
Cohesinopathies of a Feather Flock Together
Roberts Syndrome (RBS) and Cornelia de Lange Syndrome (CdLS) are severe developmental maladies that present with nearly an identical suite of multi-spectrum birth defects. Not surprisingly, RBS and CdLS arise from mutations within a single pathway—here involving cohesion. Sister chromatid tethering reactions that comprise cohesion are required for high fidelity chromosome segregation, but cohesin tethers also regulate gene transcription, promote DNA repair, and impact DNA replication. Currently, RBS is thought to arise from elevated levels of apoptosis, mitotic failure, and limited progenitor cell proliferation, while CdLS is thought to arise, instead, from transcription dysregulation. Here, we review new information that implicates RBS gene mutations in altered transcription profiles. We propose that cohesin-dependent transcription dysregulation may extend to other developmental maladies; the diagnoses of which are complicated through multi-functional proteins that manifest a sliding scale of diverse and severe phenotypes. We further review evidence that cohesinopathies are more common than currently posited.
Published in the journal:
. PLoS Genet 9(12): e32767. doi:10.1371/journal.pgen.1004036
Category:
Review
doi:
https://doi.org/10.1371/journal.pgen.1004036
Summary
Roberts Syndrome (RBS) and Cornelia de Lange Syndrome (CdLS) are severe developmental maladies that present with nearly an identical suite of multi-spectrum birth defects. Not surprisingly, RBS and CdLS arise from mutations within a single pathway—here involving cohesion. Sister chromatid tethering reactions that comprise cohesion are required for high fidelity chromosome segregation, but cohesin tethers also regulate gene transcription, promote DNA repair, and impact DNA replication. Currently, RBS is thought to arise from elevated levels of apoptosis, mitotic failure, and limited progenitor cell proliferation, while CdLS is thought to arise, instead, from transcription dysregulation. Here, we review new information that implicates RBS gene mutations in altered transcription profiles. We propose that cohesin-dependent transcription dysregulation may extend to other developmental maladies; the diagnoses of which are complicated through multi-functional proteins that manifest a sliding scale of diverse and severe phenotypes. We further review evidence that cohesinopathies are more common than currently posited.
Introduction
A shared genetic basis of developmental abnormalities
Roberts Syndrome/SC-Phocomelia (RBS) and Cornelia de Lange Syndrome (CdLS) are severe multi-spectrum developmental disorders. Patients afflicted with either RBS or CdLS present with nearly identical phenotypes that include acute long-bone growth failure (near-absence of extremities positions hands and/or feet close to the body), mental retardation, craniofacial malformation, and perturbations of heart, kidney, genital, and gastrointestinal development (Table 1). Consistent with this similar suite of phenotypes, both RBS and CdLS arise from mutations within a single pathway. Mutations in ESCO2 produce RBS [1]–[3]. ESCO2 is a member of a highly conserved acetyltransferase family (Eco1/Ctf7 in budding yeast, ESCO2/EFO2 ESCO1/EFO1 in humans) that is essential for sister chromatid tethering reactions (termed cohesion) and high fidelity chromosome segregation [4]. To date, the only known essential substrate of ESCO2 is the cohesin protein SMC3. Cohesin complex (which also contains SMC1A, MCD1/SCC1/RAD21, and Irr1/Scc3/SA1,2/STAG1,2) binding to DNA requires a deposition complex that contains SCC2/NIPBL and SCC4/MAU2 [4]. Mutations within cohesin subunits (SMC1A, SMC3, and RAD21) and cohesin auxiliary factors (NIPBL, HDAC8, and cohesin-associated PDS5/APRIN) give rise to CdLS [5]–[12]. Despite the common manifestations and genetic basis of RBS and CdLS, these developmental abnormalities are thought to arise through different responses to cohesion factor mutations: RBS through elevated levels of apoptosis and limited progenitor cell proliferation, and CdLS through transcription dysregulation. The transcription-based CdLS model is supported by findings that CdLS cells do not exhibit elevated levels of apoptosis or mitotic failure, and that chromatin-bound cohesins not only participate in sister chromatid tethering, but also i) participate in boundary elements that demarcate transcriptional domains, ii) orchestrate DNA promoter and enhancer registration, and iii) associate with transcription regulators (Figure 1). Differentiating between cohesin functions likely depends both on the timing of its enrichment to DNA and post-translational modifications—the foremost of which appears composed of an acetylation code inscribed by Eco1/Ctf7/ESCO2 [4].
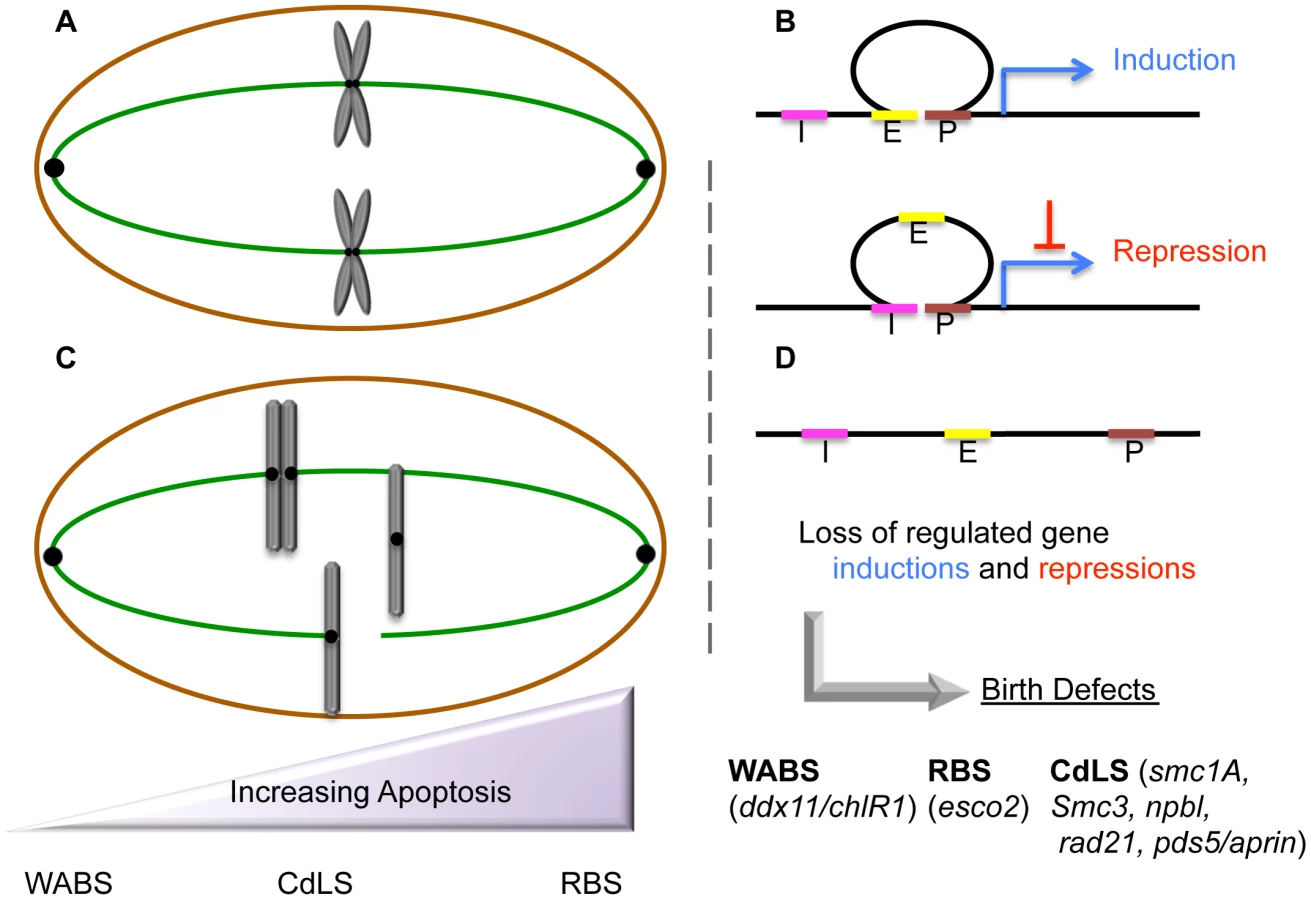

ESCO2 mutations result in elevated frequencies of apoptotic cells
Historically, the cataloging of childhood developmental disorders such as RBS was limited to physician-based descriptions. Included in RBS descriptions are cytological observations that included micronuclei, aneuploidy, and chromosomal abnormalities such as heterochromatin repulsion (HR)—often referred to as railroad track chromosomes or premature centromere separation (PCS). In humans, RBS arises through loss of both ESCO2 gene copies [1]–[3]. ESCO2 knockdown or mutation in model systems (including zebrafish, medaka, and mouse embryos) were found to recapitulate key RBS phenotypes [13]–[15]. In each case, elevated levels of apoptosis were observed in support of a model that mitotic failure induces apoptosis and further limits progenitor cell proliferation. ESCO2 depletion from either mouse neuroepithelium or zebrafish embryos targeted brain and peripheral nervous system cells for death [13]–[15], formally suggesting a mechanism through which cognitive impairment could arise in RBS patients. In contrast, ESCO2 depletion in a medaka model produced increased levels of apoptosis throughout the entire developing embryo [14]. Regardless, apoptotic loss and limited proliferation of progenitor cells remain popular mechanisms through which RBS developmental abnormalities arise, which span skeletal, organ, and cognitive defects.
CdLS: A model of transcription dysregulation in cohesinopathies
Chromosome mis-segregation, aneuploidy, and the inability to induce apoptosis are features typically associated with tumor cells—rarely with cells from phenotypically pleiotropic and diverse developmental maladies. In this light, the model in which RBS arises through elevated apoptosis and limited cell proliferation may be exceptional. Thus, it is intriguing that CdLS, the sister cohesinopathy to RBS, arises through transcriptional dysregulation and not through mitotic failure or elevated levels of apoptosis [16]. For instance, cells from CdLS patients not only undergo normal mitosis, they contain chromosome structures overtly devoid of HR/PCS phenotypes [17]–[20]. CdLS is an autosomal dominant disease (SMC1A and HDAC8 are X-linked) such that patients retain both a wildtype and dominant-mutant version of the cohesion gene [5]–[8], [12]. This heterozygosity likely accounts for the lack of PCS in CdLS patients, given that gene knockdowns (and likely homozygous mutations) of genes implicated in CdLS are either lethal or result in mitotic failure and apoptosis [21]–[23]. Based on early characterizations of NIPBL as a factor required for transcription regulation and cohesion [24], [25], numerous groups have linked cohesinopathic CdLS mutations to dysregulation of specific developmental and biochemical pathways. In genome-wide transcriptional microarrays of CdLS lymphoblastoid cell lines that harbor a mutation in NIPBL, 420 genes were differentially regulated, as compared to age and gender-matched controls. Interestingly, only modest levels of differential expression were reported (approximately 71% lower than 1.5-fold change), suggesting that CdLS phenotypes are caused by the accumulation of numerous yet small changes in expression—changes that may correlate with diminished cohesin binding near transcriptional start sites [26]. Mutations in RAD21, encoding another major subunit of the cohesin complex, result in dysregulation of the APO gene cluster—an effect also produced in IGH (Ig receptor genes), IGF2-H19 (imprinted developmental genes), and ESR1 (Estrogen receptor genes) [11], [27]–[32]. A proteomic regulation approach in CdLS cells mutated for either SMC1A or SMC3 identified 46 proteins dysregulated to a fold change of 1.3 or greater, compared to age-gender-ethnicity controls. The functions of these dysregulated proteins are distributed throughout metabolism, cytoskeleton organization, protein fate, antioxidant detoxification, and RNA processing pathways. In silico network analysis established a link between these 46 dysregulated proteins and c-MYC—a transcription factor that, when mutated, plays critical roles in both cancer progression and aberrant development. Subsequent efforts confirmed MYC dysregulation in CdLS probands, similar to that previously reported in RAD21 mutated cells, and that cohesin binding to the first exon of c-MYC is decreased in CdLS cells [31], [33].
New evidence that ESCO2 is a critical regulator of both transcription and chromosome architecture
Do the more overt phenotypes of HR/PCS, mitotic failure, and elevated levels of apoptosis in RBS mask a role for ESCO2 mutation in transcription dysregulation? In human cell lines, two recent reports document that ESCO proteins (ESCO1/EFO1 and ESCO2/EFO2 are both acetyltransferases that target SMC3 [34]–[38]) impact transcriptional regulation. ESCO2 associates with CoREST transcriptional repressor complex subunits and methyltransferases (SETDB1, G9a, and suv39h1) and demethylases (LSD1), which repress transcription through H3 modifications [39]. Knockdown of either methyltransferase SETDB1 or suv39h1or demethylase LSD1 relieved ESCO2-dependent transcription repression, suggesting that ESCO2 recruits chromatin modifiers to affect repression. Consistent with this possibility, repression was unaffected by mutation within the ESCO2 acetyltransferase domain [39]. Similar findings involving repression by LSD1 and binding of chromatin modifiers (LSD1, SETDB1, G9a, and suv39h1) were reported for ESCO1 and also in an acetyltransferase-independent fashion [40]. Thus, the ESCO/EFO family exhibits numerous functions: as an acetyltransferase that modifies cohesin (and likely other substrates) and as a scaffold through which chromatin modifiers are recruited.
Hints regarding ESCO2 targets recently were identified in human, mouse neuron, and teleost medaka models. In addition to chromatin remodeling complexes (CoREST, LSD1, HDACs), ESCO2 binds to Notch from human cell extracts [41]. Notch signaling is a major regulator of developmental programs—including organ and brain development. ESCO2 expression suppressed Notch-dependent activation, in a manner independent of ESCO2 acetyltransferase activity. Instead, ESCO2 binds directly to the Notch Intracellular Domain (NICD), a domain released upon receptor activation/cleavage, and prevents NICD activation of the transcription regulator CBF1 involved in neural progenitor cell proliferation and differentiation. ESCO2 depletion in mice inhibited neural cell differentiation (shorter neurites), while overexpression resulted in cells that exhibited longer neurites [41]. In medaka fish models, diminished ESCO2 function resulted in decreased expression of both Notch1a and Notch3 [14]. While Notch1a is a potent regulator of neuronal cell proliferation and head development, Notch3 is a vascular differentiation marker and previously linked to heart malformations in the medaka model. These studies potentially link mutated ESCO2 RBS manifestations such as microcephaly, cognitive retardation, and heart defects to transcriptional dysregulation of Notch pathways [14], [41].
ESCO2-deployment of transcriptional programs may not be surprising given early evidence that ESCO family members are critical regulators of chromatin architecture. The earliest study of the ESCO2 homolog in yeast (Eco1/Ctf7) revealed that mutations produced both cohesion and chromosome condensation defects—effects mirrored in both yeast and vertebrate cell cohesion mutations [14], [15], [42]–[45]. In zebrafish, microarray analysis confirmed alteration of gene transcripts in tissues depleted of ESCO2, a population of which overlapped with those altered in cohesin mutation [13]. Intriguingly, this shared subset of genes was dysregulated in opposition: genes repressed upon ESCO2 depletion were upregulated in cohesin mutation and vice versa. When viewed through the lens of ESCO2 as both a cohesin acetyltransferase and chromatin-remodeler scaffold, and that cohesin acetylation is read as a code through which different cell processes differentially respond [4], this apparent discrepancy is easily accommodated and suggests that transcriptional changes in either direction can alter normal development.
Overlap of cell phenotypes supports a unifying transcription-based cohesinopathy model
If our RBS transcription dysregulation model is correct, then transcriptional dysregulation in RBS cells should map to a subset of dysregulated genes in CdLS. This prediction proves true. Microarray analyses of CdLS proband patient cells identified approximately ten dysregulated genes that both accurately distinguished between controls and CdLS probands and correlated with CdLS severity. When this same gene set was tested against two RBS probands, the RBS samples were included in the CdLS group, revealing in concept a transcriptional match between CdLS and RBS gene dysregulations [26]. Is the converse relationship true: do CdLS cells exhibit elevated levels of mitotic failure and apoptosis? On the one hand, CdLS is not a recessive condition, but instead is autosomal dominant. Thus, CdLS patient cells typically retain one normal cohesion gene homolog and thus do not manifest the mitotic failure or limited cell proliferation observed in RBS patient cells in which both gene homologs are altered. However, experiments from yeast, man, and fish reveal the essential nature of these genes and that elevated levels of apoptosis can occur in response to either mutation or knockdown of SMC1A, RAD21, SMC3, or PDS5 [4], [10], [21], [23], [33], [46]–[49]. RAD21 is particularly intriguing given that caspase-dependent cleavage of RAD21 produces a C-terminal product that further promotes the apoptotic response pathway [50]. Clinical implications are intriguing as well: knockdown of RAD21 in human breast cancer or SMC1A in adenocarcinoma cells both elicited an apoptotic-type response [23], [51]. Thus, gene mutations causative for RBS and CdLS have the capacity to exhibit a sliding scale of chromosome segregation defects, elevated levels of apoptosis, and reduced cell proliferation that are superimposed on top of transcription dysregulation—the latter of which we speculate is causatively associated with developmental defects. Given that mutation of ESCO2 acetyltransferase results in RBS developmental abnormalities, might mutations in an opposing activity similarly be of clinical interest? HDAC8 (Hos1 in yeast) is a de-acetylase that opposes ESCO2 modification of SMC3 [12], [52]–[54]. Recent studies reveal that HDAC8 mutation results in transcription dysregulation—similar to NIPBL mutants in CdLS—and decreased cohesin occupancy of localization sites. Intriguingly, cohesins remain bound to chromatin in HDAC8 deficient cells, even after mitosis, which may explain the delay in anaphase and mitotic failure in these cells through which RBS is also phenocopied [12].
Cohesinopathies may not be so rare after all
The range of cohesinopathies continues to expand, projecting that the number of ESCO2-dependent maladies will increase significantly as molecular genetics continue to link mutations in ESCO2 (or ESCO1) to other multi-spectrum disorders. For instance, the majority of RBS patients exhibit significant cognitive impairment, making it difficult to exclude allele-specific ESCO2 mutations as a contributing factor in any number of cognitive syndromes. Moreover, cells from RBS patients proved indistinguishable from those of Fanconi Anemia (FA) when scored using an FA-specific diagnostic chromosome breakage test [55]. The same diagnostic assay also positively identified cells from Warsaw Breakage Syndrome (WABS) patients as FA-like. WABS presents as a multispectrum developmental malady that arises from mutations within the DNA helicase DDX11/ChlR1 [56], [57]. Chl1, the yeast homolog of ChlR1/DDX11, associates with Eco1/Ctf7 (ESCO2), and mutations in either yeast or human homolog produces significant cohesion defects [4]. Further blurring the lines between developmental maladies is the recent finding that Chl1 is critical for Scc2 (NIPBL) recruitment to DNA—conceptually linking WABS, CdLS, and RBS [58].
Genotoxic sensitivity appears to be another theme that runs throughout cohesinopathic syndromes. For instance, cells from patients afflicted with FA, WABS, CdLS, or RBS all exhibit genotoxic sensitivities [1], [3], [17], [20], [23], [55], [59], [60]. Intriguingly, all cohesinopathic cells tested to date also produce HR/PCS or other chromosomal aberrations when exposed to mitomycin [56]. Are genotoxic sensitivities a critical etiologic agent in developmental abnormalities? CdLS may be particularly instructive in that these cells exhibit some deficiencies in DNA repair, but deficiencies that are relatively limited in scope. For instance, a study that included seven CdLS patient cell lines failed to identify any that were UV sensitive, and less than half exhibited X-irradiation sensitivity (and then only at very high exposure levels). Conversely, all CdLS tested exhibit MMC sensitive [20]. A similar range of genotoxic sensitivities occurs in RBS cells [11], [61]. Thus, genotoxic sensitivities remain a useful diagnostic tool but likely are not uniquely etiologic in nature. RBS cells also exhibit DNA replication defects, but this phenotype has yet to be linked to other cohesinopathies, and the causality of this phenotype is complicated by numerous findings that Eco1/Ctf7/ESCO2 function is tightly coordinated to DNA replication fork components [4]. Intriguingly, cohesin roles during DNA damage repair change based on the proximity to the break site: break-proximal cohesion promotes DNA repair while global cohesion promotes high fidelity chromosome segregation [59]. The mechanism through which cohesins promote DNA repair and the extent that cohesinopathic genotoxic sensitivities contribute to developmental maladies remains an open question. Instead, we posit that many of the presenting manifestations (beyond developmental abnormalities) likely arise through additional roles played by those factors—a model supported by findings that DNA repair, condensation, and cohesion are differentially sensitive to changes in cohesin gene dosages including SCC2/NIPBL, MCD1/RAD21 and PDS5B [19]. In Nijmegen Breakage Syndrome (NBS) and FA patient cells, for instance, DNA damaging sensing and repair enzymes themselves are defective [62], [63]; it is the additional loss of these activities that likely correlate to the added complexities of cancers and/or anemia.
Cohesinopathies, defined here as transcription dysregulation disorders, likely encompass other maladies such as ribosomopathies (Table 1). Ribosomopathies include Diamond Blackfan anemia (DBA) and Treacher-Collins syndrome (TCS). In the case of DBA, the presenting hematologic abnormality is anemia, but this does not exclude transcription dysregulation as the basis for developmental defects also present in DBA patients. TCS, on the other hand, is distinguished primarily by craniofacial irregularities that are similar to those of cohesinopathies [64]–[66]. How are cohesinopathies and ribosomopathies linked? Early studies of Eco1/Ctf7 and cohesin function in rDNA architecture [42], [43] were quickly followed by reports of cohesin localization to rDNA and function in rDNA segregation, recombination and maintenance [67]–[71]. CdLS mouse cell transcriptomes revealed a vast array of gene dysregulations previously shown to correlate with developmental phenotypes [72], including TCOF1, which is required for ribosome biogenesis and in which mutations result in severe facial/cranial dysmorphia [73]. Even transient cohesin inactivation in yeast, and specifically during G1 prior to its role in chromosome segregation, results in transcription dyregulation—the largest class comprising ribosome biogenesis/maturation. Based on these findings, the first formal statement linking ribosomopathies (Treacher-Collins Syndrome and Diamond Blackfan anemia) to cohesinopathies was articulated [74]. More recent evidence reveals that mutation in either Ctf7/Eco1 (ESCO2) or cohesin significantly reduces both rDNA transcription and ribosome subunit productions, leading to translation initiation defects and decreased protein synthesis [74], [75]. Further support emanates from human cell studies in that ESCO2 localizes to nucleoli, a heterochromatic domain required for rRNA production [15], [61]. In human cohesinopathic cells, regulation of MYC, p53, and MDM2 are all affected by ribosome biogenesis [75]. Thus, it is tempting to speculate that cohesinopathies represent an umbrella under which ribosomopathies reside ([76] for an excellent review). In this light, testing for rRNA maturation/ribosome biogenesis defects in the roughly 35% of genetically uncharacterized CdLS patients may be of value [74], [77]. Moreover, we look forward to rigorous testing of our speculative model that many developmental maladies (RBS, WABS, NBS, FA, DBA, and TCS), currently posited as unique in etiology, may be based on transcriptional dysregulation maladies akin to CdLS, with additional levels of complexity.
Zdroje
1. VegaH, WaisfiszQ, GordilloM, SakaiN, YanagiharaI, et al. (2005) Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet 37: 468–470.
2. SchuleB, OviedoA, JohnstonK, PaiS, FranckeU (2005) Inactivation mutations in ESCO2 cause SC phocomelia and Roberts Syndrome: No phenotype-genotype correlation. Am J Hum Genet 77: 1117–1128.
3. GordilloM, VegaH, TrainerAH, HouF, SakaiN, et al. (2008) The molecular mechanism underlying Roberts syndrome involves loss of ESCO2 acetyltransferase activity. Hum Mol Genet 17: 2172–2180.
4. RudraS, SkibbensRV (2013) Cohesin codes – interpreting chromatin architecture and the many facets of cohesin function. J Cell Science 126: 31–41.
5. MusioA, SelicorniA, FocarelliML, GervasiniC, MilaniD, et al. (2006) X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet 38: 528–530.
6. TonkinET, WangTJ, LisgoS, BamshadMJ, StrachanT (2004) NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet 36: 636–641.
7. GillisLA, McCallumJ, KaurM, DeScipioC, YaegerD, et al. (2004) NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet 75: 610–623.
8. KrantzID, McCallumJ, DeScipioC, KaurM, GillisLA, et al. (2004) Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet 36: 631–635.
9. DeardorffMA, KaurM, YaegerD, RampuriaA, KorolevS, et al. (2007) Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am J Hum Genet 80: 485–494.
10. ZhangB, JainS, SongH, FuM, HeuckerothRO, et al. (2007) Mice lacking sister chromatid cohesion protein PDS5B exhibit developmental abnormalities reminiscent of Cornelia de Lange syndrome. Development 134: 3191–3201.
11. DeardorffMA, WildeJJ, AlbrechtM, DickinsonE, TennstedtS, et al. (2012) RAD21 mutations cause a human cohesinopathy. Am J Hum Genet 90: 1014–1027.
12. DeardorffMA, BandoM, NakatoR, WatrinE, ItohT, et al. (2012) HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature 489: 313–317.
13. MonnichM, KurigerZ, PrintCG, HorsfieldJA (2011) A zebrafish model of Roberts Syndrome reveals that Esco2 depletion interferes with development by disrupting the cell cycle. PLoS ONE 6: e20051 doi:10.1371/journal.pone.0020051
14. MoritaA, NakahiraK, HasegawaT, UchidaK, TaniguchiY, et al. (2012) Establishment and characterization of Roberts Syndrome and SC phocomelia model medaka (Oryzias latipes). Develop Growth Differ 54: 588–604.
15. WhelanG, KreidleE, WutzG, EgnerA, PetersJ-M, et al. (2012) Cohesin acetyltransferase Esco2 is a cell viability factor and is required for cohesion in pericentric heterochromatin. EMBO J 31: 71–82.
16. DorsettD (2011) Cohesin: genomic insights into controlling gene transcription and development. Curr Opin Genet Dev 21: 199–206.
17. RevenkovaE, FocarelliML, SusaniL, PaulisM, BassiMT, et al. (2009) Cornelia de Lange syndrome mutations in SMC1A or SMC3 affect binding to DNA. Hum Mol Genet 18: 418–427.
18. CastronovoP, GervasiniC, CeredaA, MasciadriM, MilaniD, et al. (2009) Premature chromatid separation is not a useful diagnostic marker for Cornelia de Lange syndrome. Chromosome Res 17: 763–771.
19. MehtaGD, KumarR, SrivastavaS, GhoshSK (2013) Cohesin: Functions beyond sister chromatid cohesion. FEBS Lett 587: 2299–2312.
20. VrouweMG, Elghalbzouri-MaghraniE, MeijersM, SchoutenP, GodthelpBC, et al. (2007) Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: evidence for impaired recombinational repair. Hum Mol Genet 16: 1478–1487.
21. GhiselliG (2006) SMC3 knockdown triggers genomic instability and p53-dependent apoptosis in human and zebrafish cells. Mol Cancer 5: 52.
22. SeitanVC, BanksP, LavalS, MajidNA, DorsettD, et al. (2006) Metazoan Scc4 homologs link sister chromatid cohesion to cell and axon migration guidance. PLoS Biol 4: e242.
23. ZhangY-F, JiangR, LiJ-D, ZhangX-Y, ZhaoP, et al. (2013) SMC1A knockdown induces growth suppression of human lung adenocarcinoma cells through G1/S cell cycle phase arrest and apoptosis pathways in vitro. Onco Letters 5: 749–755.
24. RollinsRA, MorcilloP, DorsettD (1999) Nipped-B, a Drosophila homologue of chromosomal adherins, participates in activation by remote enhancers in the CUT and Ultrabithorax genes. Genetics 152: 577–593.
25. RollinsRA, KoromM, AulnerN, MartensA, DorsettD (2004) Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the CUT gene. Mol Cell Biol 24: 3100–3111.
26. LiuJ, ZhangZ, BandoM, ItohT, DeardorffMA, et al. (2009) Transcriptional dysregulation in NIPBL and cohesin mutant human cells. PLoS Biol 7: e1000119 doi:10.1371/journal.pbio.1000119
27. MishiroT, IshiharaK, HinoS, TsutsumiS, AburataniH, et al. (2009) Architectural roles of multiple chromatin insulators at the human Apolipoprotein gene cluster. EMBO J 28: 1234–1245.
28. DegnerSC, WongTP, JankeviciusG, FeeneyAJ (2009) Cutting edge: developmental stage-specific recruitment of cohesin to CTCF sites throughout immunoglobulin loci during B lymphocyte development. J Immunol 182: 44–48.
29. DegnerSC, Verma-GaurJ, WongTP, BossenC, IversonGM, et al. (2011) CCCTC-binding factor (CTCF) and cohesin influence the genomic architecture of the IGH locus and antisense transcription in pro-B cells. Proc Natl Acad Sci U S A 108: 9566–9571.
30. NativioR, WendtKS, ItoY, HuddlestonJE, Uribe-LewisS, et al. (2009) Cohesin is required for higher-order chromatin conformation at the imprinted IGF2-H19 locus. PLoS Genet 5: e1000739 doi:10.1371/journal.pgen.1000739
31. StedmanW, KangH, LinS, KissilJL, BartolomeiMS, et al. (2008) Cohesins localize with CTCF at the KSHV latency control region and at cellular c-MYC and H19/IGF2 insulators. EMBO J 27: 654–666.
32. PrenzelT, KramerF, BediU, NagarajanS, BeissbarthT, et al. (2012) Cohesin is required for expression of the estrogen receptor-alpha (ESR1) gene. Epigen Chromatin 5: 1756–8935.
33. GimiglianoA, ManniniL, BianchiL, PugliaM, DeardorffMA, et al. (2012) Proteomic profile identifies dysregulated pathways in Cornelia de Lange Syndrome cells with distinct mutations in SMC1A and SMC3 genes. J Proteome Res 11: 6111–6123.
34. BellowsAM, KennaMA, CassimerisL, SkibbensRV (2003) Human EFO1p exhibits acetyltransferase activity and is a unique combination of linker histone and Ctf7p/Eco1p chromatid cohesion establishment domains. Nucleic Acids Res 31: 6334–6343.
35. HouF, ZouH (2005) Two human orthologues of Eco1/Ctf7 acetyltransferase are both required for proper sister-chromatid cohesion. Mol Biol Cell 16: 3908–3918.
36. UnalE, Heidinger-PauliJM, KimW, GuacciV, OnnI, et al. (2008) A molecular determinant for the establishment of sister chromatid cohesion. Science 321: 566–569.
37. ZhangJ, ShiX, LiY, KimBJ, JiaJ, et al. (2008) Acetylation of Smc3 by Eco1 is required for S phase sister chromatid cohesion in both human and yeast. Mol Cell 31: 143–151.
38. Rolef Ben-ShaharT, HeegerS, LehaneC, EastP, FlynnH, et al. (2008) Eco1-dependent cohesin acetylation during establishment of sister chromatid cohesion. Science 321: 563–566.
39. KimBJ, KangK-M, JungSY, ChoiH-K, SeoJ-H, et al. (2008) Esco2 is a novel corepressor that associates with various chromatin modifying enzymes. Biochem Biophys Res Comm 372: 298–304.
40. ChoiH-K, KimB-J, SeoJ-H, KangJ-S, ChoH, et al. (2010) Cohesion establishment factor, Eco1 represses transcription via association with histone demethylase, LDS1. Biochem Biophys Res Comm 394: 1063–1068.
41. LeemY-E, ChoiH-K, JungSY, KimB-J, LeeK-Y, et al. (2011) Esco2 promotes neuronal differentiation by repressing Notch signaling. Cell Signaling 23: 1876–1884.
42. SkibbensRV, CorsonLB, KoshlandD, HieterP (1999) Ctf7p is essential for sister chromatid cohesion and links mitotic chromosome structure to the DNA replication machinery. Genes Dev 13: 307–319.
43. GuacciV, KoshlandD, StrunnikovA (1997) A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell 91: 47–57.
44. SongJ, LafontA, ChenJ, WuFM, ShirahigeK, et al. (2012) Cohesin acetylation promotes sister chromatid cohesion only in association with the replication machinery. J Biol Chem 287: 34325–3436.
45. Heidinger-PauliJM, MertO, DavenportC, GuacciV, KoshlandD (2010) Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr Biol 20: 957–963.
46. RenQ, YangH, RosinskiM, ConradMN, DresserME, et al. (2005) Mutation of the cohesin related gene PDS5 causes cell death with predominant apoptotic features in Saccharomyces cerevisiae during early meiosis. Mutat Res 570: 163–173.
47. RenQ, YangH, GaoB, ZhangZ (2008) Global transcriptional analysis of yeast cell death induced by mutation of sister chromatid cohesin. Comp Funct Genomics 2008: 634283.
48. KaurM, DeScipioC, McCallumJ, YaegerD, DevotoM, et al. (2005) Precocious sister chromatid separation (PSCS) in Cornelia de Lange syndrome. Am J Med Genet 138: 27–31.
49. ZhangB, ChangJ, FuM, HuangJ, KashyapR, et al. (2009) Dosage effects of cohesin regulatory factor PDS5 on mammalian development: implications for cohesinopathies. PLoS One 4: e5232 doi:10.1371/journal.pone.0005232
50. ChenF, KamradtM, MulcahyM, ByunY, XuH, et al. (2002) Caspase proteolysis of the cohesin component RAD21 promotes apoptosis. J Biol Chem 277: 16775–16781.
51. AtienzaJM, RothRB, RosetteC, SmylieKJ, KammererS, et al. (2005) Suppression of RAD21 gene expression decreases cell growth and enhances cytotoxicity of etoposide and bleomycin in human breast cancer cells. Mol Cancer Thera 13: 361–368.
52. BeckouëtF, HuB, RoigMB, SutaniT, KomataM, et al. (2010) An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol Cell 39: 689–699.
53. BorgesV, LehaneC, Lopez-SerraL, FlynnH, SkehelM, et al. (2010) Hos1 deacetylates Smc3 to close the cohesin acetylation cycle. Mol Cell 39: 677–688.
54. XiongB, LuS, GertonJL (2010) Hos1 is a lysine deacetylase for the Smc3 subunit of cohesin. Curr Biol 20: 1660–1665.
55. van der LelijP, OostraAB, RooimansMA, JoenjeJ, de WinterJP (2010) Diagnostic overlap between Fanconi anemia and the cohesinopathies: Roberts Syndrome and Warsaw Breakage Syndrome. Anemia 2010: 565268.
56. van der LelijP, ChrzanowskaKH, GodthelpBC, RooimansMA, OostraAB, et al. (2010) Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am J Hum Genet 86: 262–266.
57. Capo-ChichiJM, BhartiSK, SommersJA, YammineT, ChoueryE, et al. (2013) Identification and biochemical characterization of a novel mutation in DDX11 causing Warsaw breakage syndrome. Hum Mutat 34: 103–107.
58. RudraS, SkibbensRV (2013) Chl1 DNA helicase regulates Scc2 deposition specifically during DNA-replication in Saccharomyces cerevisiae.. PLoS One 8: e75435 doi:10.1371/journal.pone.0075435
59. EnervaldE, LindgrenE, KatouY, ShirahigeK, StrömL (2013) Importance of Polη for damage-induced cohesion reveals differential regulation of cohesion establishment at the break site and genome-wide. PLoS Genet 9: e1003158 doi:10.1371/journal.pgen.1003158
60. HigashiTL, IkedaM, TanakaH, NakagawaT, BandoM, et al. (2013) The prereplication complex recruits XEco2 to chromatin to promote cohesin acetylation in Xenopus egg extracts. Curr Biol 22: 977–988.
61. van der LelijP, GodthelpBC, van ZonW, van GosligaD, OostraAB, et al. (2009) The cellular phenotype of Roberts Syndrome fibroblasts as revealed by ectopic expression of ESCO2. PLoS One 4: e6936 doi:10.1371/journal.pone.0006936
62. ChrzanowskaKH, GregorekH, Dembowska-BagińskaB, KalinaMA, DigweedM (2012) Nijmegen breakage syndrome (NBS). Orphanet J Rare Diseases 7: 13.
63. KeeY, D'AndreaAD (2012) Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest 122: 3799–3806.
64. NarlaA, EbertB (2010) Ribosomopathies: human disorders of ribosome dysfunction. Blood J 115: 3196–3205.
65. SchlumpJ-U, SteinA, HehrU, KarenT, Moller-HartmannC, et al. (2012) Treacher Collins syndrome: clinical implication for the paediatrician – a new mutation in a severe affected newborn and comparison with three further patients with the same mutation, and review of the literature. Eur J Pediatr 171: 1611–1618.
66. BauerM, SaldarriagaW, WolfeSA, BeckwithJB, FriasJL, et al. (2013) Two extraordinary severe cases of Treacher Collins Syndrome. Am J Med Genet 161: 445–452.
67. LalorayaS, GuacciV, KoshlandD (2000) Chromosomal addresses of the cohesin component Mcd1p. J Cell Biol 151: 1047–11056.
68. KobayashiT, HoriuchiT, TongaonkarP, VuL, NomuraM (2004) SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117: 441–453.
69. KobayashiT, GanleyAR (2005) Recombination regulation by transcription-induced cohesin dissociation in rDNA repeats. Science 309: 1581–1584.
70. HarrisonBD, HoangML, BloomK (2009) Persistent mechanical linkage between sister chromatids throughout anaphase. Chromosoma 118: 633–645.
71. GardS, LightW, XiongB, BoseT, McNairnAJ, et al. (2009) Cohesinopathy mutations disrupt the subnuclear organization of chromatin. J Cell Biol 187: 455–462.
72. KawauchiS, CalofAL, SantosR, Lopez-BurksME, YoungCM, et al. (2009) Multiple organ system defects and transcriptional dysregulation in the Nipbl(+/−) mouse, a model of Cornelia de Lange Syndrome. PLoS Genet 5: e1000650 doi:10.1371/journal.pgen.100065
73. SakaiD, TrainorPA (2008) Treacher Collins syndrome: unmasking the role of TCOF1/Treacle. Int J Biochem Cell Biol 41: 1229–1232.
74. SkibbensRV, MarzillierJ, EastmanL (2010) Cohesins coordinate gene transcriptions of related function within Saccharomyces cerevisiae. Cell Cycle 9: 1601–1666.
75. BoseT, LeeKK, LuS, XuB, HarrisB, et al. (2012) Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS Genet 8: e1002749 doi:10.1371/journal.pgen.1002749
76. GertonJL (2012) Translational mechanisms at work in the cohesinopathies. Nucleus 3: 520–525.
77. LiuJ, KrantzID (2009) Cornelia de Lange syndrome, cohesin, and beyond. Clin Genet 76: 303–314.
78. AuerbachAD (2009) Fanconi anemia and its diagnosis. Mutat Res 668: 4–10.
79. Genetics Home Reference, A Service of the National Library of Medicine, National Institutes of Health, Available: ghr.nlm.nih.gov/condition. Accessed June 2013.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 12
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- The NuRD Chromatin-Remodeling Enzyme CHD4 Promotes Embryonic Vascular Integrity by Transcriptionally Regulating Extracellular Matrix Proteolysis
- Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome Protein Expression
- The Midline Protein Regulates Axon Guidance by Blocking the Reiteration of Neuroblast Rows within the Drosophila Ventral Nerve Cord
- Tomato Yield Heterosis Is Triggered by a Dosage Sensitivity of the Florigen Pathway That Fine-Tunes Shoot Architecture