
FANCJ/BACH1 Acetylation at Lysine 1249 Regulates the DNA Damage Response
BRCA1 promotes DNA repair through interactions with multiple proteins, including CtIP and FANCJ (also known as BRIP1/BACH1). While CtIP facilitates DNA end resection when de-acetylated, the function of FANCJ in repair processing is less well defined. Here, we report that FANCJ is also acetylated. Preventing FANCJ acetylation at lysine 1249 does not interfere with the ability of cells to survive DNA interstrand crosslinks (ICLs). However, resistance is achieved with reduced reliance on recombination. Mechanistically, FANCJ acetylation facilitates DNA end processing required for repair and checkpoint signaling. This conclusion was based on the finding that FANCJ and its acetylation were required for robust RPA foci formation, RPA phosphorylation, and Rad51 foci formation in response to camptothecin (CPT). Furthermore, both preventing and mimicking FANCJ acetylation at lysine 1249 disrupts FANCJ function in checkpoint maintenance. Thus, we propose that the dynamic regulation of FANCJ acetylation is critical for robust DNA damage response, recombination-based processing, and ultimately checkpoint maintenance.
Published in the journal:
. PLoS Genet 8(7): e32767. doi:10.1371/journal.pgen.1002786
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002786
Summary
BRCA1 promotes DNA repair through interactions with multiple proteins, including CtIP and FANCJ (also known as BRIP1/BACH1). While CtIP facilitates DNA end resection when de-acetylated, the function of FANCJ in repair processing is less well defined. Here, we report that FANCJ is also acetylated. Preventing FANCJ acetylation at lysine 1249 does not interfere with the ability of cells to survive DNA interstrand crosslinks (ICLs). However, resistance is achieved with reduced reliance on recombination. Mechanistically, FANCJ acetylation facilitates DNA end processing required for repair and checkpoint signaling. This conclusion was based on the finding that FANCJ and its acetylation were required for robust RPA foci formation, RPA phosphorylation, and Rad51 foci formation in response to camptothecin (CPT). Furthermore, both preventing and mimicking FANCJ acetylation at lysine 1249 disrupts FANCJ function in checkpoint maintenance. Thus, we propose that the dynamic regulation of FANCJ acetylation is critical for robust DNA damage response, recombination-based processing, and ultimately checkpoint maintenance.
Introduction
The hereditary breast cancer associated gene product, BRCA1 is an essential tumor suppressor. To promote genomic stability, BRCA1 interacts with multiple protein partners. In particular, through its C-terminal BRCT repeats, BRCA1 directly interacts with Abraxas, CtIP and FANCJ (also known as BRIP1 or BACH1 (BRCA1-associated C-terminal helicase 1)). These BRCT-interacting proteins contribute to the function of BRCA1 in the DNA damage response (DDR). Abraxas serves to localize BRCA1 to sites of DNA damage and CtIP promotes the initiation of DNA end resection, which is critical for HR [1]–[3]. FANCJ also participates in localizing BRCA1 to sites of DNA damage, in DNA repair, and in checkpoint signaling; however, its distinct function is less clear.
Elucidating how FANCJ functions in the DDR is important, as mutations in the FANCJ gene are associated with hereditary breast cancer as well as with the rare cancer prone syndrome Fanconi anemia (FA) within the FANCJ patient complementation group (FA-J) [4]. As a DEAH-family helicase, it is expected that FANCJ metabolizes DNA substrates to facilitate DNA repair. Consistent with this idea, recombinant-FANCJ is a 5′-3′ helicase and translocase that can unwind D-loops and displace RAD51 [5]. In cells, FANCJ also localizes to sites of DNA damage. Furthermore, when FANCJ is absent, catalytically inactive, or lacks BRCA1 binding, cells display defects in double strand break repair (DSBR) and HR [6]–[9]. Recently, FANCJ was identified as a factor essential for maintaining the DNA damage induced checkpoint in response to ionizing radiation [10]. Despite these findings, FANCJ-deficient cells are only mildly sensitive to agents that induce DSBs [11].
To explain these findings, it has been proposed that FANCJ functions in DSBR, but has a more significant role in processing replication forks stalled at lesions, such as DNA interstrand crosslinks (ICLs). In support of this idea, FANCJ-null cells, similar to other FA patient cells, are extremely sensitive to agents that induce ICLs, such as cisplatin, melphalan, or mitomycin C (MMC) [7], [12], [13]. This sensitivity is reversed by complementation of FA-J cells with wild-type FANCJ (FANCJWT), but not with catalytically inactive FANCJ mutants [6], [8], [14]. Interestingly, the mechanism by which FANCJ mediates ICL processing is regulated by BRCA1 binding. HR is favored when BRCA1 binds FANCJ. When BRCA1 binding is prevented, lesion bypass is favored by a mechanism requiring the translesion synthesis polymerase polη [9]. Thus, complementation of FA-J cells with a BRCA1-interaction defective mutant FANCJS990A reverses ICL sensitivity but does not fully restore FANCJ function.
Here, we present evidence that FANCJ contributes to lesion processing by promoting a robust DDR. Essential for this function is FANCJ acetylation on a specific lysine residue. As such, preventing FANCJ acetylation suppresses DNA end resection that normally serves to engage recombination-based processing. Thus, both BRCT-interacting proteins, CtIP and FANCJ undergo DNA damage induced changes in acetylation that further regulates their function in the DDR to promote genomic stability.
Results
FANCJ is acetylated by CBP and deacetylated by HDAC3 or SIRT1
As observed for CtIP, FANCJ binds directly to the BRCT domains of BRCA1 [6], [9], [15]. Given that CtIP function is inactivated by acetylation [16], we addressed whether FANCJ was similarly modified. For this analysis, myc-tagged FANCJ was co-transfected with various Flag- or HA-tagged histone acetyltransferases. In an immunoblot probed with a pan-acetyl lysine antibody, we found that the precipitated FANCJ was acetylated only when CBP was over-expressed (Figure 1A). Moreover, FANCJ acetylation was induced by CBP in a dose dependent manner (Figure 1B).
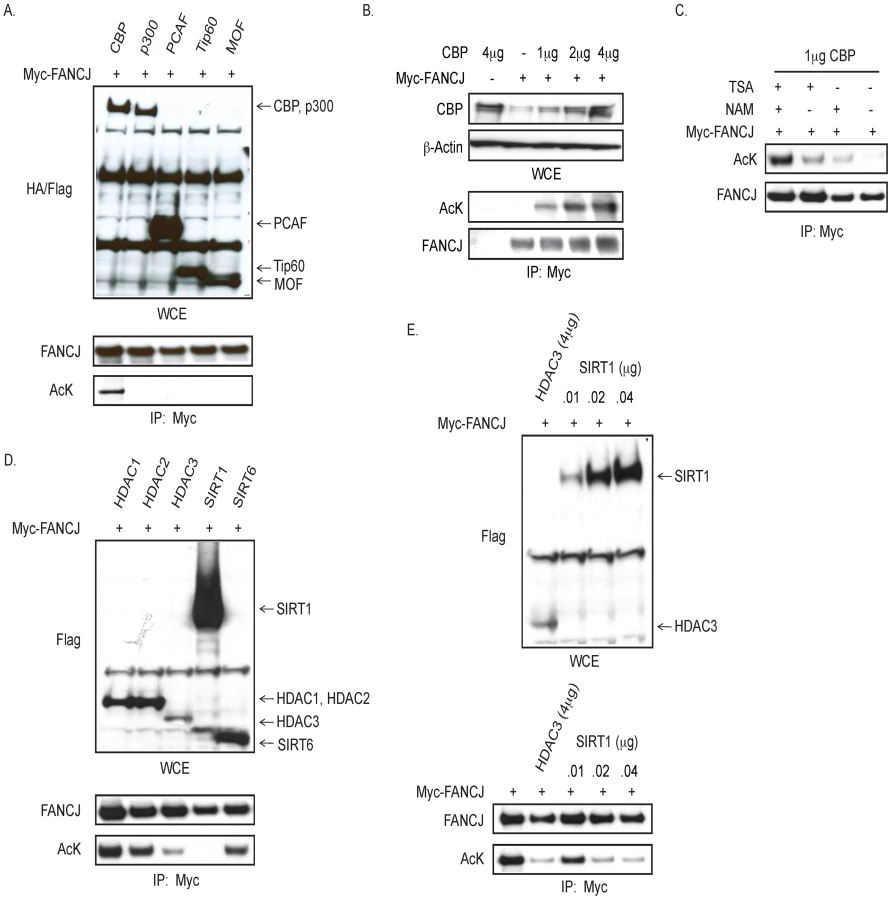
FANCJ acetylation was preserved most effectively by the inclusion of two types of deacetylase inhibitors, trichostatin-A (TSA) and nicotinamide (NAM) (Figure 1C). Thus, we considered that more than one class of histone deacetylase (HDAC) could deacetylate FANCJ. TSA inactivates class 1 and class II HDACs, whereas NAM inactivates the nicotinamide adenoine dinucleotide (NAD+)-dependent sirtuin (class III) family of HDACs (including SIRT1 to SIRT7) [17]. FANCJ acetylation was reduced more when either Flag-tagged-HDAC3 or SIRT1 were overexpressed in 293T cells than observed upon overexpression of HDAC1, HDAC2, or SIRT6 (Figure 1D). Titration of the SIRT1 expression vector transfected into 293T cells revealed that 0.01 µg of the SIRT1 construct matched the expression level of 4 µg of the HDAC3 construct. At this similar level of expression, HDAC3 more efficiently deactylated FANCJ than did SIRT1 (Figure 1E). Together, these data implicate that FANCJ can be acetylated by CBP and deacetyated by HDAC3 as well a SIRT1 when over-expressed.
FANCJ is acetylated at lysine residue 1249
To identify the FANCJ acetylation site(s), myc-tagged C-terminal FANCJ truncation mutants were co-transfected with CBP into 293T cells. By Immunoblot analysis using the pan-acetyl antibody, we found that acetylation of FANCJ required amino acids 1239 to 1249 (Figure 2A, 2C). Consistent with this region being modified, a C-terminal domain of FANCJ similar to a C-terminal p53 control was acetylated in vitro by a HAT-domain protein (Figure 2B, 2C). To determine, which of three lysine (K) residues in this C-terminal region were required for acetylation, we generated three independent FANCJ mutant constructs that converted lysines 1240, 1242, or 1249 to arginine (R). Further transfection experiments revealed that the K1249 was the dominant site for FANCJ acetylation, a lysine that is not conserved in chicken or C. elegans FANCJ species (Figure 2D, 2E).
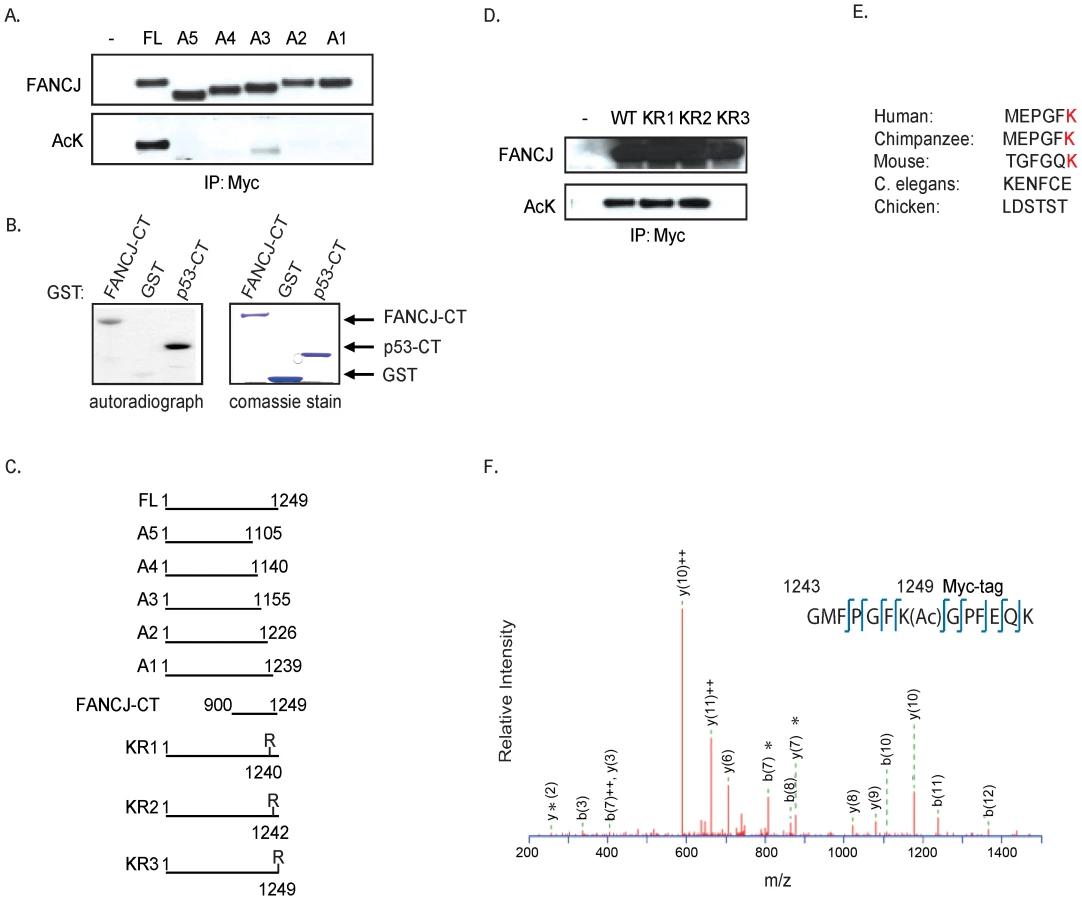
Next, we sought to provide more conclusive evidence that CBP-induced acetylation on FANCJ was at the K1249 site. We purified FANCJ from 293T cells transfected with a C-terminal myc-tagged FANCJWT or the FANCJK1249R mutant species by immunoprecipitation using a myc antibody. Isolated proteins were then digested with trypsin and subjected to tandem mass spectrometry analysis (LC-MS/MS). FANCJ-derived peptides covering the entire sequence were analyzed, and acetylation sites were identified using MASCOT search algorithm. Most of the acetylated lysine residues were detected in overlapping peptides derived from at least two independent protein preparations. In the FANCJWT, one of these sites was K1249 (Figure 2F). Interestingly, even though by antibody detection, the FANCJK1249R mutant scores unmodified as in Figure 2D; FANCJWT and FANCJK1249R mutant had three additional acetylation marks detected by mass spectrometry (Figure S1). Furthermore, the K1249R mutant had five additional acetylated lysines not found in wild-type FANCJ, suggesting that these sites are not available when K1249 is acetylated (Figure S1). Thus, immunoblot and mass spectrometry analysis confirm that the very last amino acid of FANCJ, lysine 1249 is acetylated.
FANCJ acetylation is enhanced by certain forms of DNA damage
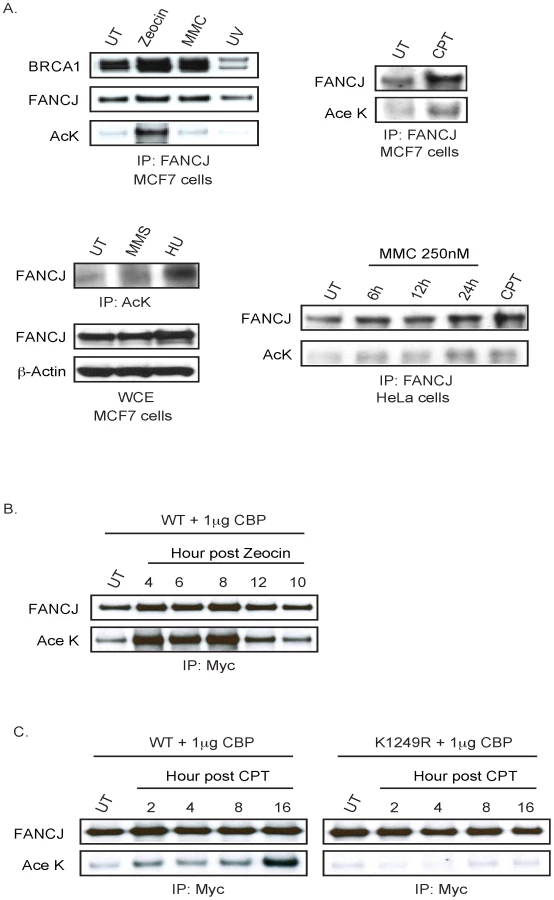
Given that DNA damage reduces CtIP acetylation [16], we addressed whether DNA damage could alter FANCJ acetylation. Endogenous FANCJ acetylation was enhanced in MCF7 cells treated with zeocin, camptothecin (CPT), or hydroxyurea (HU) as compared to ultraviolet radiation (UV), MMC, or methyl methanesulfonate (MMS) at the dose and time-post treatment analyzed (Figure 3A). Notably, zeocin had a more robust induction of FANCJ acetylation despite the dose of zeocin, CPT, or UV having similar affect on cell survival (Figure S2; data not shown). As found previously, DNA damage did not measurably alter FANCJ co-precipitation with BRCA1 with the exception of UV damage, which could reflect the UV-induced BRCA1 degradation [11], [18] (Figure 3A). DNA damage also induced FANCJ acetylation in HeLa cells, in response to not only CPT, but also MMC (Figure 3A). In response to DNA damage, we also noted that FANCJ protein levels were sometimes enhanced (Figure 3A). To clarify whether acetylation or our ability to detect acetylation was induced by DNA damage, we sought to induce DNA damage in cells in which our ability to detect FANCJ acetylation was not limiting. Indeed, the amount of acetylation on similar levels of exogenous FANCJWT achieved with low dose CBP expression was considerably enhanced following treatment with zeocin or CPT (Figure 3B, 3C). Interactions with BRCA1 and MLH1 were not required for the CBP-induced acetylation of FANCJ, because BRCA1- and MLH1-interaction-defective mutants, FANCJS990A and FANCJK141/142A were readily modified (data not shown). In contrast, following treatment with CPT, acetylation was not detected on the FANCJK1249R mutant (Figure 3C), indicating that DNA damage-induced FANCJ acetylation requires the C-terminal K1249 residue. It remains to be determined, however if FANCJ acetylation is induced by a distinct type of DNA damage.
FANCJ acetylation mutants are functional
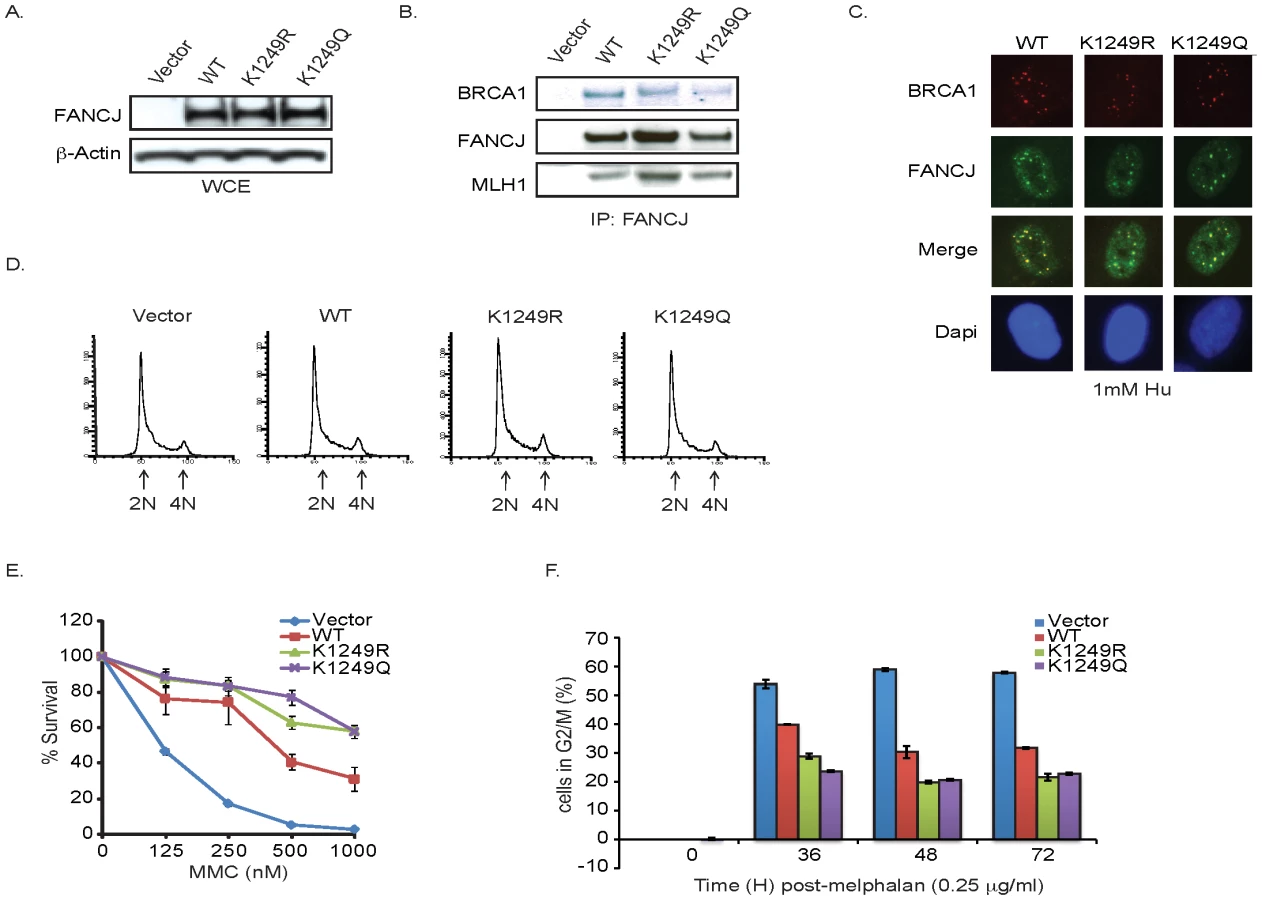
The enhanced FANCJ acetylation following DNA damage led us to hypothesize that this modification facilitated FANCJ function in DNA repair. To address this possibility, we made use of this lysine to arginine FANCJK1249R mutant that prevents acetylation and also generated a lysine to glutamine FANCJK1249Q mutant to structurally mimic acetylation. Consistent with these mutants being functional, the purified recombinant proteins displayed similar catalytic activities as FANCJWT (Figure S3). In addition, they were expressed at similar levels as FANCJWT in FANCJ-null FA-J cells (Figure 4A). Similar to FANCJWT, FANCJK1249R and FANCJK1249Q precipitated with known FANCJ interacting partners, BRCA1 and MLH1 [6], [8] (Figure 4B). In addition, the mutants co-localized with BRCA1 in response to DNA damage and the FA-J cells expressing FANCJWT or mutants had similar asynchronous cell cycle profiles (Figure 4C, 4D). The acetylation mutants also restored MMC resistance and the ability of FA-J cells to exit from an abnormal G2/M accumulation, albeit in a manner slightly more robust than FANCJWT (Figure 4E, 4F). Together, these findings suggested that the mutants were enzyme active and functional in vivo; however the mechanism by which the FANCJ mutants restore ICL resistance could be distinct from FANCJWT.
FANCJ acetylation contributes to the mechanism of lesion processing
Previously, complementation of FA-J cells with a BRCA1-binding defective mutant, FANCJS990A gave the semblance of FANCJWT function. In particular, MMC resistance was restored [8]. However, in contrast to FANCJWT, FANCJS990A provides resistance to MMC by a mechanism dependent on the DNA damage tolerance pathway. Within this tolerance pathway, translesion synthesis polymerases can bypass DNA lesions such as unhooked ICLs and intra-strand crosslinks generated by UV, but not DSBs generated by zeocin. Evidence that FANCJS990A skewed lesion processing towards DNA damage tolerance was based on several findings. First, the sensitivity to MMC in these cells was restored upon depletion of the essential tolerance factor, Rad18 or the translesion polymerase polη, but not upon depletion of the HR protein, Rad54. Second, in comparison to FANCJWT, cells expressing FANCJS990A were hyper-resistant to UV, a phenotype that was reversed upon polη-depletion. Third, in comparison to FANCJWT, FANCJS990A-expressing cells were sensitive to zeocin, indicating reduced DSBR [9]. Thus, we sought to determine whether similar to the BRCA1-binding mutant, the acetylation mutants also functioned differently from FANCJWT. To test this idea, the FA-J cell lines were left untreated or treated with increasing doses of MMC, zeocin, or UV. In comparison to the other FA-J cell lines, the FA-J cell line expressing the acetylation mutant FANCJK1249R was hyper-resistant to UV, but unable to restore normal levels of zeocin resistance. In contrast, the FA-J cell line expressing the acetylation mimic FANCJK1249Q displayed greater resistance to zeocin (Figure 5A; Figure S2). Thus, in response to UV and zeocin, cells expressing the acetylation mutants are distinct from each other as well as from cells expressing FANCJWT.
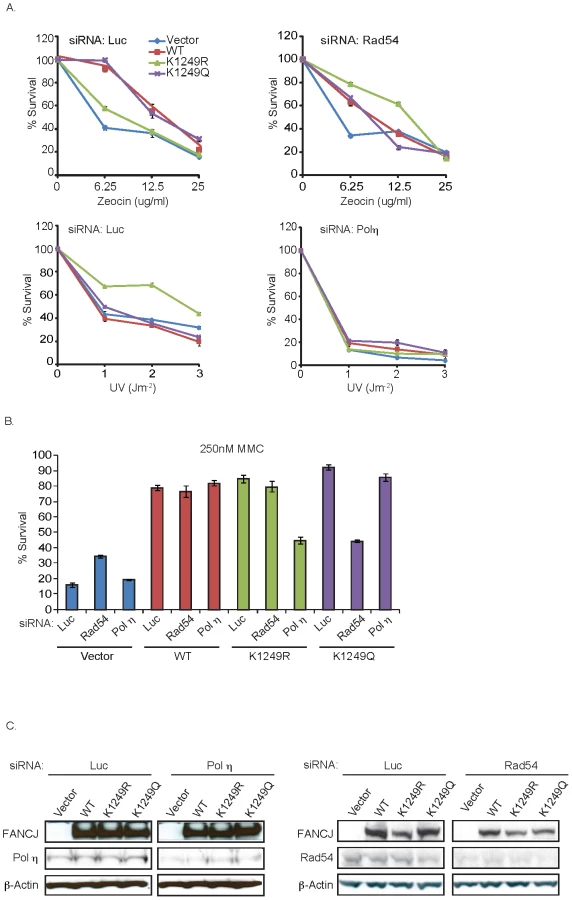
To further validate these results, we targeted recombination or DNA damage tolerance pathways by using siRNA reagents to Rad54 or polη. Significantly, depletion of Rad54 suppressed the zeocin resistance of the FA-J cell line expressing FANCJK1249Q (Figure 5A, 5C). Likewise, depletion of polη suppressed the UV hyper-resistance of the FA-J cell line expressing FANCJK1249R (Figure 5A, 5C). Furthermore, depletion of polη, but not Rad54 reversed the MMC resistance of the FA-J cell line expressing FANCJK1249R (Figure 5B). In contrast, depletion of Rad54, but not polη reduced the MMC resistance of the FA-J cell line expressing FANCJK1249Q (Figure 5B). Together, these results indicate that the acetylation of FANCJ at lysine 1249 contributes to the mechanism of lesion processing; preventing acetylation favors DNA damage tolerance and constitutive acetylation favors recombination.
FANCJ acetylation is required for a robust DDR
How could FANCJ acetylation affect lesion processing? Because both CtIP and FANCJ are acetylated and directly bind to the BRCA1-BRCT domain, we speculated that FANCJ might similarly have a role in DNA end resection. In particular, the affect of CtIP acetylation on DNA end resection was analyzed in response to CPT [16]. We found RPA foci formation at 1 h post-CPT was more robust (64% and 65%) in the FANCJWT and FANCJK1249Q FA-J cell lines as compared to vector and FANCJK1249R FA-J cell lines that had 47% and 29%, respectively (Figure 6A). Thus, as measured by RPA foci formation, FANCJWT and the acetylation mimic FANCJK1249Q were more active in DNA end resection.

RPA loading onto ssDNA also leads to its subsequent phosphorylation on Ser4 and Ser8 [3]. We found that the FA-J cell lines had a similar phosphorylation of Chk2 and γ-H2AX following exposure to two different dose of CPT, indicating that FANCJ or its ability to be acetylated is not required for DSB formation in response to CPT (Figure 6B). Likewise, at 1 h post-CPT treatment, Chk1 phosphorylation was detected (Figure 6B). In contrast, RPA phosphorylation was most robust in the CPT-treated FANCJWT and FANCJK1249Q FA-J cell lines (Figure 6B). In support of these findings, reduced RPA phosphorylation was also detected in CPT-treated FANCJ-deficient U2OS cells generated by siRNA reagents (Figure S4). Furthermore, at 4–24 h post CPT treatment, we noted diminished RPA phosphorylation in FANCJK1249R as compared with FANCJWT and FANCJK1249Q FA-J cell lines (Figure 6C). At this time, RPA phosphorylation in the FANCJK1249R FA-J cells was also reduced compared to vector FA-J cells that had gained considerable RPA phosphorylation as compared to 1 h post-CPT (Figure 6B, 6C). In the response to zeocin, which induces DSBs independent of replication, RPA phosphorylation was similar in FA-J cell lines with or without FANCJWT (Figure S5). Together, these results suggest a role for FANCJ and its acetylation in DNA end resection at stalled replication forks as induced by CPT.
To address whether the contribution of FANCJ acetylation to DNA end resection was sufficient to enhance HR, we next analyzed Rad51 foci formation. In response to CPT, we found that Rad51 foci were the most robust in FA-J cells complemented with FANCJWT or the FANCJK1249Q mutant. Instead, Rad51 foci in the FA-J cells with vector or FANCJK1249R were more anemic (Figure 7A). Furthermore, a greater number of FANCJK1249Q expressing FA-J cells were positive for Rad51 foci as quantitated between 2–16 h after CPT treatment (Figure 7A). In contrast, the γ-H2AX foci did not have a significant difference between the FA-J cells lines. Thus, a greater proportion of γ-H2AX co-staining Rad51 foci were detected in FANCJK1249Q or FANCJWT, as compared to vector or FANCJK1249R expressing FA-J cells (Figure 7A merge). Together, these findings demonstrate that in response to CPT, FANCJ and its acetylation at 1249 promote DNA end processing events that enhance RPA phosphorylation, and both RPA and Rad51 focal accumulation.
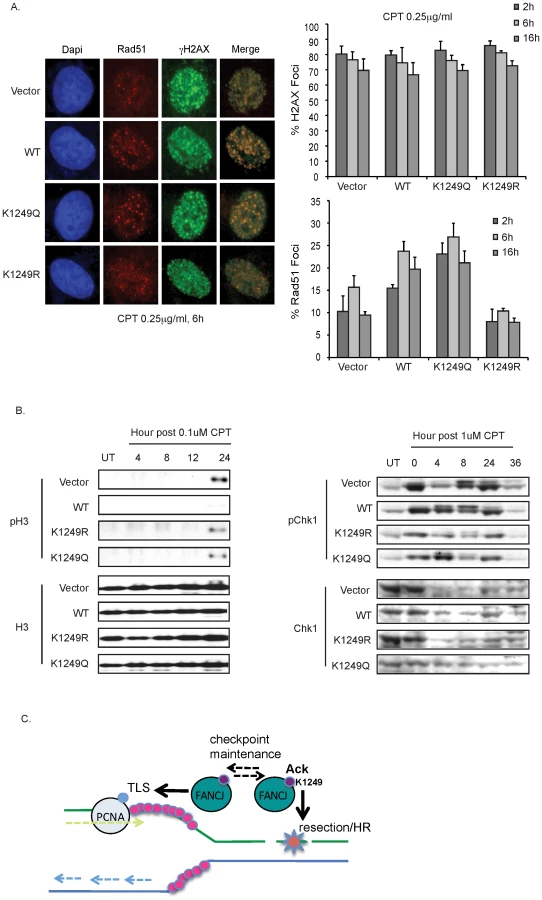
Given these findings and the recent identification that FANCJ promotes checkpoint maintenance [10], we considered that FANCJ acetylation could be essential for maintaining the checkpoint. Defects in checkpoint maintenance were evaluated by determining if CPT treated FA-J cells traversed prematurely to mitosis. FA-J cells lacking FANCJWT entered mitosis by 24 h post-CPT as indicated by a positive histone H3 phosphorylation (Figure 7B). These results are consistent with FANCJ acetylation supporting checkpoint maintenance. However, FA-J cells expressing FANCJK1249R or FANCJK1249Q also failed to maintain the checkpoint, showing H3 phosphorylation by 24 h (Figure 7B). Substantiating this finding, at time points greater than 4 h post-CPT treatment, both mutants had reduced Chk1 phosphorylation as compared to FA-J cells expressing FANCJWT (Figure 7B). Collectively, these findings suggest that FANCJ acetylation enhances the initial DDR to facilitate recombination-based repair and limit translesion synthesis. Checkpoint maintenance however, requires FANCJ and its dynamic regulation by acetylation (Figure 7C).
Discussion
Here we identify acetylation as a DNA damage-dependent regulator of the BRCA-FA protein, FANCJ. We show that acetylation at lysine 1249 is a critical regulator of FANCJ function during cellular DNA repair. We analyzed the expression of two FANCJ mutants that mimicked either the constitutive deacetylated FANCJK1249R or acetylated FANCJK1249Q protein isoforms. While the mutants functioned similar to FANCJWT in several assays and restored MMC resistance and exit from an abnormal G2/M checkpoint response to FA-J cells, the mutants were distinct from FANCJWT with respect to lesion processing. Notably, FA-J cells expressing the acetylation mutants differentially relied on repair and tolerance factors for resistance to DNA damaging agents. Our findings further demonstrate that FANCJ has the ability to potentiate HR and DNA damage induced acetylation is important for this function.
Another BRCA1-BRCT interacting protein, CtIP is acetylated and functions in DNA end resection. Thus, we considered that recombination-based lesion processing by the FANCJ acetylation mimic, FANCJK1249Q resulted from a function for FANCJ acetylation in DNA end resection. To test this idea, the FA-J cells were treated with CPT, which generates breaks in S-phase. Indeed, FA-J cells expressing the acetylation mutants were distinct in the initial response to CPT. Specifically, FA-J cells expressing the FANCJK1249Q, but not FANCJK1249R, promoted DNA end resection post-CPT exposure as measured by presentation of RPA foci or its phosphorylation at serine residues 4 and 8. Furthermore, FA-J cells expressing the FANCJK1249Q, as compared to FANCJK1249R had 2.5-fold more cells with CPT-induced Rad51 foci. This more robust DDR could reflect a role for FANCJ acetylation in loading RPA, as shown for FANCJ in response to HU [19]. Our data do not implicate a global role for FANCJ in DNA end resection given that FANCJ-deficiency did not affect the amount of RPA phosphorylation following zeocin, an agent that induces DSBs independent of replication. However, FANCJ is acetylated when cells are exposed to CPT or zeocin. Thus, DSB-induced FANCJ acetylation that is not associated with stalled or broken replication forks may contribute to some other aspect of the DDR, such as checkpoint maintenance.
In fact, we find that FANCJ as well as its acetylation are essential for checkpoint maintenance. Specifically, in the absence of FANCJ or its DNA damage induced acetylation, Chk1 phosphorylation was induced, but not maintained and correspondingly cells underwent a more rapid transit into mitosis post-CPT. Interestingly, we found that similar to FA-J cells expressing the acetylation mutant FANCJK1249R, FA-J cells expressing the acetylation mimic FANCJK1249Q failed to maintain the checkpoint despite an initial DDR to CPT. Thus, some other aspect of checkpoint signaling is perturbed in FA-J cells that express the acetylation mimic. Perhaps this mutant fails to mediate a protein interaction or act upon a DNA substrate important for checkpoint maintenance. Instead FANCJ acetylation could serve as a switch, in which acetylation and de-acetylation is essential to maintain the checkpoint (Figure 7C). Consistently, a role for FANCJ in checkpoint maintenance was reported in a recent study [10].
It follows that defects in initiating the DDR, engaging HR, and maintaining the checkpoint impact cellular DNA damage resistance. Reduced DNA repair and/or checkpoint maintenance defects could explain why FA-J cells expressing the acetylation mutant FANCJK1249R were sensitive to zeocin. Defects in repair and in maintaining the checkpoint may not increase cellular sensitivity if backup lesion processing mechanisms serve to process or bypass the lesion. Compensatory pathways could explain the lack of CPT-sensitivity in the FA-J cells with or without acetylation mutants (Figure S2). In support of this idea, our data reveal that FA-J cells expressing the acetylation mutant were resistant to DNA damage by relying on tolerance factors. As such, depletion of polη in FA-J cells expressing the non-acetylatable FANCJK1249R mutant reversed the UV and MMC resistance. Instead, FA-J cells expressing the acetylation mimic FANCJK1249Q maintained zeocin and MMC resistance in a Rad54-dependent manner. These findings suggest that the toxicity to ICLs lesions as found in cells deficient for FANCJ is avoided because FANCJ enzyme active acetylation mutants facilitate recombination in S phase or translesion synthesis bypass of unhooked ICL lesions perhaps in mitosis. In the absence of a maintained checkpoint, however recombination similar to translesion synthesis bypass is likely to be error-prone.
Previously, we found that BRCA1 binding to FANCJ altered FANCJ function in HR and translesion synthesis pathways. Indeed, we find that similar to FA-J cells expressing the acetylation FANCJK1249R mutant, FA-J cells expressing the BRCA-interaction defective mutant, FANCJS990A were hyper-resistant to UV induced damage, sensitive to zeocin induced damage, and relied on polη for MMC resistance [7]. Data also indicate that similar to FANCJK1249R, the FANCJS990A mutant fails to maintain the checkpoint. In response to melphalan treatment, FA-J cells expressing the FANCJS990A mutant, as compared to FANCJWT, underwent a reduced and more rapid G2/M checkpoint exit [7]. These similar outcomes do not reflect common defects in BRCA1 binding or acetylation. Indeed, the FANCJS990A mutant was acetylated upon co-transfection of CBP to levels similar to those observed for FANCJWT (data not shown). Moreover, co-precipitation experiments demonstrated that the FANCJK1249R mutant bound BRCA1 as well as FANCJWT. Thus, BRCA1 binding and acetylation of FANCJ may be distinct events. Nevertheless, defects in BRCA1 binding at serine 990 or acetylation at lysine 1249 could have similar outcomes for FANCJ function because both mutants fail to maintain a robust checkpoint and Rad51-based repair is reduced [9].
A stalled replication fork with exposed single stranded and double stranded regions could provide an ideal DNA substrate for FANCJ. Indeed, FANCJ requires several nucleotides for binding and metabolizing DNA [20]. FANCJ function in replication fork processing could also be similar to other 5′-3′ DNA helicase/translocases such as Ecoli RecD and yeast Rad3. Rad3 facilitates exonucleolytic degradation of DNA ends, which restricts recombination between short homologous sequences [21]. Interestingly, RecD regulates resection and recombination by changes in helicase speed, which can also facilitate a polymerase swap, in which bypass polymerases diminish fork break down [22]. Conceivably, enhanced FANCJ enzyme activity or altered substrate preference due to acetylation could generate more single-stranded DNA to elicit checkpoint responses such as RPA loading as proposed [19]. Alternatively, checkpoint maintenance could require reduced FANCJ enzyme activity so that FANCJ does not displace proteins from lesions, such as RAD51 or interacting partners BRCA1, RPA and BLM helicase [6], [23]–[25]. In this context, it is worth noting that changes in motor speed have been associated with FANCJ clinical mutants. The breast cancer associated mutant, M299I is enzyme activating and both unwinds and translocates DNA more efficiently than FANCJWT, whereas the P47A mutant is enzyme inactivating [26], [27]. Whether changes in FANCJ function derive from acetylation and/or partners that bind via this modification remains to be determined. Furthermore, based on our current data, it is unclear if distinct DNA lesions selectively induce FANCJ acetylation.
In summary, our findings indicate that FANCJ has the ability to potentiate HR through dual roles in DNA end processing and checkpoint maintenance. These two functions require FANCJ lysine 1249, a site not conserved in FANCJ orthologues such as chicken FANCJ and C. elegans Dog-1. Interestingly, unlike in human cells, FANCJ does not function in HR in chicken and C-elegans systems [28], [29]. It is not surprising that regulators of FANCJ acetylation state, HDACIII, SIRT1, and CBP have roles in DNA repair and genomic stability [30]–[32]. It remains to be determined, however, whether associated repair defects are related to failure to regulate FANCJ acetylation. Complicating this analysis, HDACIII, SIRT1, and CBP have many other histone and non-histone protein substrates that also have role in DNA repair and genomic stability. For example, SIRT1 deacetylation plays an important role in regulating the function of DNA double strand break repair proteins, such as Ku70 [33], WRN [34], and NBS1 [35]. Moreover, p300/CBP functions to regulate the activities of multiple proteins at the replication fork including PCNA [36]. CBP also regulates the activity of other helicases, including WRN [37]. Whether HDAC or HAT associated defects derive from a failure to regulate FANCJ acetylation will be an important question for future studies.
Materials and Methods
Cell lines
MCF7, HeLa, and 293T cells were grown in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin (100 U/mL each). FA-J (EUFA30-F) cells were cultured with 15% fetal bovine serum and penicillin/streptomycin (100 U/mL each). FA-J cells were infected with the POZ retroviral vector [38] containing no insert, WT, K1249R, or K1249Q FANCJ inserts. Stable FA-J POZ cell lines were selected as before [8].
Immunoprecipitation and Western blot assays
Cells were harvested, lysed, and processed for Western blot analysis as described previously using an NETN lysis buffer (20 mM Tris, 150 mM NaCl, 1 mM EDTA, and 0.5% NP-40) containing 10 mM NaF and 1 mM NaVO3 [7]. For acetylation detection, unless otherwise noted cells were lysed with 150 mM NETN buffer supplemented with 10 µM TSA and 5 mM nicotinamide. For γ-H2AX detection, cell pellets were collected and dissolved and boiled in 2× lysis buffer (50 mM Tris pH 6.8, 2% SDS, 1% B-ME). Antibodies used for immunoprecipitation (IP) and Western blot assays include FANCJ polyclonal Abs E67 [26], β-Actin (Sigma), pRPA S3/4 (Bethyl), RPA (Bethyl), pChk1 S317 (Bethyl), Chk1 (Bethyl), pChk2 (Cell signaling), Chk2 (Cell Signaling), γ-H2AX S139 (Millipore), H2AX (Bethyl), Flag (Sigma), HA (12C4), pan-acetylated lysine (Cell signaling), MLH1 (BD Bioscience), BRCA1 monoclonal (ms110), pH 3 (Millipore), H3 (Abcam), polη (Abcam), Rad54 (Abcam), Rad51 (Abcam), and Myc monoclonal (9E10).
Cell cycle progression assay
FA-J stable cell lines were either mock treated or treated with 0.25 µg/ml of melphalan (Sigma) and incubated for various times. Cells were fixed with 90% methanol in PBS overnight and then incubated 10 min with PBS containing 30 µg/ml DNase-free RNase A and 50 µg/ml propidium iodide. 1×104 cells were analyzed using a FACs Calibur instrument (Becton-Dickinson, San Jose, CA). Aggregates were gated out and the percentage of cells in G2/M was calculated using Flow Jo software.
Plasmid construction
The pCDNA3-myc.his vector (Invitrogen) was digested by Not1/Apa1 and different FANCJ fragments generated by PCR and digested by Not1/Apa1 were inserted. Primers are available upon request. Reverse primers used for K1249R-pCDNA3 and K1249RQ-pCDNA3 are 5′TTTTGGGCCCCCTAAAACCAGGAAACATGCC3′ and 5′TTTTGGGCCCCTGAAAACCAGGAAACATGCC3′, respectively. The K1249R and K1249Q pOZ vectors were generated with the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA) by using the FANCJ- pCDNA3-myc.his or FANCJ-pOZ as a template and the following primers: (K1249R-pOZ-Forward) 5′GGCATGTTTCCTGGTTTTAGGGCGGCCGCTGGAGGAGCA3′ and (K1249R-pOZ-Reverse) 5′GTCTCCTCCAGCGGCCGCCCTAAAACCAGGAAACATGCC3′; (K1239Q-pOZ-Forward) 5′GGCATGTTTCCTGGTTTTCAGGCGGCCGCTGGAGGAGAC3′ and (K1239Q-pOZ-Reverse) 5′GTCTCCTCCAGCGGCCGCCTGAAAACCAGGAAACATGCC3′; Recombinant FANCJ protein production was made in insect cells using the PVL13.2 vector as before [26]. Full-length WT FANCJ was used as a template to generate the acetylation mutants using the following primers: (K1249R-PVL132 Forward) 5′GGCATGTTTCCTGGTTTTAGGGACTACAAGGAGACG3′ and (K1249-PV132 Reverse) 5′CGTCGTCCTTGTAGTCCCTAAAACCAGGAAACATGCC3′. (K1249Q-PVL132 Forward) 5′ GGCATGTTTCCTGGTTTTCAGGACTACAAGGACGACG3′ and (K1249Q-PVL132 Reverse) 5′ CGTCGTCCTTGTAGTCCTGAAAACCAGGAAACATGCC3′. The pGEX-5X vector (GE Healthcare Life Sciences) was digested by Sal1/Not1 and the FANCJ C-terminal fragment was generated by PCR and digested by Sal1/Not1 and inserted. Primers are available upon request. All DNA constructs were confirmed by DNA sequencing.
Viability assays
Stable FA-J cell lines were untransfected or transfected with siRNA previously described against Luc, Rad54, or polη [9]. Cells were seeded onto 6 well plates and incubated overnight. Seeded cells were either untreated or treated with increasing dose of MMC (1 h, serum free), UV, CPT, (1 h, serum free), or zeocin (1 h, serum free). To assay for percent survival, cells were counted 5–8 days post infection and percent survival was calculated as before [9].
Helicase assays
Helicase assay reaction mixtures (20 µl) contained 40 mM Tris-HCl (pH 7.4), 25 mM KCl, 5 mM MgCl2, 2 mM dithiothreitol, 2% glycerol, 100 ng of bovine serum albumin/µl, 2 mM ATP, 10 fmol of 19-bp duplex DNA substrate (0.5 nM), and the concentrations of FANCJ (acetylated or non acetylated) indicated in the figures. Helicase reactions were initiated by the addition of FANCJ, and the reaction mixtures were incubated at 30°C for 15 min unless otherwise indicated. Reactions were quenched with the addition of 20 µl of 2× Stop buffer (17.5 mm EDTA, 0.3% SDS, 12.5% glycerol, 0.02% bromophenol blue, 0.02% xylene cyanol). For standard duplex DNA substrates, a 10-fold excess of unlabeled oligonucleotide with the same sequence as the labeled strand was included in the quench to prevent reannealing. Reaction products were resolved on nondenaturing 12% (19∶1 acrylamide-bisacrylamide) polyacrylamide gels, and quantitated as described previously [27].
Immnuofluorescence microscopy
Stable FA-J cell lines were seeded onto 6 well plates and incubated overnight. Cells were either untreated or treated with 1 mM HU (24 h) or 0.25 µM CPT (1 h). Cells were fixed with 3% paraformaldehyde/2% sucrose for 10 min at RT, and permeabilized with 0.5% Triton X-100 in 20 mM HEPES for 5 min on ice. Incubation with antibodies and washes were described previously [6]. For Rad51 staining, cells were fixed with 3% paraformaldehyde/2% sucrose for 10 min at RT, permeabilized with ice-cold methanol for 30 min, and blocked with 4% BSA for 1 h. Staining was as described previously [6].
In vitro acetylation assay
The acetyltransferase assays were performed in 30 µl of reaction, which includes reaction buffer (50 mM HEPES (ph 8.0), 10% glycerol, 1 mM DTT, 1 mM PMSF, 10 mM Na-butyrate), 1 µL [3H]-acetyl-CoA, 1 µl recombinant HAT domain of p300 (gift of Dr. Luo), and recombination FANCJ-CT or p53-CT [39]. Reaction were carried out at 30°C for 1 h and separated by SDS-PAGE, analyzed by autoradiography. Concentrations of recombinant proteins were determined by comassie staining from Invitrogen.
In gel digestion
Gel bands containing FANCJ1 were de-stained twice with 25 mM ammonium bicarbonate in 50% acetonitrile for 30 min in 37°C, reduced with 7.6 mg/ml dithiothreitol at 60°C for 10 min, and alkylated with 18.6 mg/mL iodoacetamide at room temperature for 1 hour. The bands were then washed twice with 25 mM ammonium bicarbonate in 50% acetonitrile for 15 min at 37°C prior to shrinking with 50 µL acetonitrile for 10 min at room temperature. 100 ng trypsin (Promega) was added to each sample and 25 mM ammonium bicarbonate was added until the gels were fully swollen (∼10–50 µL) and the digestion proceeded overnight at 30°C. Following digestion, peptide extracts were transferred into new tubes and the gels were further extracted with 50 µL of 50% acetonitrile containing 5% formic acid (v/v) and following 15 min were added to the initial extracts. The latter process was repeated for a total of three extractions. Extracts were then dried on a SpeedVac and reconstituted in 20 µL of 0.1% formic acid for LC-MS/MS analysis.
LC/MS/MS
Tryptic peptides (2 µL) were directly loaded at 4 µL/min for 7 min onto a custom-made trap column (100 µm I.D. fused silica with Kasil frit) containing 2 cm of 200 Å, 5 µm Magic C18AQ particles (Michrom Bioresources). Peptides were then eluted using a custom-made analytical column (75 µm I.D. fused silica) with gravity-pulled tip and packed with 25 cm 100 Å, 5 µm Magic C18AQ particles (Michrom). Peptides were eluted with a linear gradient from 100% solvent A (0.1% formic acid∶acetonitrile (95∶05)) to 35% solvent B (acetonitrile containing 0.1% formic acid) in 35 min at 300 nL/min using a Proxeon Easy nanoLC system directly coupled to a LTQ Orbitrap Velos mass spectrometer (Thermo Scientific) [40]. Data were acquired using a data-dependent acquisition routine of acquiring one mass spectrum from m/z 350–2000 in the Orbitrap (resolution 60,000) followed by tandem mass spectrometry scans in the LTQ linear ion trap of the 10 most abundant precursor ions found in the mass spectrum. Charge state rejection of singly-charged ions and dynamic exclusion was utilized to minimize data redundancy and maximize peptide identification [41].
Data analysis
The raw data files were processed and searched against the human index of the SwissProt database (version 09/21/11) containing both the mutant and wild-type forms of FANCJ1 with Mascot (version 2.3.02; Matrix Science) using parent mass tolerances of 15 ppm and fragment mass tolerances of 0.5 Da. Full tryptic specificity with 2 missed cleavages was used and variable modifications of acetylation (protein N-term and lysine), pyro-glutamination (N-term glutamine), and oxidation (methionine), and fixed modification of carbamidomethylation (cysteine) were considered. Mascot search results were also loaded into Scaffold (Version 3.3.1; Proteome Software) for comparative analyses using spectral counting of tandem mass spectra and full annotation of the data [42].
Supporting Information
{kind=link}
Zdroje
1. SobhianBShaoGLilliDRCulhaneACMoreauLA 2007 RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science 316 1198 1202
2. WangBMatsuokaSBallifBAZhangDSmogorzewskaA 2007 Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 316 1194 1198
3. SartoriAALukasCCoatesJMistrikMFuS 2007 Human CtIP promotes DNA end resection. Nature 450 509 514
4. CantorSBGuillemetteS 2011 Hereditary breast cancer and the BRCA1-associated FANCJ/BACH1/BRIP1. Future oncology 7 253 261
5. WuYSuhasiniANBroshRMJr 2009 Welcome the family of FANCJ-like helicases to the block of genome stability maintenance proteins. Cell Mol Life Sci 66 1209 1222
6. CantorSBBellDWGanesanSKassEMDrapkinR 2001 BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 105 149 160
7. LitmanRPengMJinZZhangFZhangJ 2005 BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8 255 265
8. PengMLitmanRXieJSharmaSBroshRMJr 2007 The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. Embo J 26 3238 3249
9. XieJLitmanRWangSPengMGuillemetteS 2010 Targeting the FANCJ-BRCA1 interaction promotes a switch from recombination to poleta-dependent bypass. Oncogene 29 2499 2508
10. Cotta-RamusinoCMcDonaldER3rdHurovKSowaMEHarperJW 2011 A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 332 1313 1317
11. PengMLitmanRJinZFongGCantorSB 2006 BACH1 is a DNA repair protein supporting BRCA1 damage response. Oncogene 25 2245 2253
12. LevitusMWaisfiszQGodthelpBCde VriesYHussainS 2005 The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet 37 934 935
13. LevranOAttwoollCHenryRTMiltonKLNevelingK 2005 The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet 37 931 933
14. WuYBroshRMJr 2009 FANCJ helicase operates in the Fanconi Anemia DNA repair pathway and the response to replicational stress. Curr Mol Med 9 470 482
15. YuXWuLCBowcockAMAronheimABaerR 1998 The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem 273 25388 25392
16. KaidiAWeinertBTChoudharyCJacksonSP 2010 Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 329 1348 1353
17. MarksPAMillerTRichonVM 2003 Histone deacetylases. Curr Opin Pharmacol 3 344 351
18. ZhanQJinSNgBPlisketJShangaryS 2002 Caspase-3 mediated cleavage of BRCA1 during UV-induced apoptosis. Oncogene 21 5335 5345
19. GongZKimJELeungCCGloverJNChenJ 2010 BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Molecular cell 37 438 446
20. GuptaRSharmaSDohertyKMSommersJACantorSB 2006 Inhibition of BACH1 (FANCJ) helicase by backbone discontinuity is overcome by increased motor ATPase or length of loading strand. Nucleic Acids Res 34 6673 6683
21. BailisAMMainesSNegrittoMT 1995 The essential helicase gene RAD3 suppresses short-sequence recombination in Saccharomyces cerevisiae. Mol Cell Biol 15 3998 4008
22. DillinghamMSKowalczykowskiSC 2008 RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72 642 671, Table of Contents
23. SommersJARawtaniNGuptaRBugreevDVMazinAV 2009 FANCJ uses its motor ATPase to disrupt protein-DNA complexes, unwind triplexes, and inhibit rad51 strand exchange. J Biol Chem
24. GuptaRSharmaSSommersJAKennyMKCantorSB 2007 FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood 110 2390 2398
25. SuhasiniANRawtaniNAWuYSommersJASharmaS 2011 Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom's syndrome. The EMBO journal 30 692 705
26. CantorSDrapkinRZhangFLinYHanJ 2004 The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci U S A 101 2357 2362
27. GuptaRSharmaSSommersJAJinZCantorSB 2005 Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem 280 25450 25460
28. BridgeWLVandenbergCJFranklinRJHiomK 2005 The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet 37 953 957
29. YoudsJLBarberLJWardJDCollisSJO'NeilNJ 2008 DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol 28 1470 1479
30. BhaskaraSHiebertSW 2011 Role for histone deacetylase 3 in maintenance of genome stability. Cell Cycle 10 727 728
31. HaigisMCSinclairDA 2010 Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 5 253 295
32. IyerNGOzdagHCaldasC 2004 p300/CBP and cancer. Oncogene 23 4225 4231
33. JeongJJuhnKLeeHKimSHMinBH 2007 SIRT1 promotes DNA repair activity and deacetylation of Ku70. Exp Mol Med 39 8 13
34. LiKCastaAWangRLozadaEFanW 2008 Regulation of WRN protein cellular localization and enzymatic activities by SIRT1-mediated deacetylation. J Biol Chem 283 7590 7598
35. YuanZZhangXSenguptaNLaneWSSetoE 2007 SIRT1 regulates the function of the Nijmegen breakage syndrome protein. Mol Cell 27 149 162
36. NaryzhnySNLeeH 2004 The post-translational modifications of proliferating cell nuclear antigen: acetylation, not phosphorylation, plays an important role in the regulation of its function. J Biol Chem 279 20194 20199
37. LiKWangRLozadaEFanWOrrenDK 2010 Acetylation of WRN protein regulates its stability by inhibiting ubiquitination. PLoS One 5 e10341 doi:10.1371/journal.pone.0010341
38. NakataniYOgryzkoV 2003 Immunoaffinity purification of mammalian protein complexes. Methods Enzymol 370 430 444
39. GuWRoederRG 1997 Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90 595 606
40. OlsenJVSchwartzJCGriep-RamingJNielsenMLDamocE 2009 A dual pressure linear ion trap Orbitrap instrument with very high sequencing speed. Molecular & cellular proteomics : MCP 8 2759 2769
41. SecondTPBlethrowJDSchwartzJCMerrihewGEMacCossMJ 2009 Dual-pressure linear ion trap mass spectrometer improving the analysis of complex protein mixtures. Analytical chemistry 81 7757 7765
42. SearleBC 2010 Scaffold: a bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 10 1265 1269
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 7
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- Guidelines for Genome-Wide Association Studies
- The Role of Rice HEI10 in the Formation of Meiotic Crossovers
- Identification of Chromatin-Associated Regulators of MSL Complex Targeting in Dosage Compensation
- GWAS Identifies Novel Susceptibility Loci on 6p21.32 and 21q21.3 for Hepatocellular Carcinoma in Chronic Hepatitis B Virus Carriers