evolutionary constraint and hence function.
The base composition of genomic sequences varies widely, both across species and along chromosomes [2,3]. For instance, the genomic GC-content of cellular organisms ranges from 13% to about 75% [4,5], with vast intra-genomic heterogeneity. These large-scale variations in base composition affect all parts of genomes, intergenic regions and genes—including all three codon positions [6]—and hence cannot be simply explained by selective constraints on the encoded proteins. Determining the underlying causes (selective or neutral) of these variations in GC-content is a major issue in genetics: if they result from selection, it implies that the genomic base composition per se is an important trait that contributes to the fitness of organisms; conversely, if these “genomic landscapes” are largely shaped by non-adaptive molecular processes, then characterizing these processes is essential for the reliable detection of selection (see e.g. [7]).
In mammals, the analysis of polymorphism data and substitution patterns along genomes demonstrated that the evolution of GC-content is driven by recombination, which tends to increase the probability of fixation of AT→GC mutations [8,9]. The impact of recombination on base composition in these genomes is most probably due to a phenomenon known as GC-biased gene conversion (gBGC), which favours G/C nucleotides at polymorphic sites in the conversion of intermediates of recombination (see review in [10]). Although gBGC as a process is unrelated to natural selection, it affects the probability of fixation of alleles in patterns similar to selection [11]. It has been shown to be an important confounding factor, which can mimic some marks of positive selection [7,12] and interfere with selection by actively promoting the fixation of deleterious alleles [13,14]. The process of gBGC has been observed directly in meiosis products from yeast and human [15,16], and there is ample evidence, based on the analysis of relationships between recombination rate and substitution patterns within genomes, that this process affects many other eukaryotes [17–19].
In Bacteria and Archaea, several environmental factors potentially affecting genomic GC-content have been proposed (such as the availability of oxygen or nitrogen in the environment, growth temperature, or the variety of environments encountered by an organism, see for instance [20] and ref. therein). Because these effects are weak and the nature of the selective pressures remain elusive, the major force driving genomic GC-content has long been considered to be mutational bias [21]. Recently however, two independent analyses have shown that in virtually all Bacteria, independently of their genomic GC-content, there is an excess of G/C→A/T mutations [22,23]. This suggests that an unknown process, selective or neutral, is opposing this universal mutational bias by favouring the fixation of G/C alleles Previously, an analysis of a large number of E. coli genomes had suggested a possible role of gBGC, based on the link between GC-content, recombination and the organization of the chromosome in this species [24]. However Hildebrand et al. [23] observed that the excess of G/C→A/T mutations was still present after removing datasets with evidence of recombination. Moreover they found no correlation between GC-content and recombination rate across bacterial species. They therefore concluded that this force could not be gBGC and hence that selection was driving an increase of genomic GC in Bacteria. The nature of this selective advantage remains however mysterious, though various hypotheses have been proposed [25,26].
Here we argue that the analyses performed by Hildebrand et al. [23] are not conclusive regarding the gBGC hypothesis, and we present evidence that variations in GC-content observed in Bacteria are influenced by gBGC. One pervasive signature of gBGC is that genomic regions undergoing high recombination rates will also acquire a high GC-content [6]. We thus studied the relationship between recombination and GC-content in 20 groups of Bacteria and one group of Archaea. This dataset covers a wide range of clades representative of the bacterial diversity. To avoid problems inherent to comparisons of recombination rates among species (such as differences in polymorphism, genome samples, population size, mutation rates, an other life history factors), we examined the intragenomic variability for both recombination and GC-content.
We show that in a wide variety of bacterial species, genes with evidence of recombination have a higher GC-content. We further show that this bias towards G/C nucleotides in recombining genes cannot be explained by selection on codon usage, and could interfere with the selection for AT-ending optimal codons. These two observations strongly suggest that homologous recombination, via gBGC, is a crucial factor universally influencing the nucleotide content of genes and genomes. If confirmed, gBGC can account for several pervasive yet unexplained features of bacterial genomes. Finally, we emphasize that because gBGC has the ability to both mimic and interfere with natural selection, gBGC must be considered by future studies geared at understanding processes driving bacterial genome evolution.
Results
A universal relationship between recombination and GC% in Bacteria
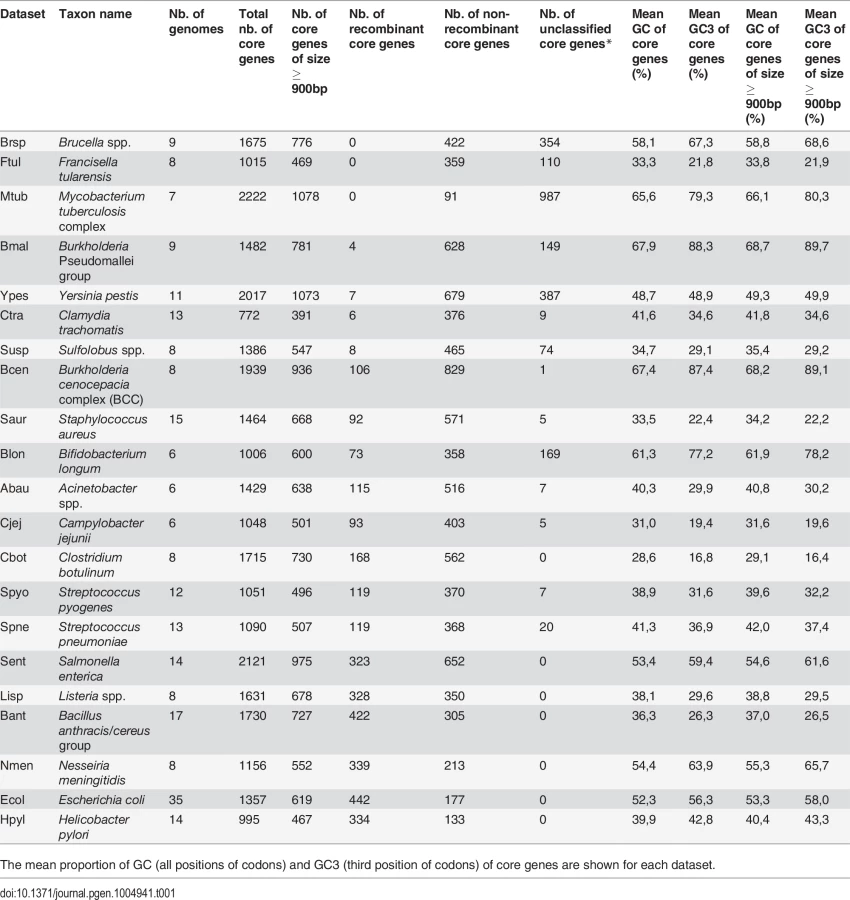
In Bacteria, recombination occurs in the form of gene conversion (i.e. unidirectional transfer of genetic material from a donor sequence towards a homologous recipient sequence). To detect past gene conversion events in bacterial species, it is necessary to compare closely related genomes. We therefore selected in the database of homologous gene families HOGENOM (release 6) [27] all groups of closely related species or strains encompassing at least 6 sequenced genomes. This dataset contains 20 bacterial groups and one archaeal group. For each gene family represented in these groups, we computed i) the average GC-content at different positions of codons and ii) the index of recombination provided by PHI [28] based on alignments of standardized length (see methods for details). PHI is a rapid method for detecting recombination in multiple alignments at the scale of the gene, which has been shown to be more robust than most methods to variations in recombination rates, sequence divergence and population dynamics [28]. We used this test to determine if homologous gene families had experienced gene conversion events among members of the taxa of interest. One important feature of this test is that it measures whether there is sufficient phylogenetic signal in an alignment to tell if recombination has occurred. Only alignments with sufficient signal, whether recombinant or non-recombinant were retained for tests in the remaining of this study. We also used three other approaches for detecting recombination, and these confirm the robustness of our conclusions (see Methods and Supplementary Material).
In Eukaryotes, a general relationship between various estimates of recombination rate and the GC% of genes has been documented and provides indirect evidence for gBGC. Our first goal was to test this prediction in Bacteria and Archaea. To exclude a potential effect of the number of genes in the alignment on our estimates of recombination (because alignments with more sequences are expected to give more power to detect recombination), we focused on single-copy genes of the core genome (i.e. genes that are present in only one copy and found in each genome of a group). In 7 of the 21 groups, the proportion of single-copy genes of the core genome with evidence for recombination was very low (<2% of all gene alignments tested), suggesting that these species are clonal or nearly so (Table 1; shaded datasets in Fig. 1). Indeed, the Burkholderia pseudomalei group, Chlamydia trachomatis, Francisella tularensis, Mycobacterium tuberculosis and Yersinia pestis are species known to be pathogenic clonal complexes with low polymorphism and probably very low recombination [29–33], while Brucella spp. and Sulfolobus spp. are likely composed of ecologically isolated clades, because of their respective lifestyle as obligate intracellular pathogen or ecotypes endemic of hot springs [34–36]. In 11 of the 14 remaining groups, we found a significant positive difference in average GC-content at all and/or at the third position of codons (GC3) between recombinant and non-recombinant genes (Fig. 1). In these 11 species, the difference in GC3 is always larger than that at all positions, suggesting that the effect of recombination on gene composition is stronger at synonymous positions (probably because of purifying selection on protein sequences). Two notable exception to this pattern are i) the bacterial species Helicobacter pylori, where GC-content seems to be lower in recombining genes and ii) the Bacillus anthracis/cereus group, where GC at all positions and GC3 display opposite patterns, with GC3 being higher in recombining genes. Consistent results are obtained using alternative recombination detection methods (S1 Fig.).

Selection on optimal codons cannot explain the association of GC with recombination
Recombination is known to enhance the efficacy of selection by breaking linkage between neighboring selected sites. It is therefore possible that selection is more efficient in recombining genes. This effect (referred to as Hill-Robertson interference) should theoretically be more pronounced in the case of selection on codon usage [37], which is relatively weak compared to selection on amino acid sequences. Thus recombination—in the absence of any gBGC—can potentially explain the pronounced effect observed on GC3: if Hill-Robertson interference leads to a higher frequency of optimal codons in highly recombining genes, and if optimal codons tended to be GC-rich, this effect could explain a relationship between GC-content and recombination. The “selection model” sketched above predicts that the frequency of all optimal codons (both GC-ending and AU-ending) should increase with recombination. In contrast, a model incorporating the effect of gBGC predicts that GC-ending codons (not specifically optimal codons) should be enriched in recombining regions, and that AU-ending codons (and possibly AU-ending optimal codons if gBGC is strong enough to override selection on codon usage) should display the opposite pattern. We therefore looked specifically at the frequency of the different types of codons, i.e. optimal and non-optimal, in recombining and non-recombining genes.
There is a debate over the best way to define optimal codons, based on their over-representation in either ribosomal protein genes (RP), or genes with the highest codon bias (HCB) [38–40]. We therefore analyzed the frequency of GC-ending and AU-ending optimal codons (FopGC and FopAU) and non-optimal codons (FnopGC and FnopAU) according to both RP and HCB definitions (Fig. 2, S3 Fig., and S4 Fig., respectively). The higher GC3 of recombining genes means that GC-ending codons are over-represented in recombining genes, but this is true for optimal GC-ending codons in only 2 (RP optimal codons) or 4 (HCB optimal codons) species out of 11. This effect is hence essentially due to non-optimal codons (FnopGC is significantly higher in recombining genes than non-recombining genes in respectively 9 and 8 species for RP and HCB definitions). Moreover, optimal AU-ending codons are significantly depleted in recombining genes for 8 (resp. 5) species for RP (resp. HCB) codons. In fact, only two species, S. pyogenes and Nesseiria meningitidis (using the HCB method—only S. pyogenes using the RP method) exhibit a pattern partially compatible with the selection hypothesis presented above. All species display either an increase of FnopGC and/or a decrease of FopAU in recombining genes, a fact that cannot be explained by a higher efficiency of selection. This pattern excludes the possibility of pervasive selection for codon usage promoting a better adaptation to the pool of tRNA for genes in regions of high recombination, but is compatible with the predictions of gBGC.

Intergenic regions display a pattern that is consistent with gBGC
In principle, gBGC should affect all genomic regions where recombination occurs, including intergenic regions. Intergenes are generally shorter than coding regions. Furthermore, they evolve more rapidly and hence are more difficult to align. Hence, the methods that we used to detect recombination in coding regions cannot be applied with intergenes. We therefore used the recombinant or non-recombinant status of the neighboring protein-coding genes as a proxy of the status of the intergenes. In 11 of the 14 taxonomic groups, we observed that intergenes flanked by recombining genes have a higher GC-content than intergenes flanked by non-recombining genes (S2 Fig.). The difference in GC-content between the two classes is weaker than that observed in coding regions, and when considered individually, only one comparison is statistically significant (Streptococcus pyogenes, p < 0.01). This is possibly because the prediction of recombination status of intergenic regions is indirect (based on the status of flanking genes) and therefore less accurate than that of coding regions. However, the number of cases where the difference in GC-content between the two classes is positive (11 out of 14 comparisons) is significantly higher than expected by chance (Chi-squared test: p = 0.03).
Quantifying the impact of recombination on bacterial genome evolution
To quantify the relationship between recombination and base composition, we first analyzed the genome of S. pyogenes, one of the species for which the signature of gBGC is strong (Fig. 1, see also Sup. Mat.). We used ClonalOrigin [41] to compute the population-scaled recombination rate (rho) for each gene of the core genome. The correlation between rho and the GC-content at third codon position of each gene (GC3) is slight but significant (R2 = 0.034, p<10−4, n = 478). Interestingly, when we exclude genes for which the estimate of rho is less reliable (about 10% of the data, see Sup. Mat.), the correlation strongly increases (R2 = 0.087; p<10−9, n = 437). Due to the low amount of data available in a gene-scale alignment, the measure of rho is expected to be noisy. Thus, the observed correlation is probably an underestimate. To try to get more robust estimates of rho, we binned the dataset into 20 groups of genes according to their GC3, and we computed the correlation between the average GC3 and the average rho of each bin. Using this approach, we observed a strong correlation between the GC3 and recombination (R2 = 0.60; Fig. 3A).

For a comparison, we performed a similar analysis in humans: we randomly selected 600 human genes (which corresponds to the average number of genes analyzed in our bacterial data sets), binned the data set into 20 groups of genes according to their GC3, and we computed the correlation between the average GC3 and the average recombination rate as obtained from population-wide surveys [42]. The average correlation (computed after repeating the random sampling 1,000 times) is R2 = 0.55 (with 95% of the R2 values in the interval [0.26–0.78]; one representative example is presented in Fig 3A).
Using this binning approach, we noted that the proportion of genes in a bin that are detected as recombinant by PHI is strongly correlated with the average value of rho (R2 = 0.70; p<10−5). This suggests that this index (hereafter referred to as PREC, for ‘proportion of recombinant’) is a good proxy for the average recombination rate in a bin. In fact, we observed that GC3 correlates more strongly with PREC (R2 = 0.68; Fig. 3B) than with rho. Given that the computation of rho with ClonalOrigin is extremely time consuming, we decided to use PREC to evaluate the correlation between GC3 and recombination rate in other species. Correlations were positive and significant for 11 of the 14 species, with R2 values ranging from 0.24 to 0.68, and we did not observe any significant negative correlation (Fig. 3B). This shows that the correlations between GC-content and recombination rate in bacteria are of similar magnitude to what is observed in humans.
Discussion
The link between GC-content and recombination is independent from constraints on protein coding genes
Our results suggest that recombination affects the GC-content of genes in most bacterial phyla. We analyzed genes of the core genome to compare the base composition of genes with or without evidence of recombination. In 11 of the 14 species in which a significant amount of recombination could be detected, we observed that the GC-content (measured either at the third codon position or along the entire coding region) is higher among recombining genes compared to other (hereafter labeled as “non-recombining”) core genes.
Several hypotheses have been proposed to explain the variations in GC-content among bacterial genomes [25]. Several studies have revealed that the genomic GC-content of bacterial genomes is always higher than what would be predicted from mutational bias [22,23,43]. Hence, it seems inescapable that some other evolutionary force is driving the genomic GC-content towards higher values in virtually all bacterial species, except maybe for the most AT-rich genomes [22,43]. Recombination is known to enhance the efficiency of selection by breaking linkage among sites. It is therefore conceivable that our results merely reveal a universal selective pressure favoring GC-rich alleles. But the mechanism underlying such selection would have to be acting more efficiently on synonymous sites than non-synonymous sites because the difference of GC% between recombining and non-recombining genes is higher at the third position of codons. This excludes potential selection on amino-acid content. One selectable trait that may influence synonymous positions is codon usage. If optimal codons tended to be GC-rich, recombination could drive GC% higher by favoring the adaptation of genes to better translation efficiency. However, we observed a higher GC-content in recombining genes even in species favoring A/U-ending codons (Fig. 2). Moreover, A/U-ending and G/C-ending codons show opposite relationships with recombination in most species, irrespective of their optimality. These observations suggest that the evolutionary force explaining our results is also largely independent from selection on codon usage. This conclusion is further supported by the fact that the relationship between GC-content and recombination is also observed in intergenic regions (S2 Fig.).
In fact, as suggested by Hershberg and Petrov [22], who observed that the intergenic regions of bacterial genomes also have higher GC% than expected from their mutational pattern, it seems likely that the process is unlinked to gene expression or function. Hence, either there is selection acting simultaneously on each nucleotide of a bacterial genome to become G or C, or GC-biased gene conversion, which has now been observed in a variety of Eukaryotes is also at work in Bacteria.
gBGC effect on nucleotide composition is manifest within genomes
The hypothesis that gBGC plays a role in bacterial genome evolution has been considered previously [23]. Hildebrand et al. analyzed the correlation between genome-wide measures of recombination rate (scaled by effective population size) with genomic GC-content among 34 species, covering different bacterial phyla. As they did not find any significant correlation, they concluded that there was no evidence of gBGC in Bacteria [23]. However, we argue here that this observation is not conclusive. In fact, the strength of gBGC depends on four variables: the effective population size (Ne), the rate of recombination per bp per generation (r), the length of conversion tracts (L) and the intensity of the repair bias (b0) (for review, see [6]). In a haploid organism, the population-scaled gBGC coefficient is:

Similar to selection, the impact of gBGC on genome evolution depends on its intensity relative to genetic drift, and becomes negligible when B ≪ 1. There is evidence that besides Ne and r, both L and b0 can vary strongly across species. For example, in budding yeast, when a GC/AT heterozygote site is involved in a gene conversion event, the GC-allele is transmitted with a probability pGC = 0.507 (which is significantly higher than the expected Mendelian transmission ratio; [15]), whereas in humans, a recent analysis of gene conversions tracts associated to non-crossover recombination showed that GC-alleles are transmitted with a probability pGC = 0.70 [16]. Thus, the parameter b0 (b0 = 2 ∙ pGC – 1) is about 30 times higher in humans than in yeast. Conversely, gene conversion tracts are on average about 4 times longer in yeast than in mammals [15,44]. Thus, for a same population-scaled recombination rate (Ne r), the intensity of gBGC would be about 7 times stronger in humans than in yeast. This example illustrates that because of variations in L and b0, the gBGC model does not necessarily predict a good correlation between population-scaled recombination rate and GC-content across species. In fact, to test the predictions of the gBGC model, it is more appropriate to investigate correlations between base composition and recombination rate within genomes, so that the other parameters (Ne, L and b0) can be controlled for.
The impact of gBGC on bacterial genome evolution is quantitatively strong
The observations presented previously are qualitatively consistent with the gBGC model. However, they do not provide a quantification of the impact of recombination on bacterial genomes: to what extent might this model account for the strong variations of GC-content observed across bacterial species? The gBGC model predicts that, all else being equal, the present-day GC-content of a genome should directly reflect its average recombination rate over long evolutionary time. To test this prediction, it is important to take into account two difficulties. First, recombination rates measured in extant populations reflect recent events (more recent than the coalescent time, i.e. of the order of Ne generations), and hence may not correspond to the average recombination rate over times necessary for genomic GC-content to evolve significantly (i.e. inter-species divergence times). Second, the precision in the estimate depends on the physical scale at which recombination is measured. To illustrate these points let us consider the human genome, where the impact of gBGC is well documented [6]. At the gene scale, the correlation between present-day recombination rate (measured in a 10-kb window, centered on the middle of the gene, using HapMap genetic map [42]), and the gene GC-content (at third codon position) is significant but quite weak (R2 = 0.035, p<10−10). However, at 1Mb scale the correlation is much stronger (R2 = 0.15; [9]). Furthermore, when GC-content variations and recombination rates are measured over the same evolutionary time period, the correlation becomes very strong (1Mb scale: R2 = 0.64; [45]).
To test whether the impact of gBGC in bacteria was comparable to what is observed in mammals, we first focused on Streptococcus pyogenes, one of the species for which the signature of gBGC is strong (Fig. 1, see also Sup. Mat.). We computed the population-scaled recombination rate (rho) for each gene of the core genome, using ClonalOrigin [41]. The correlation between rho and the GC-content at third codon position of each gene (GC3) is higher than what is observed in humans (R2 = 0.087; p<10−9). This result is remarkable, given that recombination rates are measured here at the gene scale (typically about 1kb).
To go further, we binned the data set into 20 groups of genes according to their GC3, and we computed the correlation between the average GC3 and the average rho of each bin. Our reasoning is that by computing average values, we should get estimates of rho that are more robust to measurement noise and to possible temporal variations in recombination rates. Using this approach, we observed a strong correlation between the GC3 and recombination (R2 = 0.60; Fig. 3A). To investigate the amplitude of this relation in the other bacterial species studied here, we used PREC (an index based on the proportion of genes in a bin that are detected as recombinant by PHI), which provides a good estimate of the average recombination rate in a bin, and is much easier to compute than ClonalOrigin’s rho. We observed a significant correlation for 11 of the 14 species, and these significant correlations were positive in all cases, with R2 values comprised between 0.24 and 0.68 (on average R2 = 0.43; Fig. 3B). Thus, in many bacteria, the average recombination rate in a bin is a good predictor of its average GC-content. We performed an analogous analysis in human genes using jackknife sampling of the dataset to scale it to the size of bacterial datasets. The average correlation observed in humans is R2 = 0.55 (Fig 3A). Hence, in many bacteria, the intensity of the relationships between GC-content and recombination is comparable to that observed in humans, where the impact of gBGC on base composition is known to be strong [45]. This is consistent with the hypothesis that on the long term, the gBGC process can have a major influence on the evolution of base composition in bacteria.
A dynamic model of the evolution of nucleotide composition: long-term equilibrium vs. short-term disequilibrium
If the base composition of a genome is at evolutionary equilibrium then, by definition, the number of A/T→G/C substitutions must be equal to the number of G/C→A/T substitutions. Hildebrand and colleagues [23] noted that in a large majority of bacterial genomes (94/149), the number of G/C→A/T changes (inferred from the comparison of closely related organisms) exceeds the number of A/T→G/C changes. Given that genomic base composition strongly fluctuates over long evolutionary times (as demonstrated by the wide distribution of GC-content across bacterial species), it is not surprising that many genomes are not at equilibrium. However, what is unexpected is that this non-stationarity predominantly leads to loosing GC-content: a priori, at the scale of the entire bacterial biodiversity, one would expect to observe as many GC-increasing genomes as GC-decreasing genomes. One possible explanation is that the observed excess G/C→A/T changes among closely related genomes corresponds to polymorphic mutations, which eventually do not reach fixation because either selection or gBGC favors GC-alleles over AT-alleles [23]. Hildebrand and colleagues observed an excess of G/C→A/T changes even in bacterial genomes that show no evidence of recent recombination population-wise (i.e. Ner = 0) [23]. They therefore rejected the hypothesis that the fixation bias could be due to gBGC. However, this conclusion relies on one important assumption: that the Ner parameter measured in extant populations reflects the long-term average recombination rate. In fact it is expected that Ne (and hence Ner) should fluctuate over time, as populations go through periods of bottlenecks and expansion. Immediately after a bottleneck, Ner would be close to 0, and hence genomes should accumulate G/C→A/T substitutions. However, on the long term, this can be compensated by an increase in GC-content when the effective population size becomes larger (and hence B > 1). Thus, the base composition of genomes may remain above the mutational equilibrium on the long term, even if many lineages go through periods during which Ner is null (and hence B = 0). Interestingly, the rare species for which the long-term recombination rate is effectively null (typically endosymbiotic bacteria), generally have very AT-rich genomes [46], as predicted by the gBGC hypothesis.
Universal mutational bias and selection on the strength of gBGC
Several lines of evidence suggest that the pattern of spontaneous mutations is biased towards AT nucleotides, in Eukaryotes as well as Prokaryotes [22,23,47]. Under such a bias, it is expected that the selective pressure to reduce mutation rate should generally favor a GC-biased DNA repair machinery. Recombination and repair are tightly linked processes that use many common pathways. In yeast, the analysis of conversion tracts in meiotic product indicates that the conversion bias is most probably due to the mismatch repair machinery (MMR) [48]. The MMR components involved in homologous recombination (MutS, MutL) are generally conserved between Bacteria and Eukaryotes. Hence, in Bacteria as well as Eukaryotes, gBGC could be the secondary effect of a selection for biased repair mechanisms. However, population genetics model show that when gBGC is strong, it drives the fixation of deleterious mutations [49]. Thus, one interesting hypothesis is that in highly recombining species, selection might favor unbiased repair mechanisms (i.e., values of b0 close to 0 in equation (1)), so as to limit the deleterious consequences of strong gBGC. H. pylori is notorious for being highly recombining [50,51], as confirmed by our results. It is interesting that we have found no evidence for gBGC in this species, and recombining genes even seem to have slightly lower GC-content than non recombining genes. This trend is however relatively weak because there is no significant trend of variation of recombination rates among classes of homogeneous GC-content, as shown in Fig. 3B. One possible explanation for the absence of gBGC in H. pylori could hence be that b0 is null in this species, in compensation of a very high rate of recombination.
Although the MMR pathway is a good candidate as a molecular source of gBGC in Bacteria, the association of gBGC with MutSL genes is not straightforward. These genes are absent from three of our genome datasets, C. jejuni, H. pylori and Bifidobacterium longum, resulting from ancestral losses in Delta-Proteobacteria and Actinobacteridae, respectively [52]. In H. pylori, we indeed find no evidence of gBGC while the genomes are recombining at high frequency [50,51]. In C. jejuni and B. longum, however, we observe patterns similar to the other bacterial datasets that are in support of the existence of gBGC, indicating that it does not depend on the presence of a typical MutSL complex. The existence of gBGC in Bacteria and Eukaryotes however suggests that it may have been present in the last universal common ancestor of all cellular life forms (LUCA). Unfortunately, the only archaeal dataset matching our criteria was a group of Sulfolobus sp. genomes for which our analysis showed few evidence of recombination (Table 1), in agreement with the previously described isolation of endemic clades in this group [36].
We do not claim that gBGC is the unique determinant of base composition in bacterial genome: in fact there is evidence that mutation patterns vary significantly among species [22], and these variations are expected to contribute to differences in genome base composition. However, the model we propose provides a simple explanation for several important results of comparative bacterial genomics. First, gBGC can explain why bacterial genomes can maintain a high GC-content, even though mutation is universally AT-biased [22,23]. Second, gBGC can explain some of the intragenomic heterogeneity in GC-content observed in bacterial genomes. Indeed, we observe that genes with evidence of recombination display on average substantially higher GC-content than other genes. This observation also suggests that the probability of recombination is variable among genes in the genome, as proposed under some speciation models [53]. Furthermore, given that recently acquired genes tend to be AT-rich, gBGC would contribute to their progressive enrichment in GC-content [54,55].
gBGC, a new component of the neutral theory of evolution
The variations of GC-content in Bacteria have long remained unexplained. The results presented here highlight a strong relationship between the GC-content of genes and their history of recombination. This result, and the observation that bacterial genomes are generally above the GC-content predicted from their mutational bias towards AT, are fully consistent with the existence of gBGC. To explain our results under a selective model, one would have to hypothesize: i) that all bacterial species are under the same selection for higher GC throughout their genome, ii) that this selective pressure affects all positions of a genome, independently of gene function and expression, and iii) that the efficiency of selection varies with recombination (Hill-Robertson interference). Incidentally, if the correlations between GC-content and recombination (Fig. 3B) were due to Hill-Robertson interference, this would imply that all regions in a genome (except possibly the most GC-rich) are mal-adapted. We favor the gBGC model because it is much more parsimonious, and it relies on mechanisms that have already been uncovered in Eukaryotes. Ultimately, it will be possible to test experimentally the existence of gBGC by analyzing recombination products in bacteria.
Our discovery is important because gBGC has been shown to interfere with the efficiency of selection in Eukaryotes, and to lead to false positives in the search for regions under positive selection in a genome. The prevalence—if not universality—of this phenomenon underlines the importance of incorporating gBGC in the set of evolutionary forces to be considered when searching for signature of adaption in genomes.
Materials and Methods
Genomic datasets
We used the HOGENOM database [27] to select sets of genome sequences comprising at least six closely related strains or species. The selection of closely related genomes was based on a genomic distance derived from the HOGENOM6 database. For each pair of genomes present in the database, we computed the best identity score, as obtained using BLAST, within each family of homologous proteins as defined in HOGENOM, and averaged these scores to obtain a global similarity score s. We then took 1-s as a distance and selected groups of genomes with at least 6 members and a distance lower than 0.15. This criterion left 21 groups of species representing a variety of bacterial and archeal species (Table 1). For each gene family, CDSs were extracted using ACNUC Python API [56] and re-aligned with MUSCLE [57] using default parameters. GC% and codon frequencies were computed using custom Python scripts.
Detection of recombining genes
Detection of recombination based on multiple alignments is expected to be sensitive to both the number of sequences aligned, and the total length of the alignment. For this reason, we used only the universal unicopy genes of each species, and selected the 900 central positions of each nucleotide alignment (remaining positions as well as genes shorter than this threshold were ignored in our analysis). We used the software Phipack implementing the PHI test [28] (parameters: window size of 100bp, p-value computed from 1,000 permutations) to test if a gene family alignment contained evidence for recombination. Only families for which the site permutation test could be performed were considered, i.e. where the phylogenetic signal was sufficient to accept or reject the hypothesis of recombination. An alignment was determined to be “recombinant” if the p-value of the permutation test was lower than 0.05. To confirm the results obtained with PHI, we also used three other tests of recombination: NSS [58], MaxChi2 [59] and Geneconv [60]. NSS and MaxChi2 statistics were computed together with PHI (as implemented in Phipack package) using the same permutations to compute p-values. A consensus of PHI, NSS and MaxChi2 was done, classifying as recombinant or non-recombinant those gene families consistently detected by all those three methods as recombinant or non-recombinant, respectively; families with disagreeing results were discarded for the consensus analysis. Geneconv was run with the parameters “-GScale = 1 -Numsims = 10000 -Maxsimglobalpval = 0.05”, and a gene family was considered recombinant when at least one significant global recombinant fragment was reported; other families were considered non-recombinant. As Geneconv differs from the other methods on the nature of the reported evidence, it was not considered when defining a consensus classification. Individual tests and their consensus yielded quite different classifications of gene families, but all led to qualitatively very similar results (see S1 Fig., S3 Fig. and S4 Fig.). To test for the presence of gBGC, we performed Student’s t-tests comparing the mean GC-content (at all or at each separate codon position) of recombinant versus non-recombinant core gene family sets. All alignments as well as the results of the various tests for recombination are available at ftp://pbil.univ-lyon1.fr/pub/datasets/DAUBIN/Lassalle_PloS_Genet_2015
Estimation of recombination rate
We used ClonalOrigin [41] to estimate the recombination rate on full-length core gene family alignments of 900bp or more. As ClonalOrigin inferences are highly demanding in computation time and power, we only performed this analysis on the moderately sized dataset of S. pyogenes genomes (Spyo). The program was used with default parameter weights, running the MCMC with 1,000,000 burn-in generations and 500,000 generations sampled every 1,000 (command line was: ‘warg -a 1,1,0.1,1,1,1,1,1,0,0,0 -x 1000000 -y 500000 -z 1000’). Families for which this task could not be finished in less than 1 month computation were discarded, yielding a total dataset of 478 gene families (out of 496 core ≥900bp-long ones). A number of genes also exhibited very high variance in their estimates of rho and their exclusion yielded better correlations with GC3.
Recombination inference in intergenes
Intergenic regions are evolving fast, leading to inaccurate alignments that are not amenable to robust detection of recombination as performed on coding sequences. We thus chose to test the relationship of intergenic GC% with recombination in a gene-centered way: for each core protein-coding gene tested with PHI, we considered non-coding regions up to 400bp on both sides of a CDS and averaged their GC%, provided they were of a size larger than 50bp each (to avoid stochastic errors due to too small number of observed nucleotides). A measure of intergenic GC% per core gene family was obtained as the mean over all genomes of the previous values, and was associated to the recombinant/non-recombinant status of the gene family for further testing.
Frequencies of optimal codons
Optimal codons for each amino acid were computed by comparing synonymous codon frequencies within CDSs encoding ribosomal proteins (as defined from HOGENOM family annotations) versus all other CDSs (RP method). Under the hypothesis of selection for highly expressed genes to be adapted to the tRNA pool, codons statistically enriched in ribosomal proteins (Chi-squared test based on a 2×2 contigency table of counts of occurrence at a focal codon against those of its synonyms in ribosomal vs. other protein-coding genes, with p-value < 0.001) were considered as “optimal”; others were classified as “non-optimal”. (S2 Table). We then computed the absolute frequency of optimal (Fop) and non-optimal codons (Fnop) over all coding sequences. Fop and Fnop were calculated separately for codons pooled by composition at the third position, i.e. ending in A/U or G/C. As there is a debate on whether this method is appropriate to define optimal codons [38–40], we also used an alternative definition and took optimal codons datasets from a previous exhaustive survey of Hershberg and Petrov [38] (HCB method). A set of optimal codons was selected when determined in Hershberg and Petrov [38] study for several strains with a strong consensus, i.e. when >60% documented strains agreed on the preferred codon and remaining strains preferred a codon with the same composition (A/U or G/C) at the third position (found in all datasets but A. baumanii) (S3 Table).
Supporting Information
Zdroje
1. Doolittle WF (2013) Is junk DNA bunk? A critique of ENCODE. Proc Natl Acad Sci 110 : 5294–5300. doi: 10.1073/pnas.1221376110 23479647
2. Bernardi G, Olofsson B, Filipski J, Zerial M, Salinas J, et al. (1985) The mosaic genome of warm-blooded vertebrates. Science 228 : 953–958. 4001930
3. Sueoka N (1962) ON THE GENETIC BASIS OF VARIATION AND HETEROGENEITY OF DNA BASE COMPOSITION. Proc Natl Acad Sci 48 : 582–592. 13918161
4. McCutcheon JP, Moran NA (2010) Functional convergence in reduced genomes of bacterial symbionts spanning 200 My of evolution. Genome Biol Evol 2 : 708–718. doi: 10.1093/gbe/evq055 20829280
5. Pagani I, Liolios K, Jansson J, Chen I-MA, Smirnova T, et al. (2012) The Genomes OnLine Database (GOLD) v.4: status of genomic and metagenomic projects and their associated metadata. Nucleic Acids Res 40: D571–D579. doi: 10.1093/nar/gkr1100 22135293
6. Duret L, Galtier N (2009) Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet 10 : 285–311. doi: 10.1146/annurev-genom-082908-150001 19630562
7. Ratnakumar A, Mousset S, Glémin S, Berglund J, Galtier N, et al. (2010) Detecting positive selection within genomes: the problem of biased gene conversion. Philos Trans R Soc Lond B Biol Sci 365 : 2571–2580. doi: 10.1098/rstb.2010.0007 20643747
8. Spencer CC, Deloukas P, Hunt S, Mullikin J, Myers S, et al. (2006). The influence of recombination on human genetic diversity. PLoS Genetics, 2:e148. doi: 10.1371/journal.pgen.0020148 17044736
9. Duret L, Arndt PF (2008) The Impact of Recombination on Nucleotide Substitutions in the Human Genome. PLoS Genet 4: e1000071. doi: 10.1371/journal.pgen.1000071 18464896
10. Webster MT, Hurst LD (2012) Direct and indirect consequences of meiotic recombination: implications for genome evolution. Trends Genet 28 : 101–109. doi: 10.1016/j.tig.2011.11.002 22154475
11. Nagylaki T (1983) Evolution of a finite population under gene conversion. Proc Natl Acad Sci U S A 80 : 6278–6281. 6578508
12. Galtier N, Duret L (2007) Adaptation or biased gene conversion? Extending the null hypothesis of molecular evolution. Trends Genet TIG 23 : 273–277. doi: 10.1016/j.tig.2007.03.011 17418442
13. Galtier N, Duret L, Glémin S, Ranwez V (2009) GC-biased gene conversion promotes the fixation of deleterious amino acid changes in primates. Trends Genet TIG 25 : 1–5. doi: 10.1016/j.tig.2008.10.011 19027980
14. Necşulea A, Popa A, Cooper DN, Stenson PD, Mouchiroud D, et al. (2011) Meiotic recombination favors the spreading of deleterious mutations in human populations. Hum Mutat 32 : 198–206. doi: 10.1002/humu.21407 21120948
15. Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM (2008) High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 454 : 479–485. doi: 10.1038/nature07135 18615017
16. Williams A, Geneovese G, Dyer T, Truax K, Jun G, et al. (2014) Non-crossover gene conversions show strong GC bias and unexpected clustering in humans. bioRxiv: 009175. doi: 10.1101/009175
17. Capra JA, Pollard KS (2011) Substitution patterns are GC-biased in divergent sequences across the metazoans. Genome Biol Evol 3 : 516–527. doi: 10.1093/gbe/evr051 21670083
18. Escobar JS, Glémin S, Galtier N (2011) GC-biased gene conversion impacts ribosomal DNA evolution in vertebrates, angiosperms, and other eukaryotes. Mol Biol Evol 28 : 2561–2575. doi: 10.1093/molbev/msr079 21444650
19. Pessia E, Popa A, Mousset S, Rezvoy C, Duret L, et al. (2012) Evidence for widespread GC-biased gene conversion in eukaryotes. Genome Biol Evol 4 : 675–682. doi: 10.1093/gbe/evs052 22628461
20. Foerstner KU, von Mering C, Hooper SD, Bork P (2005) Environments shape the nucleotide composition of genomes. EMBO Rep 6 : 1208–1213. doi: 10.1038/sj.embor.7400538 16200051
21. Sueoka N (1988) Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci U S A 85 : 2653. 3357886
22. Hershberg R, Petrov DA (2010) Evidence That Mutation Is Universally Biased towards AT in Bacteria. PLoS Genet 6: e1001115. doi: 10.1371/journal.pgen.1001115 20838599
23. Hildebrand F, Meyer A, Eyre-Walker A (2010) Evidence of Selection upon Genomic GC-Content in Bacteria. PLoS Genet 6. doi: 10.1371/journal.pgen.1001107 20838593
24. Touchon M, Hoede C, Tenaillon O, Barbe V, Baeriswyl S, et al. (2009) Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5: e1000344. doi: 10.1371/journal.pgen.1000344 19165319
25. Rocha EPC, Feil EJ (2010) Mutational Patterns Cannot Explain Genome Composition: Are There Any Neutral Sites in the Genomes of Bacteria? PLoS Genet 6: e1001104. doi: 10.1371/journal.pgen.1001104 20838590
26. Raghavan R, Kelkar YD, Ochman H (2012) A selective force favoring increased G+C content in bacterial genes. Proc Natl Acad Sci 109 : 14504–14507. doi: 10.1073/pnas.1205683109 22908296
27. Penel S, Arigon A-M, Dufayard J-F, Sertier A-S, Daubin V, et al. (2009) Databases of homologous gene families for comparative genomics. BMC Bioinformatics 10: S3. doi: 10.1186/1471-2105-10-S6-S3 19534752
28. Bruen TC, Philippe H, Bryant D (2006) A Simple and Robust Statistical Test for Detecting the Presence of Recombination. Genetics 172 : 2665–2681. doi: 10.1534/genetics.105.048975 16489234
29. Ussery DW, Kiil K, Lagesen K, Sicheritz-Pontén T, Bohlin J, et al. (2009) The genus burkholderia: analysis of 56 genomic sequences. Genome Dyn 6 : 140–157. doi: 10.1159/000235768 19696499
30. Joseph SJ, Didelot X, Gandhi K, Dean D, Read TD (2011) Interplay of recombination and selection in the genomes of Chlamydia trachomatis. Biol Direct 6 : 28. doi: 10.1186/1745-6150-6-28 21615910
31. Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, et al. (1999) Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc Natl Acad Sci 96 : 14043–14048. doi: 10.1073/pnas.96.24.14043 10570195
32. Keim P, Johansson A, Wagner DM (2007) Molecular Epidemiology, Evolution, and Ecology of Francisella. Ann N Y Acad Sci 1105 : 30–66. doi: 10.1196/annals.1409.011 17435120
33. Supply P, Marceau M, Mangenot S, Roche D, Rouanet C, et al. (2013) Genomic analysis of smooth tubercle bacilli provides insights into ancestry and pathoadaptation of Mycobacterium tuberculosis. Nat Genet 45 : 172–179. doi: 10.1038/ng.2517 23291586
34. Wattam AR, Williams KP, Snyder EE, Almeida NF Jr, Shukla M, et al. (2009) Analysis of ten Brucella genomes reveals evidence for horizontal gene transfer despite a preferred intracellular lifestyle. J Bacteriol 191 : 3569–3579. doi: 10.1128/JB.01767-08 19346311
35. Whitaker RJ, Grogan DW, Taylor JW (2003) Geographic Barriers Isolate Endemic Populations of Hyperthermophilic Archaea. Science 301 : 976–978. doi: 10.1126/science.1086909 12881573
36. Reno ML, Held NL, Fields CJ, Burke PV, Whitaker RJ (2009) Biogeography of the Sulfolobus islandicus pan-genome. Proc Natl Acad Sci 106 : 8605–8610. doi: 10.1073/pnas.0808945106 19435847
37. McVean GA, Charlesworth B (2000) The effects of Hill-Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics 155 : 929–944. 10835411
38. Hershberg R, Petrov DA (2009) General Rules for Optimal Codon Choice. PLoS Genet 5: e1000556. doi: 10.1371/journal.pgen.1000556 19593368
39. Wang B, Shao Z-Q, Xu Y, Liu J, Liu Y, et al. (2011) Optimal Codon Identities in Bacteria: Implications from the Conflicting Results of Two Different Methods. PLoS ONE 6: e22714. doi: 10.1371/journal.pone.0022714 21829489
40. Hershberg R, Petrov DA (2012) On the Limitations of Using Ribosomal Genes as References for the Study of Codon Usage: A Rebuttal. PLoS ONE 7: e49060. doi: 10.1371/journal.pone.0049060 23284622
41. Didelot X, Lawson D, Darling A, Falush D (2010) Inference of Homologous Recombination in Bacteria Using Whole Genome Sequences. Genetics 186 : 1435–1449. doi: 10.1534/genetics.110.120121 20923983
42. Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449 : 851–861. doi: 10.1038/nature06258 17943122
43. Balbi KJ, Rocha EPC, Feil EJ (2009) The Temporal Dynamics of Slightly Deleterious Mutations in Escherichia coli and Shigella spp. Mol Biol Evol 26 : 345–355. doi: 10.1093/molbev/msn252 18984902
44. Cole F, Baudat F, Grey C, Keeney S, de Massy B, et al. (2014) Mouse tetrad analysis provides insights into recombination mechanisms and hotspot evolutionary dynamics. Nat Genet 46 : 1072–1080. doi: 10.1038/ng.3068 25151354
45. Munch K, Mailund T, Dutheil JY, Schierup MH (2014) A fine-scale recombination map of the human-chimpanzee ancestor reveals faster change in humans than in chimpanzees and a strong impact of GC-biased gene conversion. Genome Res 24 : 467–474. doi: 10.1101/gr.158469.113 24190946
46. Moran NA (2002) Microbial minimalism: genome reduction in bacterial pathogens. Cell 108 : 583–586. 11893328
47. Lynch M (2010) Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci 107 : 961–968. doi: 10.1073/pnas.0912629107 20080596
48. Lesecque Y, Mouchiroud D, Duret L (2013) GC-Biased Gene Conversion in Yeast Is Specifically Associated with Crossovers: Molecular Mechanisms and Evolutionary Significance. Mol Biol Evol 30 : 1409–1419. doi: 10.1093/molbev/mst056 23505044
49. Glémin S (2010) Surprising fitness consequences of GC-biased gene conversion: I. Mutation load and inbreeding depression. Genetics 185 : 939–959. doi: 10.1534/genetics.110.116368 20421602
50. Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, et al. (1998) Free recombination within Helicobacter pylori. Proc Natl Acad Sci 95 : 12619–12624. doi: 10.1073/pnas.95.21.12619 9770535
51. Falush D, Kraft C, Taylor NS, Correa P, Fox JG, et al. (2001) Recombination and mutation during long-term gastric colonization by Helicobacter pylori: Estimates of clock rates, recombination size, and minimal age. Proc Natl Acad Sci 98 : 15056–15061. doi: 10.1073/pnas.251396098 11742075
52. Lin Z, Nei M, Ma H (2007) The origins and early evolution of DNA mismatch repair genes-multiple horizontal gene transfers and co-evolution. Nucleic Acids Res 35 : 7591–7603. doi: 10.1093/nar/gkm921 17965091
53. Retchless AC, Lawrence JG (2007) Temporal fragmentation of speciation in bacteria. Science 317 : 1093–1096. doi: 10.1126/science.1144876 17717188
54. Daubin V, Lerat E, Perrière G (2003) The source of laterally transferred genes in bacterial genomes. Genome Biol 4: R57. doi: 10.1186/gb-2003-4-9-r57 12952536
55. Daubin V, Ochman H (2004) Bacterial Genomes as New Gene Homes: The Genealogy of ORFans in E. coli. Genome Res 14 : 1036–1042. doi: 10.1101/gr.2231904 15173110
56. Gouy M, Gautier C, Attimonelli M, Lanave C, di Paola G (1985) ACNUC—a portable retrieval system for nucleic acid sequence databases: logical and physical designs and usage. Comput Appl Biosci CABIOS 1 : 167–172. 3880341
57. Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5 : 113. doi: 10.1186/1471-2105-5-113 15318951
58. Jakobsen IB, Easteal S (1996) A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Comput Appl Biosci CABIOS 12 : 291–295. 8902355
59. Smith JM (1992) Analyzing the mosaic structure of genes. J Mol Evol 34 : 126–129. 1556748
60. Sawyer S (1989) Statistical tests for detecting gene conversion. Mol Biol Evol 6 : 526–538. 2677599
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2015 Číslo 2
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Genomic Selection and Association Mapping in Rice (): Effect of Trait Genetic Architecture, Training Population Composition, Marker Number and Statistical Model on Accuracy of Rice Genomic Selection in Elite, Tropical Rice Breeding Lines
- Discovery of Transcription Factors and Regulatory Regions Driving Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling
- Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
- Proteotoxic Stress Induces Phosphorylation of p62/SQSTM1 by ULK1 to Regulate Selective Autophagic Clearance of Protein Aggregates
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy