
Depletion of Retinoic Acid Receptors Initiates a Novel Positive Feedback Mechanism that Promotes Teratogenic Increases in Retinoic Acid
Normal embryonic development and tissue homeostasis require precise levels of retinoic acid (RA) signaling. Despite the importance of appropriate embryonic RA signaling levels, the mechanisms underlying congenital defects due to perturbations of RA signaling are not completely understood. Here, we report that zebrafish embryos deficient for RA receptor αb1 (RARαb1), a conserved RAR splice variant, have enlarged hearts with increased cardiomyocyte (CM) specification, which are surprisingly the consequence of increased RA signaling. Importantly, depletion of RARαb2 or concurrent depletion of RARαb1 and RARαb2 also results in increased RA signaling, suggesting this effect is a broader consequence of RAR depletion. Concurrent depletion of RARαb1 and Cyp26a1, an enzyme that facilitates degradation of RA, and employment of a novel transgenic RA sensor line support the hypothesis that the increases in RA signaling in RAR deficient embryos are the result of increased embryonic RA coupled with compensatory RAR expression. Our results support an intriguing novel mechanism by which depletion of RARs elicits a previously unrecognized positive feedback loop that can result in developmental defects due to teratogenic increases in embryonic RA.
Published in the journal:
. PLoS Genet 9(8): e32767. doi:10.1371/journal.pgen.1003689
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003689
Summary
Normal embryonic development and tissue homeostasis require precise levels of retinoic acid (RA) signaling. Despite the importance of appropriate embryonic RA signaling levels, the mechanisms underlying congenital defects due to perturbations of RA signaling are not completely understood. Here, we report that zebrafish embryos deficient for RA receptor αb1 (RARαb1), a conserved RAR splice variant, have enlarged hearts with increased cardiomyocyte (CM) specification, which are surprisingly the consequence of increased RA signaling. Importantly, depletion of RARαb2 or concurrent depletion of RARαb1 and RARαb2 also results in increased RA signaling, suggesting this effect is a broader consequence of RAR depletion. Concurrent depletion of RARαb1 and Cyp26a1, an enzyme that facilitates degradation of RA, and employment of a novel transgenic RA sensor line support the hypothesis that the increases in RA signaling in RAR deficient embryos are the result of increased embryonic RA coupled with compensatory RAR expression. Our results support an intriguing novel mechanism by which depletion of RARs elicits a previously unrecognized positive feedback loop that can result in developmental defects due to teratogenic increases in embryonic RA.
Introduction
Improper retinoic acid (RA) signaling during development can cause congenital malformations that affect the forelimbs, ocular, cardiovascular, respiratory, urogenital and nervous systems [1]–[4]. Despite almost a century of investigation, the mechanisms underlying many congenital defects due to fluctuations in RA signaling are still not understood. RA acts as a ligand for RA receptors (RARs), members of the nuclear hormone family of transcription factors [5]. Work using disparate embryonic models has provided critical insight into the molecular mechanisms and developmental requirements of RAR function in vertebrate embryos [6]–[12]. In addition, RAR deficiency and inappropriate RA signaling are associated with numerous types of cancers [13]. In the majority of cases, the mechanism by which loss of RARs promote tumorigenesis is not understood. Therefore, understanding the roles of RARs during development will help elucidate the mechanisms underlying congenital defects, and possibly cancers, caused by inappropriate RA signaling [3], [4].
RA signaling employs a number of feedback mechanisms in order to maintain appropriate levels in the embryo and tissues. The best characterized feedback mechanism is through regulation of the RA producing [retinol dehydrogenases (RDHs) and retinaldehyde dehydrogenases (Aldh1a)] and degrading (Cyp26) enzymes. Specifically, increased RA signaling inhibits the expression of the RA producing enzymes, while promoting Cyp26a1 expression. Conversely, decreased RA signaling promotes expression of the RA producing enzymes, while inhibiting Cyp26a1 expression [14]–[18]. While modulation of RA signaling also affects the expression of other factors that control RA signaling [5], [19], less well understood are feedback mechanisms that may influence RAR expression. RA response elements (RAREs) have been found in murine RARα2 and RARβ2 promoters and RARβ2 has been shown to be RA responsive 20–22. However, if decreases in RA signaling, in particular due to loss of RAR expression, lead to compensatory expression of other RARs is less clear. While initial studies of mouse RAR KO mice suggested that there was not compensatory RAR expression in RAR deficient mice [11], [12], more recent studies using siRNA to deplete RARα have challenged this model and suggested that there may be compensatory RAR expression in RARα deficient embryos [23]. Therefore, if there are RA feedback mechanisms that influence RAR expression and how the employment of these feedback mechanisms impact embryonic development are not well understood.
Here, we find that depletion of RARαb1, a previously unrecognized yet conserved zebrafish RARα splice variant, causes an increase in CM specification and heart size, which is due to the triggering of a feedback mechanism that surprisingly promotes increased RA signaling from surplus embryonic RA and compensatory RAR expression. Our results provide insight into a newly recognized positive feedback mechanism that we posit resists fluctuations in RA signaling due to perturbation in RAR expression. However, if improperly maintained, the positive feedback can result in RA induced congenital defects. Altogether, the results from this study significantly enhance our understanding of the feedback mechanisms that are used to maintain appropriate RA signaling levels and previously unexplored mechanisms that potentially underlie congenital defects.
Results
RARαb1 deficient embryos have enlarged hearts and increased CM specification
In contrast to the studies of RARs in mice [9]–[12], depletion of RARs has not been able to recapitulate all of the consequences of loss of RA signaling in zebrafish [8], which prompted us to determine if additional conserved RAR variants exist in zebrafish beyond what has already been reported [24]. We cloned a previously unrecognized RARα splice variant that is orthologous to human, mouse and Xenopus RARα1 termed RARαb1 (Figure 1A–1C). The previously cloned zebrafish RARα homologs RARαa and RARαb are teleost specific paralogs and both are orthologous to the splice variant 2 found in tetrapods (Figure 1B, 1D) [24]. Both rarαb1 and rarαb2 are expressed maternally and zygotically (Figure 1E), with ubiquitous expression until the tailbud stage (Figure S1A–S1I). After the tailbud stage, their expression patterns deviate (Figure 1F–1H and Figure S1J–S1O).

We used a translation blocking morpholino (MO) to examine the function of RARαb1 (Figure 1B). By 48 hours post-fertilization (hpf), RARαb1 deficient embryos had enlarged hearts with increased CM number and expression of CM marker genes myl7, vmhc and amhc (Figure 2A, 2B, 2M, 2N and Figure S2A–S2D). Similar increases in CM number were also found at 55 hpf (Figure S3A–S3C), suggesting the major addition of surplus CMs occurs during earlier stages of development. Consistent with this idea, we observed an expansion of CM differentiation (myl7, vmhc, and amhc) and progenitor (nkx2.5 and hand2) marker expression in RARαb1 deficient embryos at earlier stages via in situ hybridization (ISH) and quantitative real-time PCR (qPCR; Figure 2C–2L, 2O–2Q). Injecting the RARαb1 MO along with rarαb1 mRNA that lacks the 5′UTR MO binding sequence is able to rescue the increased heart size, supporting the specificity of the phenotype (Figure S4A–S4D). Together, these results suggest that RARαb1 deficient embryos have increased CM specification, number and heart size.

Depletion of zebrafish RARαb paralogs promotes RA signaling
The increased atrial and ventricular CM number in RARαb1 deficient embryos are reminiscent of RA signaling deficient embryos [25], [26]. Therefore, we examined hoxb5b expression, which functions downstream of RA signaling to restrict atrial CM number [26] and is likely a direct target of RARs (Figure S5A–S5D). Unexpectedly, we found that hoxb5b expression was increased in RARαb1 deficient embryos (Figure 3A–3C). While this was initially perplexing, our recent studies showed that Hoxb5b overexpression is able to mimic many of the teratogenic effects of RA treatment [27]. Therefore, we asked if the increases in hoxb5b expression in RARαb1 deficient embryos could be a cause of the enlarged hearts. While depletion of hoxb5b alone using a low concentration of hoxb5b MO does not affect CM number (Figure S6A–S6C), we found that concurrent depletion of RARαb1 and Hoxb5b largely restored heart morphology, CM differentiation marker expression, and CM number relative to the RARαb1 deficient embryos (Figure 3F–3N), suggesting that the increased CM number in RARαb1 deficient embryos is in part a consequence of the increased hoxb5b expression.
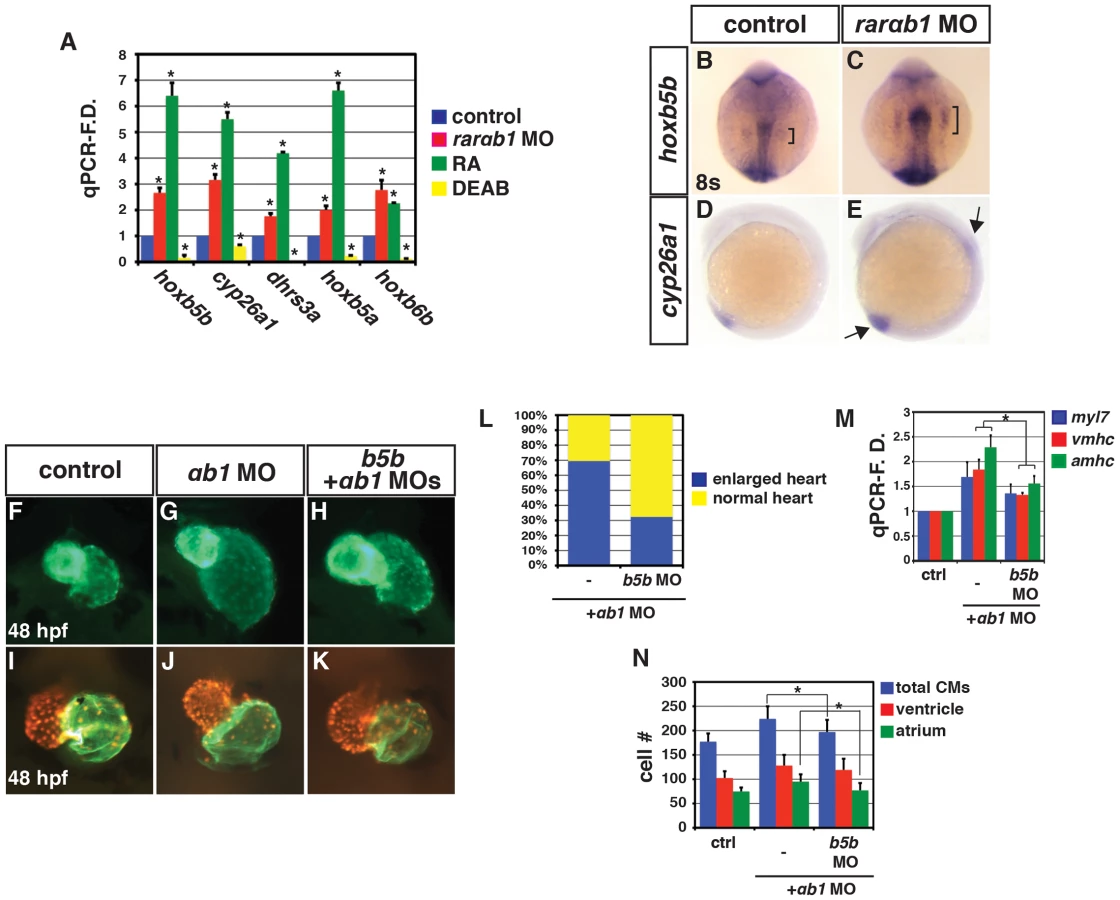
We next examined the expression of additional RA signaling responsive genes. Similar to hoxb5b, we found that the expression of additional RA signaling responsive genes, including cyp26a1, dhrs3a, hoxb6b and hoxb5a, was increased in RARαb1 deficient embryos (Figure 3A). Comparing RA responsive gene expression in RA treated and RARαb1 deficient embryos, we found that the trends were similar, but that RA treatment typically induced a greater increase in expression (Figure 3A). Conversely, treatment with DEAB, an antagonist of the RA producing enzyme Aldh1a, inhibited RA responsive gene expression (Figure 3A). These findings indicate that RARαb1 depletion paradoxically results in increased expression of RA signaling responsive genes.
We next wanted to determine if increases in RA signaling responsive genes were specific to RARαb1 depletion, so we examined RA responsive gene expression in RARαb2 deficient embryos. Previous studies found that RARαb2 deficient embryos lack forelimbs (pectoral fins) and tbx5a expression [8], [28], which we confirmed (Figure S7A, S7C, S7D, S7F, S7H, S7I). However, similar to RARαb1 depletion (Figure 3A and Figure 4A), RARαb2 deficient embryos had increased expression of RA signaling responsive genes (Figure 4A). While the previous studies found a loss of forelimbs, defects in heart development were not reported. Despite the loss of forelimbs and increase in RA signaling responsive genes, we did not observe an increase in heart size, CM number or CM gene expression (Figure S8A–S8D). Therefore, although eliciting similar increases in RA signaling responsive gene expression, individual depletion of RARαb1 and RARαb2 results in distinct defects.
#obr:4#
To determine the functional consequences of concurrent RARαb1 and RARαb2 depletion, we co-injected a suboptimal dose of each MO. Unfortunately, co-injection of an optimal dose of each MO resulted in significant non-specific toxicity even when injected along with p53 MO. However, concurrent depletion of the RARαbs using suboptimal MO doses resulted in a dramatic increase in RA signaling responsive genes, above what was seen with depletion of RARαb1 and RARαb2 alone using the optimal MO doses (Figure 4A). Additionally, there was an anterior shift of hoxb5a expression in the spinal cord of RARαb1+2 deficient embryos, suggesting the spinal cords are posteriorized (Figure S9A–S9E). Increased RA signaling inhibits aldh1a2 expression through a negative feedback mechanism 16–18. Although aldh1a2 expression in individual RARαb1 and RARαb2 deficient embryos was not suppressed (Figure 4B), aldh1a2 expression was decreased in embryos depleted for both RARαb variants (Figure 4B). To corroborate the increases in endogenous RA signaling responsive genes, we used the RA signaling reporter line Tg(12XRARE-ef1a:EGFP)sk72 29. Again, co-depletion of both RARαbs resulted in a greater expansion of egfp expression, compared to the individual depletion of each RARαb (Figure 4D–4H). Therefore, these experiments support the hypothesis that the RARαb1+2 deficient embryos are sensing more significant increases in RA signaling than embryos deficient for either RARαb variant alone.
We next examined the consequences of this functional interaction on heart development. We found that the hearts of RARαb1+2 deficient embryos had increased atrial size, CM number, and a dramatic increase in amhc expression (Figure 4I, 4L–4N and Figure S10A–S10D). Significant effects on CM number or heart size were not found when using a suboptimal dose of either RARαb1 or RARαb2 MO alone (Figure 4I–4K, 4M), though we did find a modest increase in CM marker gene expression in the RARαb1 deficient embryos (Figure 4N). Interestingly, in RARαb1+2 deficient embryos we found more significant increases in atrial CM number and amhc expression (Figure 4M, 4N), which were remarkably similar to the consequences of modest increases in RA signaling due to RA treatment 27. Increased RA signaling can also inhibit forelimb development 17 and RARαb1 deficient embryos also have smaller forelimbs and a modest reduction of tbx5a expression (Figure S7A, S7B, S7D, S7F, S7G, S7I). A functional interaction with the RARαb variants that resulted in loss of forelimbs was also observed (Figure S7D, S7E). Therefore, concurrent depletion of RARαb variants elicits increases in RA signaling with heart and forelimb phenotypes that are strikingly similar to increases in RA signaling caused from RA treatment.
Rarαb1 Deficient Embryos Have Increased Embryonic Ra
We sought to understand the mechanism underlying the increase in RA signaling in RARαb deficient embryos. In the absence of RA, RARs are thought to interact with transcriptional co-repressors, while binding of RA converts the RARs to transcriptional activators 1,5. A previous study in Xenopus suggested that RARs are required as transcriptional repressors in some developmental contexts 6. However, our gain-of-function analysis did not support that these zebrafish RARs function as transcriptional repressors (Figure S11A–S11L), consistent with what we have reported previously 29. However, Manshouri et al. 23 found a compensatory increase in the expression of other RARs when using siRNA to deplete RARα in mice. Similarly, we found that the expression of other zebrafish RARs 24 was increased in RARαb deficient embryos (Figure 4C and Figure S12A–S12L), suggesting that compensatory RAR expression is a conserved response to depletion of RARα homologs in vertebrates. Although Manshouri et al. 23 proposed the compensatory RAR expression was RA signaling dependent, our results suggest that the expression of most RARs is potentially regulated independent of RA signaling (Figure 4C), because the effects on RAR expression did not parallel modulation of RA signaling using RA and DEAB. While we observed compensatory expression of other RARs in RARαb deficient embryos, it is difficult to conclude that increased RAR expression is the sole cause of the increase in RA signaling since overexpression of RARs in zebrafish embryos does not produce significant positive or negative effects on RA responsive gene expression (Figure S11A–S11J) 29. Nevertheless, our results suggest that when depleting RARαbs in zebrafish embryos compensatory RARs are present that can mediate RA signaling.
Because we did not have evidence that RARs act as transcriptional repressors or that the increased expression of RARs alone contributes to the increases in RA signaling in RARαb deficient embryos, we hypothesized that the depletion of RARs may trigger an increase in embryonic RA. Although aldh1a2 expression was suppressed in RARαb1+2 deficient embryos similar to when embryos sense increases in RA signaling (Figure 4B) 16–18, the expression of rdh10a and rdh10b, which control a limiting step in RA production in vertebrates by generating retinal from retinol 14,15, was increased in RARαb1 and RARαb1+2 depleted embryos (Figure 4B and Fig. S13A–S13C). Interestingly, rdh10b expression, which was not sensitive to modulation of RA signaling, was increased in RARαb deficient embryos (Figure 4B). Therefore, our results suggest that depletion of RARαbs triggers an increase in RA through promoting rdh10 expression.
In addition to inhibiting aldh1a2 expression, increased RA signaling promotes a negative feedback mechanism that limits RA levels by positively regulating Cyp26a1 expression 16–18. Since we observe an increase in cyp26a1 expression in RARαb1 deficient embryos (Figure 3A, 3D, 3E and Figure 4A), which was also consistent with the hypothesis that there is increased embryonic RA, we postulated that the increased Cyp26a1 may be protecting the RARαb1 deficient embryos from teratogenic increases in embryonic RA. Therefore, we concurrently depleted RARαb1 and Cyp26a1 to determine if there was a functional interaction indicative of increased embryonic RA. For these experiments, a suboptimal dose of cyp26a1 MOs (Figure S14A–S14E) was used to more easily discern a functional interaction. In either the RARαb1 or Cyp26a1 deficient embryos alone, we never observed absence of the MHB or defects in tail elongation (Figure 5A–5C, 5E–5G). However, co-depletion of RARαb1 and Cyp26a1 resulted in a loss of the MHB and truncated tails (Figure 5D, 5H), similar to increases in RA signaling 17,19,29,30. Furthermore, we found that RARαb1+Cyp26a1 deficient embryos had dismorphic hearts with a specific reduction in ventricular CM number compared to controls embryos hearts (Figure 5I–5L, 5Q), which interestingly resembles the trend we previously found in embryos with intermediate increases in RA signaling 27.
#obr:5#
Although one interpretation of the functional interaction of RARαb1 and Cyp26a1 depletion is that there is increased embryonic RA levels in these embryos, we wanted to further test this hypothesis using additional assays. First, we sought to use a distinct readout of embryonic RA, so we made a novel stable transgenic RA sensor line which incorporated the RARαb ligand binding domain (RLBD) fused to the Gal4 DNA binding domain (GDBD) expressed under the β-actin promoter (Figure S15A–S15G) 31. Previous studies have found that similar GDBD fusions with nuclear hormone receptor LBDs create an effective reporter of nuclear hormone activity 6,32,33. We observed a dramatic increase in reporter expression when RARαb1 and Cyp26a1 were depleted together in Tg(β-actin:GDBD-RLBD); Tg(UAS:EGFP) embryos (Figure 5M–5P, 5R) 34. Second, our hypothesis predicted that reducing embryonic RA levels should be able to rescue teratogenic phenotypes found in RARαb1+Cyp26a1 and RARαb1 deficient embryos. Consistent with this hypothesis, DEAB treatment of RARαb1+Cyp26a1 deficient embryos was able to rescue the loss of MHB (Figure 6A–6J). Additionally, treatment of RARαb1 deficient embryos with DEAB partially rescue the enlarged heart phenotype and restored atrial CM number (Figure 6K–6O). Lastly, our hypothesis predicts that exogenous treatment with a concentration of RA that causes a minor increase in RA signaling should result in aberrant heart phenotypes that are similar to RARαb1 deficient embryos. Indeed, embryos treated with low concentrations of exogenous RA (lower than we had reported using previously 27) had enlarged hearts with an increase in both atrial and ventricular CM number at 48 hpf (Figure 6P–6R). Altogether, our results suggest that increases in embryonic RA, coupled with compensatory RAR expression, contribute to the developmental defects found in RARαb1 deficient embryos.
#obr:6#
Discussion
Together, our study supports a novel paradigm whereby RARαb depletion elicits a positive feedback mechanism that can result in teratogenic increases in RA signaling. Importantly, our work highlights that loss and gain of RA signaling can cause similar developmental defects. RA signaling is required to restrict CM specification 25,26, while high increases in RA signaling can eliminate CM specification (Figure 7A) 27. However, our present findings suggest that low increases in RA signaling, achieved when treating embryos with µM concentrations of RA or through RARαb depletion, can also promote increases in both atrial and ventricular CM specification (Figure 7A). As we found previously, modest, but slightly higher increases of RA signaling can promote atrial CM specification without significantly affecting ventricular CM specification 27, which is strikingly similar to what we found with concurrent depletion of the RARαb variants here (Figure 7A). Moreover, intermediate increases in RA signaling can inhibit ventricular CM specification, which is similar what we observed when concurrently depleting RARαb1 and Cyp26a1 (Figure 7A). It also appears that modulation of Hox activity downstream of both gain and loss RA signaling is at least partially responsible for the increases in CM specification, suggesting the hypothesis that the similar effects on CM number are actually due to opposite perturbations of anterior-posterior patterning within the ALPM. Therefore, our analysis corroborates and extends previous observations that there are differential effects on atrial and ventricular CM populations as there is a progressive increase from low to intermediate levels of RA signaling in the early embryo.
#obr:7#
It is interesting that depletion of RARα homologs using MOs in zebrafish, presented in this study, and Xenopus 6 elicit similar phenotypic responses. In Xenopus embryos, RARα depletion alone results in loss of the MHB 6. While depletion of RARαb1 alone does not result in MHB defects in zebrafish embryos, we have found that RARαb1+Cyp26a1 deficient embryos completely lack the MHB. Taken together, these results suggest that the underlying consequences of increased RA signaling due to depletion of RARα homologs are likely conserved at least in Xenopus and zebrafish embryos, but that in Xenopus perhaps the role of Cyp26 enzymes in protecting the brain has been lost. Despite similarities in the phenotypes that both point to an increase in RA signaling in RARα and RARαb deficient Xenopus and zebrafish embryos, our results contrast with the model proposed by Koide et al. 6, which concluded that RARs are required to function as transcriptional repressors. Importantly, the tools used in the previous study, including dominant-negative RARs, transcriptional co-repressors, and inverse agonists, would not have allowed them to distinguish between a transcriptional de-repressive model and the positive feedback mechanism involving the production of excess RA supported here.
In addition to the phenotypic similarities when depleting RARα homologs in Xenopus and zebrafish, depletion of zebrafish RARαbs results in compensatory RAR expression similar to RARα depletion in mice 23, supporting the hypothesis that this feedback response to RARα deficiency is conserved in vertebrates. Importantly, the response to RAR depletion is likely different than complete ablation of RARs. RAR KO mice have not been reported to have compensatory increases in other RARs 11,12, suggesting that a complete loss of RAR expression may cause a breakdown of this feedback loop. However, when considering the probability that RAR expression would be completely lost vs. depleted, we postulate that insults resulting in depletion of RAR expression would be much more likely. Consistent with this idea, variable levels of RAR expression deficiency, which in the case of RARβ can be due to epigenetic silencing, is commonly observed in a variety of cancers 13.
Given the conserved feedback mechanisms already recognized that limit fluctuations in RA signaling in vertebrates 16,17,19,23, it seems logical that a conserved mechanism that senses RAR deficiency would also exist to prevent loss of RA signaling. We propose that this newly recognized positive feedback mechanism would be more suitable to prevent transient deficiency in RARs. As demonstrated here, persistent RARαb depletion can result in a hypervigilant response of RA signaling and RA-induced teratogenic defects. Overall, these data provide insight into a previously unappreciated RAR-dependent positive feedback mechanism (Figure 7B), which is active during development. Further elucidation of this RA signaling feedback mechanism may illuminate the etiology of poorly understood RA-insensitive cancers 13,23 and congenital defects 1,3.
Materials And Methods
Ethics Statement
All zebrafish husbandry and experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Cincinnati Children's Hospital Medical Center.
Zebrafish Husbandry And Transgenic Lines
Zebrafish (Danio rerio) were raised and maintained as previously described 35. The following transgenic lines were used: Tg(-5.1myl7:DsRed-NLS) 36, Tg(-5.1myl7:EGFP)twu26 37, Tg(12XRARE-ef1a:EGFP)sk72 29,Tg(β-actin:GDBD-RLBD)cch1 (was created using the Gateway/Tol2 system 38 and additional characterization is reported in 31), Tg(UAS:EGFP) 34, and Tg(UAS:nfsB-mcherry) 39.
Ish
Whole-mount ISH was carried out using standard procedures 40. All probes except rarαb1 (accession number: KF030797) and rarαb2 were reported previously. myl7 (formerly called cmlc2; ZDB-GENE-991019-3), amhc (ZDB-GENE-031112-1), vmhc (ZDB-GENE-991123-5), nkx2.5 (ZDB-GENE-980526-321), hand2 (ZDB-GENE-000511-1), hoxb5a (ZDB-GENE-980526-70), hoxb5b (ZDB-GENE-000823-6), dhrs3a (ZDB-GENE-040801-217), cyp26a1 (ZDB-GENE-990415-44), rarαb1/2 (which recognizes both isoforms and was formerly called rarαb 24; ZDB-GENE-980526-72), rarαa (ZDB-GENE-980526-284), rarγa (ZDB-GENE-980526-531), rarγb (ZDB-GENE-070314-1), rdh10a (ZDB-GENE-070112-2242), tbx5a (ZDB-GENE-030909-7), eng2a (ZDB-GENE-980526-167), egr2b (formerly called krox20; ZDB-GENE-980526-283), egfp (accession number: JQ064510.1), and mcherry (accession number: JN795134.1).
Mo And Mrna Injections
The rarαb1 MO (5′-TGCAGGTCATCCGTAATGCCCGATC) was designed to the 5′ UTR of rarαb1. Additional MOs targeting another region of the 5′ UTR and the donor splice junction, which saturated the available MO target sites, were also tried. However, injection of these MOs resulted in significant toxicity and were not able to be used for analysis. Sequences to the rarαb2 and hoxb5b MOs were reported previously 8,26. The total amount of rarαb1 MO injected was 16 ng. The total amount of rarαb2 MO injected was 7 ng. The suboptimal doses used to test genetic interactions were half these concentrations. The amount of hoxb5b MO used was 0.25 ng. A cocktail of 4 ng cyp26a1 MO1 (5′-TCTTATCATCCTTACCTTTTTCTTG) and 2 ng cyp26a1 MO2 (5′-TAAAAATAATACACTACCTGCAAAC) produced a phenotype similar to gir mutants 17. Suboptimal doses used in experiments were 0.9 ng (cyp26a1 MO1) and 0.45 ng of (cyp26a1 MO2). For all injection experiments, 3 ng of p53 MO were used to help suppress non-specific MO-induced cell death 41. For experiments, the total amount of MO injected was always kept constant by equilibrating the concentrations with Standard Control MO (Gene Tools).
Capped mRNA was made using a Message Machine Kit (Ambion). 150 pg of mRNA was used for over-expression of all mRNAs in all experiments.
Cell Culture And Luciferase Assay
Luciferase reporter assays were performed in HEK 293 cells as previously described 29.
Western Blot Analysis And Chip
Western blots were performed as previously described 29. Mouse monoclonal anti-myc antibody (Covance) was used for both Western blot analysis and ChIP experiments. The dynabeads (Invitrogen) ChIP protocol was adapted from the Dorsky Lab (University of Utah) ZFIN Protocol. qPCR was used to quantify the enrichment of the fragment containing the RARE (DR5) in embryos injected with the myc-rarαb1 mRNA with respect to control uninjected embryos.
Comparison Of Genomic Sequences
The genomic sequence flanking zebrafish hoxb5b (−8 to +8 kb) was compared with the corresponding region for Hoxb5 in mouse using mVista. NHR SCAN was used to identify binding sites for nuclear receptor.
Identification Of Rarαb1 And Rt-Pcr
Rarαb1 was identified by using BLAST against the zebrafish genome (Ensemble_V7) with the human and mouse RARα1 A domains. MacVector was used for sequences alignments. For RT-PCR, primer pairs were designed so that they specifically recognized rarαb1 and rarαb2 (Figure 1B). Primer sequences are available upon request.
Cloning
The full-length coding sequence for rarαb1 was cloned into pCS2p+. The rarαb2-pCS2p+ construct used for overexpression was reported previously 29. The myc tagged RARαb1 was made using the pCS2+MT vector. For rarαb1 and rarαb2 probes, 536 base pairs (bps) of rarαb1 and 443 bps of rarαb2, which include the 5′ untranslated region (UTR) and the specific A domains with no overlap, were cloned (Figure 1B). These fragments were cloned into pGEM-T easy (Promega).
Qpcr
Total RNA was isolated from 25 embryos, homogenized in TRIzol (Ambion) and collected using Pure link RNA Micro Kit (In Vitrogen). 1 µg or 0.5 µg RNA was used for cDNA synthesis using the ThermoScript Reverse Transcriptase kit (Invitrogen). Quantitative real time PCR (qPCR) for myl7, amhc, vmhc, nkx2.5, hand2, hoxb5b, hoxb5a, hoxb6b, dhrs3a, cyp26a1, aldhh1a2, rdh10a, rdh10b, rarαa, rarαb1, rarαb2, rarγa and rarγb, egfp and mcherry was performed using standard PCR conditions in a Bio-Rad CFX PCR machine with Power SYBR Green PCR Master Mix (Applied Biosystems). Expression levels were standardized to ef1α expression and all the data were analyzed using the 2−ΔΔCT Livak Method. All experiments were performed in a biological triplicate. Primer sequences are available upon request.
Area And Length Measurements
Areas of myl7, vmhc and amhc expressing cells were measured using ImageJ and statistical analysis was performed as previously described 26. Length of egfp expression and distance between hoxb5b and egr2b were measured also using ImageJ and statistical analysis was performed as previously described.
Imaging Of Zebrafish Heart And Cell Counting
Immunohistochemistry, cell counting and statistical analysis were done as previously described 26.
Ra And Deab Treatment
RA and DEAB, treatment of embryos was done as previously described 26,27. Embryos that have been used for gene expression analysis at 8 somites were treated with 1 µM DEAB, an Aldh1a2 inhibitor, beginning at 40% epiboly or with 1 µM RA for 1 hr beginning at 40% epiboly. For analysis of the effects of low concentrations of RA on heart development, embryos were treated with 0.05 µM RA for 1 hr beginning at 40% epiboly and harvested at 48 hpf. For rescue experiments related to the heart phenotype of RARαb1 deficient embryos, embryos were treated with 0.025 µM DEAB beginning at 40% epiboly until 24 hpf. For rescue experiments related to the MHB in RARαb1+Cyp26a1 deficient embryos, embryos were treated with 0.25 µM DEAB.
Statistical Analysis
To assess whether the means of two groups are statistically different from each other, we applied the Student's t-test. A p value of <0.05 was considered statistically significant.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. NiederreitherK, DolleP (2008) Retinoic acid in development: towards an integrated view. Nature reviews Genetics 9: 541–553.
2. RhinnM, DolleP (2012) Retinoic acid signalling during development. Development 139: 843–858.
3. LammerEJ, ChenDT, HoarRM, AgnishND, BenkePJ, et al. (1985) Retinoic acid embryopathy. N Engl J Med 313: 837–841.
4. RizzoR, LammerEJ, ParanoE, PavoneL, ArgyleJC (1991) Limb reduction defects in humans associated with prenatal isotretinoin exposure. Teratology 44: 599–604.
5. BastienJ, Rochette-EglyC (2004) Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 328: 1–16.
6. KoideT, DownesM, ChandraratnaRA, BlumbergB, UmesonoK (2001) Active repression of RAR signaling is required for head formation. Genes & development 15: 2111–2121.
7. LiP, PashmforoushM, SucovHM (2010) Retinoic acid regulates differentiation of the secondary heart field and TGFbeta-mediated outflow tract septation. Developmental Cell 18: 480–485.
8. LinvilleA, RadtkeK, WaxmanJS, YelonD, SchillingTF (2009) Combinatorial roles for zebrafish retinoic acid receptors in the hindbrain, limbs and pharyngeal arches. Developmental Biology 325: 60–70.
9. LohnesD, MarkM, MendelsohnC, DolleP, DierichA, et al. (1994) Function of the retinoic acid receptors (RARs) during development (I). Craniofacial and skeletal abnormalities in RAR double mutants. Development 120: 2723–2748.
10. MendelsohnC, LohnesD, DecimoD, LufkinT, LeMeurM, et al. (1994) Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120: 2749–2771.
11. LohnesD, KastnerP, DierichA, MarkM, LeMeurM, et al. (1993) Function of retinoic acid receptor gamma in the mouse. Cell 73: 643–658.
12. LufkinT, LohnesD, MarkM, DierichA, GorryP, et al. (1993) High postnatal lethality and testis degeneration in retinoic acid receptor alpha mutant mice. Proceedings of the National Academy of Sciences of the United States of America 90: 7225–7229.
13. SopranoDR, QinP, SopranoKJ (2004) Retinoic acid receptors and cancers. Annu Rev Nutr 24: 201–221.
14. SandellLL, LynnML, InmanKE, McDowellW, TrainorPA (2012) RDH10 oxidation of Vitamin A is a critical control step in synthesis of retinoic acid during mouse embryogenesis. PLoS One 7: e30698.
15. SandellLL, SandersonBW, MoiseyevG, JohnsonT, MushegianA, et al. (2007) RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Genes & development 21: 1113–1124.
16. Dobbs-McAuliffeB, ZhaoQ, LinneyE (2004) Feedback mechanisms regulate retinoic acid production and degradation in the zebrafish embryo. Mechanisms of Development 121: 339–350.
17. EmotoY, WadaH, OkamotoH, KudoA, ImaiY (2005) Retinoic acid-metabolizing enzyme Cyp26a1 is essential for determining territories of hindbrain and spinal cord in zebrafish. Developmental Biology 278: 415–427.
18. NiederreitherK, McCafferyP, DragerUC, ChambonP, DolleP (1997) Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenase type 2 (RALDH-2) gene during mouse development. Mechanisms of Development 62: 67–78.
19. CaiAQ, RadtkeK, LinvilleA, LanderAD, NieQ, et al. (2012) Cellular retinoic acid-binding proteins are essential for hindbrain patterning and signal robustness in zebrafish. Development 139: 2150–2155.
20. de TheH, Vivanco-RuizMM, TiollaisP, StunnenbergH, DejeanA (1990) Identification of a retinoic acid responsive element in the retinoic acid receptor beta gene. Nature 343: 177–180.
21. HoffmannB, LehmannJM, ZhangXK, HermannT, HusmannM, et al. (1990) A retinoic acid receptor-specific element controls the retinoic acid receptor-beta promoter. Molecular Endocrinology 4: 1727–1736.
22. LeroyP, NakshatriH, ChambonP (1991) Mouse retinoic acid receptor alpha 2 isoform is transcribed from a promoter that contains a retinoic acid response element. Proceedings of the National Academy of Sciences of the United States of America 88: 10138–10142.
23. ManshouriT, YangY, LinH, StassSA, GlassmanAB, et al. (1997) Downregulation of RAR alpha in mice by antisense transgene leads to a compensatory increase in RAR beta and RAR gamma and development of lymphoma. Blood 89: 2507–2515.
24. WaxmanJS, YelonD (2007) Comparison of the expression patterns of newly identified zebrafish retinoic acid and retinoid X receptors. Developmental dynamics : an official publication of the American Association of Anatomists 236: 587–595.
25. KeeganBR, FeldmanJL, BegemannG, InghamPW, YelonD (2005) Retinoic acid signaling restricts the cardiac progenitor pool. Science 307: 247–249.
26. WaxmanJS, KeeganBR, RobertsRW, PossKD, YelonD (2008) Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Developmental Cell 15: 923–934.
27. WaxmanJS, YelonD (2009) Increased Hox activity mimics the teratogenic effects of excess retinoic acid signaling. Developmental dynamics : an official publication of the American Association of Anatomists 238: 1207–1213.
28. HeX, YanYL, EberhartJK, HerpinA, WagnerTU, et al. (2011) miR-196 regulates axial patterning and pectoral appendage initiation. Developmental Biology 357: 463–477.
29. WaxmanJS, YelonD (2011) Zebrafish retinoic acid receptors function as context-dependent transcriptional activators. Developmental Biology 352: 128–140.
30. MartinBL, KimelmanD (2010) Brachyury establishes the embryonic mesodermal progenitor niche. Genes & development 24: 2778–2783.
31. MandalA, RydeenA, AndersonJ, SorrellMR, ZygmuntT, et al. (2013) Transgenic retinoic acid sensor lines in zebrafish indicate regions of available embryonic retinoic acid. Developmental dynamics : an official publication of the American Association of Anatomists
32. TiefenbachJ, MollPR, NelsonMR, HuC, BaevL, et al. (2010) A live zebrafish-based screening system for human nuclear receptor ligand and cofactor discovery. PLoS One 5: e9797.
33. AllenbyG, BocquelMT, SaundersM, KazmerS, SpeckJ, et al. (1993) Retinoic acid receptors and retinoid X receptors: interactions with endogenous retinoic acids. Proceedings of the National Academy of Sciences of the United States of America 90: 30–34.
34. AsakawaK, SusterML, MizusawaK, NagayoshiS, KotaniT, et al. (2008) Genetic dissection of neural circuits by Tol2 transposon-mediated Gal4 gene and enhancer trapping in zebrafish. Proceedings of the National Academy of Sciences of the United States of America 105: 1255–1260.
35. Westerfield M (1993) The zebrafish book : a guide for the laboratory use of zebrafish (Brachydanio rerio). Eugene, OR: M. Westerfield. 1 v. (unpaged)
36. MablyJD, MohideenMA, BurnsCG, ChenJN, FishmanMC (2003) heart of glass regulates the concentric growth of the heart in zebrafish. Current biology : CB 13: 2138–2147.
37. HuangCJ, TuCT, HsiaoCD, HsiehFJ, TsaiHJ (2003) Germ-line transmission of a myocardium-specific GFP transgene reveals critical regulatory elements in the cardiac myosin light chain 2 promoter of zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists 228: 30–40.
38. KwanKM, FujimotoE, GrabherC, MangumBD, HardyME, et al. (2007) The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Developmental Dynamics 236: 3088–3099.
39. DavisonJM, AkitakeCM, GollMG, RheeJM, GosseN, et al. (2007) Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Developmental Biology 304: 811–824.
40. OxtobyE, JowettT (1993) Cloning of the zebrafish krox-20 gene (krx-20) and its expression during hindbrain development. Nucleic Acids Res 21: 1087–1095.
41. RobuME, LarsonJD, NaseviciusA, BeiraghiS, BrennerC, et al. (2007) p53 activation by knockdown technologies. PLoS genetics 3: e78.
42. JarinovaO, HatchG, PoitrasL, PrudhommeC, GrzybM, et al. (2008) Functional resolution of duplicated hoxb5 genes in teleosts. Development 135: 3543–3553.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 8
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- Chromosomal Copy Number Variation, Selection and Uneven Rates of Recombination Reveal Cryptic Genome Diversity Linked to Pathogenicity
- Genome-Wide DNA Methylation Analysis of Systemic Lupus Erythematosus Reveals Persistent Hypomethylation of Interferon Genes and Compositional Changes to CD4+ T-cell Populations
- Associations of Mitochondrial Haplogroups B4 and E with Biliary Atresia and Differential Susceptibility to Hydrophobic Bile Acid
- A Role for CF1A 3′ End Processing Complex in Promoter-Associated Transcription