
Myeloid Dendritic Cells Induce HIV-1 Latency in Non-proliferating CD4 T Cells
Latently infected resting CD4+ T cells are a major barrier to HIV cure. Understanding how latency is established, maintained and reversed is critical to identifying novel strategies to eliminate latently infected cells. We demonstrate here that co-culture of resting CD4+ T cells and syngeneic myeloid dendritic cells (mDC) can dramatically increase the frequency of HIV DNA integration and latent HIV infection in non-proliferating memory, but not naïve, CD4+ T cells. Latency was eliminated when cell-to-cell contact was prevented in the mDC-T cell co-cultures and reduced when clustering was minimised in the mDC-T cell co-cultures. Supernatants from infected mDC-T cell co-cultures did not facilitate the establishment of latency, consistent with cell-cell contact and not a soluble factor being critical for mediating latent infection of resting CD4+ T cells. Gene expression in non-proliferating CD4+ T cells, enriched for latent infection, showed significant changes in the expression of genes involved in cellular activation and interferon regulated pathways, including the down-regulation of genes controlling both NF-κB and cell cycle. We conclude that mDC play a key role in the establishment of HIV latency in resting memory CD4+ T cells, which is predominantly mediated through signalling during DC-T cell contact.
Published in the journal:
. PLoS Pathog 9(12): e32767. doi:10.1371/journal.ppat.1003799
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003799
Summary
Latently infected resting CD4+ T cells are a major barrier to HIV cure. Understanding how latency is established, maintained and reversed is critical to identifying novel strategies to eliminate latently infected cells. We demonstrate here that co-culture of resting CD4+ T cells and syngeneic myeloid dendritic cells (mDC) can dramatically increase the frequency of HIV DNA integration and latent HIV infection in non-proliferating memory, but not naïve, CD4+ T cells. Latency was eliminated when cell-to-cell contact was prevented in the mDC-T cell co-cultures and reduced when clustering was minimised in the mDC-T cell co-cultures. Supernatants from infected mDC-T cell co-cultures did not facilitate the establishment of latency, consistent with cell-cell contact and not a soluble factor being critical for mediating latent infection of resting CD4+ T cells. Gene expression in non-proliferating CD4+ T cells, enriched for latent infection, showed significant changes in the expression of genes involved in cellular activation and interferon regulated pathways, including the down-regulation of genes controlling both NF-κB and cell cycle. We conclude that mDC play a key role in the establishment of HIV latency in resting memory CD4+ T cells, which is predominantly mediated through signalling during DC-T cell contact.
Introduction
Antiretroviral therapy (ART) for the treatment of HIV has led to a substantial reduction in morbidity and mortality; however, ART cannot cure HIV and life-long treatment is required. This is directly due to the persistence of long-lived latently infected cellular reservoirs, that include microglia, astrocytes, macrophages and naïve T cells [1]–[4], however, resting memory CD4+ T cells [5]–[7], are considered to be the major contributors. Latently infected resting CD4+ T cells are found in blood and tissue sites, including lymphoid tissue and the gastrointestinal tract [7]–[10]. The frequency of latently infected cells is up to ten times higher in tissue than in blood in HIV-infected patients or SIV-infected macaques on suppressive ART [8], [10].
It is unclear how latency is established in vivo. However, in vitro, latency can be established following survival of an activated CD4+ T cell that returns to a resting state carrying integrated virus [5], [11]–[13]. Alternatively, latency has also been established following direct infection of resting cells in the presence of chemokines or following spinoculation [14]–[19]. Dendritic cells (DC) are found throughout the body and interact closely with resting CD4+ T cells within lymphoid tissues. Therefore, given the high frequency of latently infected cells in lymphoid tissue, we hypothesised that latency in resting CD4+ T cells may result from interactions with DC as CD4+ T cells recirculate through lymphoid tissue. Using a novel model of resting CD4+ T cells co-cultured with primary DC, we demonstrate that myeloid DC (mDC) induce post-integration latency in resting memory CD4+ T cells, which required close DC-T cell contact.
Results
Myeloid DC promote HIV latency in non-proliferating CD4+ T cells
Resting CD4+ T cells and syngeneic DC (including the two major blood DC subpopulations, plasmacytoid (pDC) and myeloid (mDC) DC) were sorted from the blood of healthy donors (Fig. S1). Seminaphtharhodafluor-1 (SNARF)-labelled resting CD4+ T cells were cultured either alone or co-cultured with DC at a DC: T cell ratio of 1: 10. Following 24 hours of culture, cells were infected with a CCR5-tropic enhanced green fluorescent protein (EGFP)-reporter virus, NL(AD8)-nef/EGFP (multiplicity of infection, MOI 0.5), and cultured for 5 days (Fig. 1 A). Cells were then analysed for expression of EGFP by flow cytometry to quantify productive infection (Fig. 1 B).
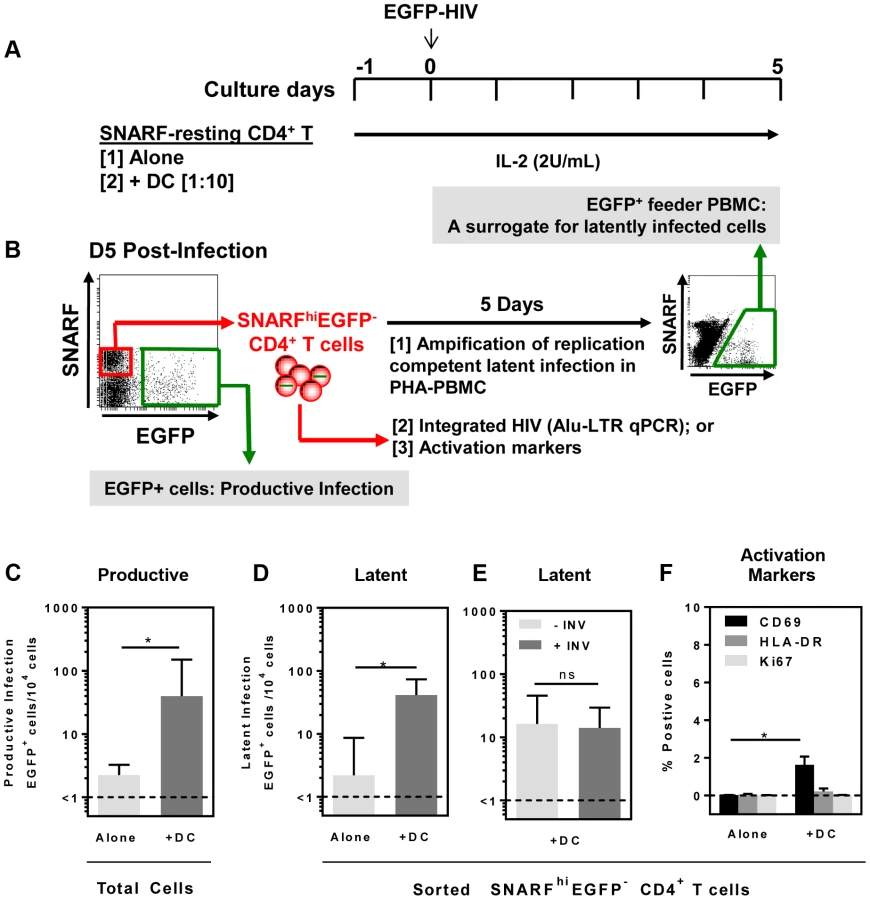
In the DC-CD4+ T cell co-cultures, we detected a spreading productive infection with the number of infected cells 5 days post-infection significantly greater (median (IQR) = 40 (31, 150) EGFP+ cells/104 cells; n = 5) compared to CD4+ T cells cultured alone (1.5 (1, 2.5) EGFP+ cells/104 cells, p = 0.03; Fig. 1 C). These results were consistent with previous work demonstrating enhanced productive infection of CD4+ T cells in the presence of DC [20], [21].
At day 5 post-infection, non-proliferating (SNARFhi) CD4+ T cells that were not productively infected (EGFP−) were sorted (purity was always >99%). Latent virus was quantified in the SNARFhiEGFP− CD4+ T cells upon stimulation with phytohaemagglutinin (PHA) in the presence of feeder peripheral blood mononuclear cells (PBMC) following a further 5 days of culture (Fig. 1 B). The number of EGFP+ cells following stimulation was, therefore, a surrogate measure for the number of latently infected cells in the SNARFhiEGFP− CD4+ T cells. When SNARFhiEGFP− CD4+ T cells were sorted from cultures infected in the absence of DC, few latently infected cells were detected (2 (1, 8.5) EGFP+ cells/104 cells; n = 5). In contrast, when SNARFhiEGFP− CD4+ T cells were sorted from the DC-T cell co-cultures a significant increase in the number of latently infected cells was observed (41 (28, 73) EGFP+ cells/104 cells; p = 0.03; n = 5; Fig. 1 D). Furthermore, when infections were performed in the presence of the protease inhibitor indinavir, there was no significant difference in the number of latently infected cells, as measured by EGFP expression following co-culture of SNARFhiEGFP− with PHA and feeder PBMC (Fig. 1 E). This confirms that a productive, spreading infection was not required to establish latency. Together, these results demonstrate that DC facilitate latent HIV infection in non-proliferating CD4+ T cells.
We next asked whether DC-T cell co-culture had activated the SNARFhi CD4+ T cells, which allowed for HIV entry. Sorted SNARFhiEGFP− CD4+ T cells that were co-cultured with DC for 5 days showed signs of early activation with increased expression of CD69 (1.5% (0.1, 2.1); p = 0.02; n = 4; Fig. 1 F). However, these cells did not express either HLA-DR or Ki67 (Fig. 1 F). As expected, resting CD4+ T cells that were cultured alone did not express any of the activation markers. These results confirmed that the sorted SNARFhiEGFP− cells were non-proliferating, partially activated CD4+ T cells.
To determine whether mDC or pDC were facilitating latency in resting CD4+ T cells, we next co-cultured sorted mDC and pDC with SNARF-labelled resting CD4+ T cells for 24 hours prior to infection, and experiments were performed as described above. While productive infection was enhanced in both the mDC and pDC-T cell co-cultures (Fig. 2 A), latent infection was only identified in the SNARFhiEGFP− CD4+ T cells that had been co-cultured with mDC (33 (19, 51) EGFP+ cells/104 cells; n = 5) following re-stimulation with PHA and feeder PBMC (Fig. 2 B) or following direct activation with anti-CD3/CD28, together with IL-7 and the integrase inhibitor L8, which allowed the detection of post-integration latency (Fig. 2 C). To further confirm that post-integration latency was established in resting CD4+ T cells co-cultured with mDC, we used a real time PCR assay to quantify integrated HIV DNA. Integrated HIV DNA was present in SNARFhiEGFP− CD4+ T cells sorted from the mDC co-cultures (1100 (686, 4960) HIV DNA copies/106 cells, n = 3; Fig. 2 D), but not in the CD4+ T cells sorted from the pDC co-cultures or the CD4+ T cells cultured alone (both <330 HIV DNA copies/106 cells). Similar results were observed with a nef competent EGFP-reporter virus (Fig. 2 E), demonstrating that the establishment of mDC-induced latency was not dependent on Nef.
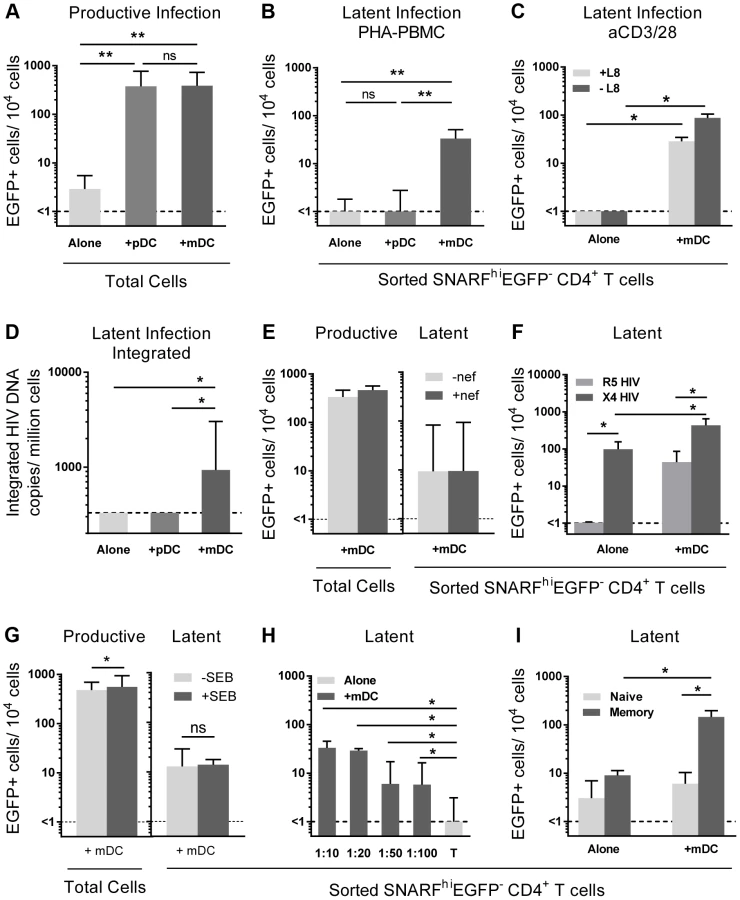
Unlike experiments that utilised R5 EGFP HIV, when experiments were performed with an X4 EGFP reporter virus, latent infection was detected in the resting CD4+ T cells cultured alone (87 (51, 155) EGFP+ cells/104 cells; n = 4; Fig. 2 F). However, latency was still significantly enhanced in the non-proliferating CD4+ T cells in the presence of mDC (468 (213, 621) EGFP+ cells/104 cells) when compared to the T cells cultured alone.
In some experiments we added a low dose of Staphylococcus enterotoxin B (SEB; 10 ng/mL) to the mDC-T cell co-cultures to enhance productive infection and increase cognate interactions between mDC and T cells. In the presence of SEB we observed a significant increase in the level of productive infection; however, there was no difference in the level of latent infection (Fig. 2 G). Finally, we cultured mDC and T cells together at ratios ranging from 1∶10 to 1∶100 to determine the minimum interaction required between mDC and T cells to induce latency. We found that latency could still be established at a ratio of DC: T cells as low as 1∶100 (Fig. 2 H).
Taken together, these results demonstrated that in vitro mDC and not pDC facilitated post-integration latency in non-proliferating CD4+ T cells.
Myeloid DC induce latency in memory CD4+ T cells
We have previously shown that memory CD4+ T cells and not naïve CD4+ T cells are susceptible to latent infection following chemokine exposure [14]. To determine whether mDC-induced T cell latency occurred in memory or naïve CD4+ T cells, we separated the SNARFhiEGFP− CD4+ T cells into CD45RO+ (memory) and CD45RO− (naïve) fractions prior to culture with feeder PBMC. In these experiments, latent infection was detected at significantly higher levels in the CD45RO+ memory CD4+ T cell fraction (146 (14, 197) EGFP+ cells/104 cells; Fig. 2 I).
Depletion of CD69 expressing cells has no effect on DC-induced latency
A proportion of non-proliferating SNARFhiEGFP− cells sorted from DC-T cell co-cultures (Fig. 1 F and 3 A) expressed CD69. Therefore, in order to exclude the possibility that we were only detecting infection of the cells showing early signs of activation, we depleted CD69+ cells from the SNARFhiEGFP− T cells at day 5 post-infection prior to co-culture with PHA and feeder PBMC. We found no significant difference in the level of latency following depletion of the CD69+ cells (Fig. 3 B). CD69 expression can be transient, therefore, to confirm that we had not missed cells that expressed CD69, which had then been down-regulated; we measured the expression of CD69 over time following co-culture with mDC and infection with HIV. We demonstrated that CD69 expression peaked at day 2 and remained elevated out to day 5 post-infection (data not shown). These results demonstrate that the subpopulation of CD4+ T cells that were partially activated and expressing CD69 were not preferentially latently infected following mDC-T cell co-culture.

HIV infection of mDC-T cell co-cultures leads to an increase in chemokine and cytokine expression
To determine why mDC and not pDC led to the establishment of latency, we compared cytokine levels in pDC-T cell and mDC-T cell co-cultures 5 days following HIV infection using bead arrays for known DC-secreted cytokines. Supernatants collected from HIV-infected mDC-T cell co-cultures compared to the HIV-infected pDC-T cell co-cultures had significantly increased expression of IL-6 (p = 0.002), IL-10 (p = 0.01) and CXCL9 (p = 0.002; Fig. 4 A). TNF-alpha was also up-regulated in the mDC-T cell compared to pDC-T cell co-cultures but the difference was not statistically significant (Fig. 4 A). As expected, IFN-alpha was detected at high levels in the pDC-T cell co-cultures but not in the mDC-T cell co-cultures (Fig. 4 A; p = 0.01), and latency was not established in pDC-T cell co-cultures even in the presence of neutralising antibodies to IFN-alpha (Fig. 4 B). Interestingly, when equal numbers of pDC and mDC were added to resting CD4+ T cells latency was reduced when compared to T cells cultured only with mDC (Fig. 4 C). This suggests that while pDC themselves do not induce latency, they are able to inhibit the establishment of latency mediated by mDC.
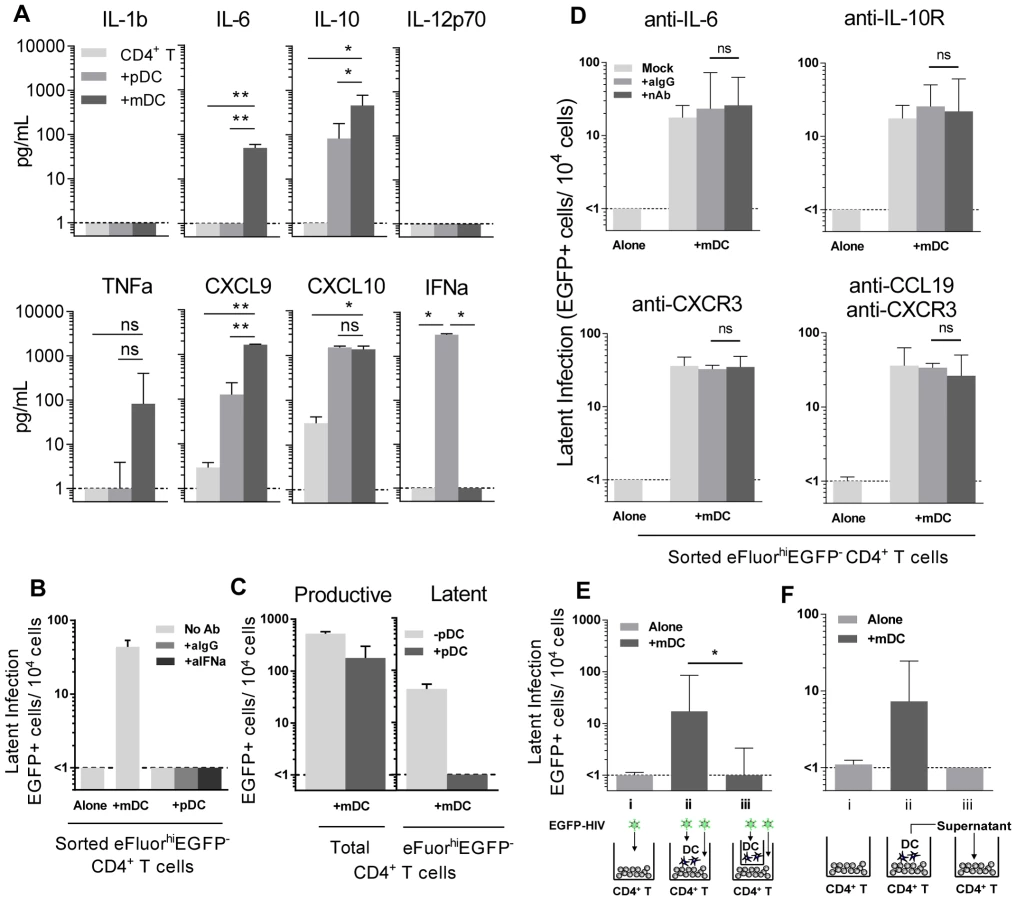
Blocking Abs to IL-6, IL-10R, CXCR3 and CCL19 have no effect on DC-induced latency
To determine whether the soluble factors that were differentially expressed in mDC-T cell co-cultures compared to pDC-T cell co-cultures were contributing to the establishment of mDC-induced latency, neutralising antibodies (nAb), to either the soluble factor or its receptor, were added to the T cells prior to co-culture with DC and the addition of HIV. Specific nAbs or an anti-IgG control were added to eFluor670 (alternative proliferation dye to SNARF)-labelled resting memory (CD45RO+) CD4+ T cells prior to co-culture with mDC, and again following infection, and latency determined as described in Fig. 1 (Fig. 4 D).
When nAbs to IL-6, the IL-10 receptor (IL-10R) or CXCR3 (CXCL9 and CXCL10 receptor) were added to the mDC-T cell co-cultures, no significant decrease in the number of latently infected CD4+ T cells was observed when compared to cultures where control anti-IgG was added. As we had previously shown that the chemokine CCL19 can condition resting CD4+ T cells allowing for enhanced entry and integration of HIV [14], we also added anti-CCL19, either alone or in combination with anti-CXCR3, to the mDC-T cell co-cultures. However, we did not detect a significant decrease in the number of latently infected cells. The activity of these nAbs was confirmed by their ability to block STAT3 signalling (aIL-6, aIL-10R) or chemokine induced migration (aCXCR3, aCCL19; Fig. S2).
DC-induced latency requires close proximity of DC and CD4+ T cells
To determine whether DC-T cell contact was required to establish latency in resting CD4+ T cells, we co-cultured mDC and resting CD4+ T cells separated by a 0.4 µm membrane transwell. Following 24 hours of culture, HIV was added to the mDC in the upper chamber and the CD4+ T cells in the lower chamber. Without mDC-T cell contact, the establishment of latency was significantly inhibited (<1 EGFP+ cell/104 cells) when compared to co-cultures without membranes (18 (10, 85) EGFP+ cell/104 cells; n = 5; p = 0.03; Fig. 4 E).
To further elucidate whether soluble factors other than those previously inhibited were involved in DC-induced latency, we added supernatant from infected mDC-T cell co-cultures to uninfected resting CD4+ T cells and then infected these cells with EGFP-HIV. Media changes were performed daily using supernatant from infected mDC-T cell co-cultures. Under these conditions the resting CD4+ T cells would be exposed to any soluble factors and free viral particles present in the mDC-T cell co-cultures but would not have any contact with the mDC. Latency was not detected in these cultures (Fig. 4 F), providing further evidence that DC-T cell contact, and not a soluble factor, was required for the establishment of DC-induced latency.
Direct DC-T cell signalling can occur following interactions between several cell surface receptors. In particular, interactions between lymphocyte function associated antigen-1 (LFA-1; composed of CD11a and CD18) on T cells and intercellular adhesion molecule 1 (ICAM-1) on DC are involved in DC-T cell adhesion [22] and subsequent T cell activation via formation of an immunological synapse [23]. To inhibit DC-T cell clustering, we used blocking antibodies to CD18 (10–20 µg/mL). Blocking of CD18 significantly inhibited, but did not eliminate, DC-T cell clustering, as observed by microscopy (data not shown). Following incubation with anti-CD18, there was no effect on the number of productively infected cells, however, we observed a significant decrease in the number of latently infected cells from the mDC-T cell co-cultures (20 (8, 34) latently infected cells/104 cells), when compared to cells cultured without anti-CD18 (25 (17, 48) latently infected cells/104 cells; p = 0.03) or in the presence of control anti-IgG (32 (22, 36) latently infected cells/104 cells; n = 6; p = 0.03; Fig. 5 A). However, when resting CD4+ T cells were stimulated with soluble ICAM-1 and anti-IgG there was no increase in latency observed (Fig. 5 B), suggesting that ICAM-LFA signalling alone does not induce latency in this model system. Furthermore, in the presence of anti-CD18 the number of latently infected cells from the mDC-T cell co-cultures remained greater than the CD4+ T cells cultured alone (<1 latently infected cell/104 viable cells; p = 0.01) suggesting that the effect of anti-CD18 was most likely due to the partial decrease in clustering/DC-T cell contact.
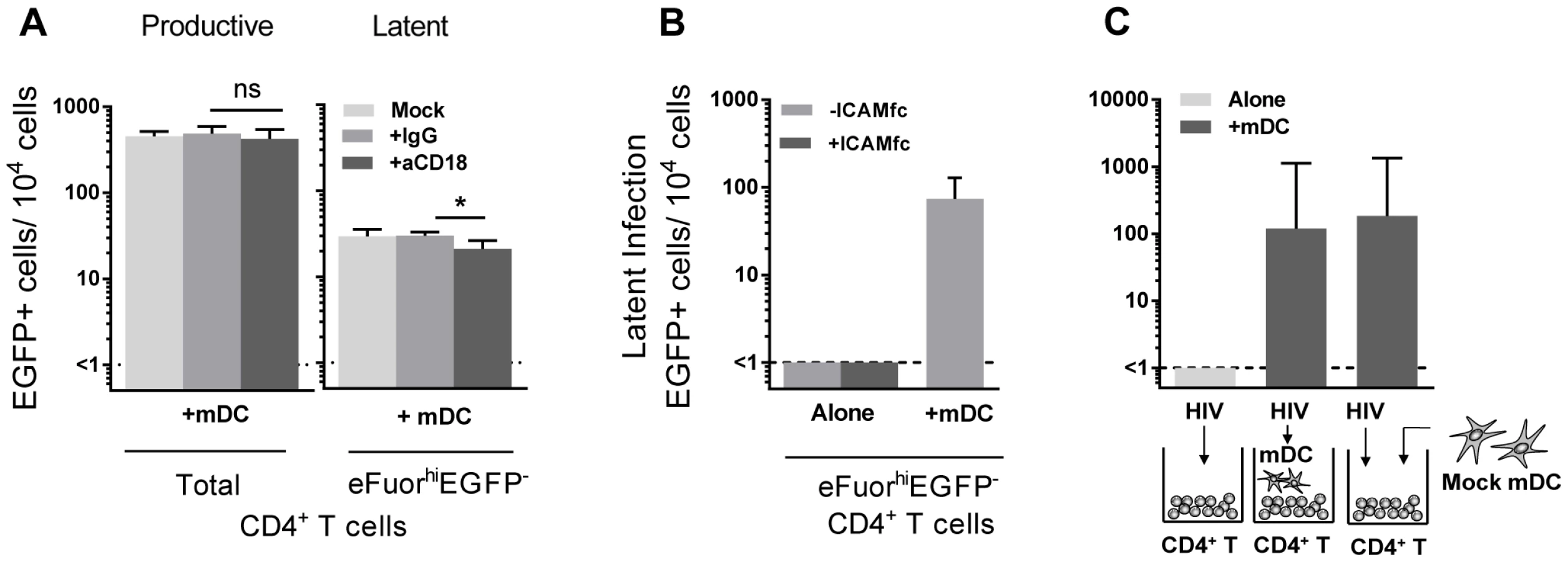
In order to determine whether mDC transfer of HIV was involved in the establishment of latency we performed experiments where we added virus to resting CD4+ T cells, washed off virus and added uninfected mDC to the CD4+ T cells (Fig. 5 C). Under these conditions we were still able to detect latency in the non-proliferating CD4+ T cells. Together, these results indicate that cell-cell contact plays a role in DC-induced T cell latency but that the mDC were not required to be infected and then transfer HIV to the resting CD4+ T cells.
Multiple genes are differentially expressed in latently infected, non-proliferating CD4+ T cells co-cultured with DC
To determine the effect of DC on gene transcription in latently infected resting CD4+ T cells, SNARF-labelled resting CD4+ T cells, from four independent donors, were cultured either alone or with syngeneic bulk blood DC at a 1∶10 ratio for 24 hours prior to infection with NL(AD8)-nef/EGFP. In these experiments, we included IL-7 (10 ng/mL) in all cultures to increase cell survival of the resting cells and infections were performed at an MOI of 5 to ensure a high frequency of latently infected cells. Mock infections were performed in parallel with media alone. Non-proliferating (SNARFhi) CD4+ T cells that were not productively infected (EGFP−) were sorted 5 days post-infection and lysed for either the detection of HIV DNA by real-time PCR or RNA for microarray studies (Fig. 6 A).

Infection was confirmed in the resting CD4+ T cells following co-culture with DC, in 4 independent experiments, by detection of HIV DNA (3×104 (7.4×103, 5.7×105) copies/106 cells; Fig. 6 B). Changes in gene expression were quantified in the sorted SNARFhiEGFP− CD4+ T cells using Illumina oligonucleotide microarrays. To identify genes expressed in DC-induced latency, we compared the expression profiles of non-proliferating, latently infected CD4+ T cells (HIV T (+DC)) to mock infected CD4+ T cells (Mock T (+DC)) that had been co-cultured with DC. In order to control for the effect of virus or DC alone, we first subtracted the gene expression profiles of control cells, which were T cells that had been cultured alone that were either uninfected (Mock T) or exposed only to virus (HIV T).
A scatter plot (Fig. 7 A), representing the common (genes that fall on the diagonal) and differentially expressed genes (genes that fall off the diagonal) from this comparison, highlighted the significant differences in gene expression between latently infected cells and controls (r = 0.77). Additionally, this plot showed that several of the genes that discriminate latently infected cells from control cells were genes downstream of type I interferons, including interferon-induced protein with tetratricopeptide repeats 1 (IFIT-1), interferon alpha-inducible protein 27 (IFI27), and 2′-5′-oligoadenylate synthetase 1 (OAS1).
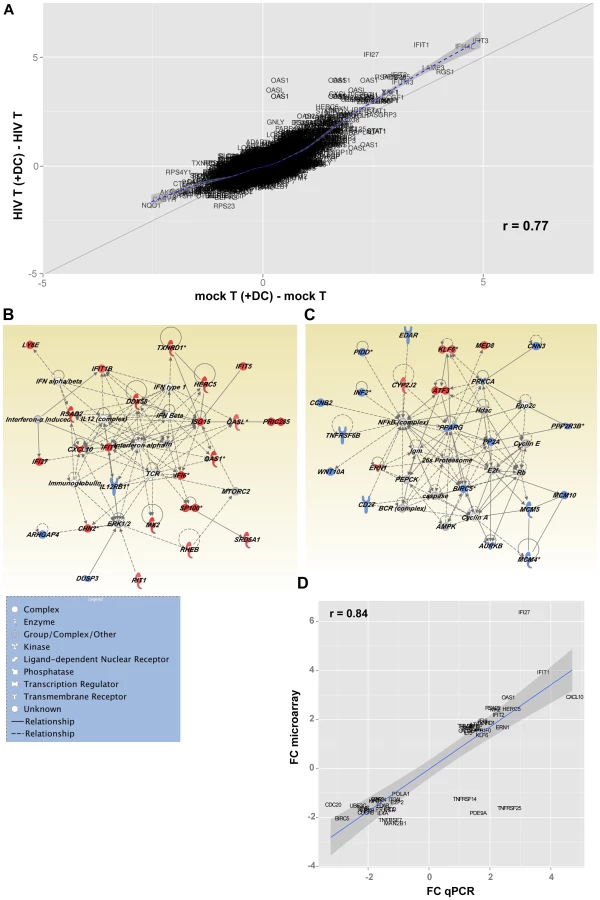
Inhibition of cell cycle entry in latently infected, non-proliferating CD4+ T cells
Heatmaps of the top 100 genes (Fig. S3) confirmed the de novo induction of genes encompassing several biological and metabolic pathways in T cells exposed both to DC and virus. These included transcripts of the Interferon pathway, genes involved in the regulation of cell cycle entry and mitosis, as well as receptor and effector molecules of cell survival and apoptosis.
Network analysis (Fig. 7 B and C) showed that two major pathways were regulated in T cells following co-culture with DC exposed to HIV. Exposure of CD4+ T cells to HIV and DC led to the up-regulation of genes downstream of type I interferon by nucleotide sensors. Figure 7 B confirms the wide-ranging impact of the up-regulation of the type I Interferon pathway, as several molecules with antiviral activity were up-regulated, including ISG15 ubiquitin-like modifier (ISG15), and DEAD box polypeptide 58 (DDX58; also known as RIG-I). Genes involved in actin polymerisation, and the organisation of microtubules, were also up-regulated as a consequence of interferon pathway up-regulation. Additionally, the interferon pathway intercepted with the mammalian target of rapamycin complex 2 (mTORC2) pathway, which plays an important role in autophagy and T cell survival [24] (Fig. 7 B; Table S1).
Network analysis confirmed the negative impact of exposure of CD4+ T cells to DC and virus on the NF-κB pathway as well as several cellular metabolic pathways (fatty oxidation and glucose metabolism) regulated by peroxisome proliferator-activated receptor gamma (PPARG; Fig. 7 C). Inhibition of the NF-κB transcriptional network, which plays a significant role in HIV transcription, led to the down-regulation of protein kinase C alpha (PRKCA). This observation confirms the quiescence of these cells and may also be a step towards the induction of HIV latency. Triggering of a transcriptional program leading to T cell quiescence was confirmed by the increased expression of Kruppel-like factor 6 (KLF6), a gene with anti-proliferative functions [25], [26], as well as activating transcription factor 3 (ATF3) that has been recently been shown to negatively regulate activating protein 1 (AP-1)-mediated HIV transcription. Additionally, we observed a down-regulation of several molecules involved in DNA replication such as members of the minichromosome maintenance (MCM) complex (MCM4, MCM5, and MCM10) and the aurora kinase (Fig. 7 C). Pathways controlling pyrimidine and purine synthesis were also expressed at lower levels in cells exposed to virus and DC highlighting a reduced availability of nucleotides for cell division (Table S1). The down-regulation of NF-κB resulted in the decreased expression of several molecules that play a critical role in T cell survival, including CD27 (TNFRSF7), baculoviral IAP repeat containing 5 (BIRC5/survivin) and tumor necrosis factor receptor superfamily, member 6b (TNFRSF6B/DCR3), a decoy receptor that inhibits Fas ligand and LIGHT-mediated signalling [27]. In order to confirm the differential expression of genes in these different populations of cells, we used a highly quantitative PCR approach and showed a strong correlation between gene expression data measured by either gene array or PCR (Fig. 7 D and Table S2).
Taken together, results of transcriptional profiling highlighted the impact of two major transcriptional nodes in the inhibition of viral replication and the induction of latency. The up-regulation of Type I Interferons and their downstream target genes could trigger several genes endowed with antiviral activities and would also impact cell proliferation, survival and metabolic processes. Concomitantly, the down-regulation of NF-κB will lead to T cell quiescence and decreased levels of activation, both of which are required for HIV replication.
Discussion
The study of latently infected resting CD4+ T cells ex vivo from HIV-infected patients on ART is greatly limited by the low frequency of latently infected cells and the lack of a distinctive surface marker to distinguish latently infected from uninfected cells. Here we demonstrate that latency can be efficiently established via direct infection of non-proliferating CD4+ T cells in the presence of DC. Using primary blood DC and resting CD4+ T cells we have demonstrated that: [1] co-culture of resting memory CD4+ T cells with DC can establish latent infection; [2] mDC but not pDC mediate this effect; [3] close cell-cell proximity is required between DC and T cells; and [4] multiple cell cycle genes were altered in non-proliferating CD4+ T cells, containing latently infected cells. These novel findings provide a potential pathway for the establishment and maintenance of latent infection in resting CD4+ T cells that recapitulates the likely events within lymphoid tissues in HIV-infected patients in vivo.
Previous studies have explored the ability of DC to enhance productive HIV infection within DC-CD4+ T cell co-cultures [28]–[31]; however, we are the first to present data demonstrating the ability of specific subpopulations of DC to induce latency in resting CD4+ T cells in these co-cultures. Using this model we clearly demonstrated that following co-culture of mDC with resting memory CD4+ T cells, post-integration latency was established. This was demonstrated by inducible virus (established both in the presence and absence of indinavir) and detectable integrated HIV DNA in T cells cultured with mDC but not those cultured alone following infection with an R5 EGFP reporter virus. While integrated R5 HIV DNA was only detected following co-culture with mDC in our model of latency, it was similar to that previously reported for resting CD4+ T cells infected in isolation with a wild type X4 NL4.3 virus [32]. Resting CD4+ T cells express very low levels of CCR5 in contrast to expressing very high levels of CXCR4. Additionally, we saw significantly higher levels of latency (∼100 fold) in our T cells cultured alone when we used an X4 EGFP reporter virus compared to an R5 EGFP virus.
Unlike memory CD4+ T cells, we were unable to detect latency in naïve CD4+ T cells following mDC co-culture. It is possible that the differential establishment of latency in resting naïve and memory T cells was due to differences in their cortical actin density and actin dynamics as previously suggested by others [33]. While mDC induced T cell latency in this model, pDC did not. Interestingly, pDC played an active inhibitory role in establishing latency, when co-cultures were performed with equal numbers of pDC and mDC (Fig. 4 C). One potential explanation for the difference between co-cultures of bulk DC and T cells (where latency was established) and DC-T cell co-cultures containing purified equal numbers of pDC and mDC (where latency was inhibited) could potentially be that the number of pDC present in the bulk DC (roughly 1 pDC to 3 mDC) was too low to inhibit latency. How pDC actively suppress the establishment of latency is unknown, but it does not appear to be mediated by IFN-alpha.
Establishment of mDC-induced latency was not dependent on DC-T cell transfer of HIV, as latency was still detected when T cells were infected in isolation and uninfected mDC added only after virus had been washed off. Nor was it dependent on the amount of virus replication, because while only mDC were able to induce latent infection, similar levels of productive infection were observed in both the pDC and mDC co-cultured CD4+ T cells (Fig. 2 A). Furthermore, we found that addition of SEB to the culture model enhanced productive infection but did not increase latent infection (Fig. 2 G). Together, these data provide evidence that the establishment of latency in the non-proliferating CD4+ T cells when co-cultured with mDC was not simply due to higher viral exposure in these cultures.
These results differ from a previous study that also looked at direct infection of resting CD4+ T cells, which concluded that DC had no effect on the integration levels of R5 or X4 virus in either naïve or memory CD4+ T cells [34]. However, in this study, although primary DC were used (defined as BDCA-1+ and BDCA-4+ cells), total DC were present at a frequency of only 0.89% and therefore the frequency of mDC may have been too low to demonstrate an effect of mDC on the infection of resting CD4+ T cells. Additionally, contrary to our data, a recent study has reported the ability of monocyte derived DC (MDDC) to activate latent infection in T cells [35]. A key difference was that latency in this study was unusually established in proliferating CD4+ T cells and not non-proliferating T cells as in our study. Furthermore, MDDC, as opposed to primary DC, were utilised in this study. MDDC have multiple significant functional and lineage differences to primary DC as we have recently demonstrated using detailed sorting and gene expression analyses [36].
In this study we utilised total blood CD11c+ mDC, which consist of at least three different subsets, a major SLAN (6-sulfo LacNAc+), an intermediate CD1c+ (BDCA-1) and a minor CD141+ (BDCA-3) population, each with different functional properties [37]–[40]. However, it is currently unclear whether one or more of these subsets is responsible for inducing latency in resting CD4+ T cells.
We have previously demonstrated that multiple chemokines, including CCL19, CXCL9 and CXCL10, can condition resting CD4+ T cells allowing for the establishment of HIV latency [14], [17]. However, blocking CCL19 and CXCR3, the receptor for CXCL9, 10 and 11, had a minimal impact on DC-induced latency (Fig. 4 D). While it is possible that there may be involvement of chemokines other than those inhibited, given that latency was not detected in resting CD4+ T cells infected in the presence of infected mDC-T cell culture supernatants, this is unlikely (Fig. 4 F). Rather, our data supports an essential role for direct DC-T cell interactions or DC-T cell signalling as mDC-induced latency was prevented when the mDC were cultured in transwells above the resting CD4+ T cells (Fig. 4 E). Unlike our previous work that was performed in the absence of productive infection [14], latency following DC-T cell co-culture was established in the presence of productive infection, which may more accurately mimic the establishment of latency in acute infection in vivo. Therefore, in the presence of productive infection it is possible that there are alternative pathways that lead to the establishment and maintenance of latency in resting CD4+ T cells.
Interactions between ICAM-1, found on DC, and LFA-1, found on T cells, strengthen DC-T cell adhesion and play a key role in the formation of the immunological synapse [41]. We have shown that clustering, facilitated by ICAM-1-LFA-1 interactions, contributed to DC-induced T cell latency, as latency was significantly reduced, but not eliminated, when blocking antibodies to CD18/LFA-1 were added to the DC-T cell co-cultures (Fig. 5 A). However, interactions between ICAM-1 and LFA-1 alone were not sufficient to induce T cell latency in the absence of mDC (Fig. 5 B), therefore, the reduction in latency observed in the presence of anti-CD18 was most likely due to the reduction in DC-T cell clustering rather than specific LFA-ICAM signalling events. As there are numerous other molecules involved in DC-T cell clustering, such as LFA-3 and CD2, additional signalling pathways should be explored as potential mediators of DC-induced HIV latency. Interestingly, a recent paper has demonstrated enrichment of latency in CD2 expressing T cells from HIV-infected patients on ART [42].
Transcriptional profiling experiments served to highlight changes in cellular gene expression in resting non-proliferating CD4+ T cells that contained latently infected CD4+ T cells. We showed significant differences in gene expression between resting CD4+ T cells from HIV and mock infected DC-T cell cultures. However, it is important to note that, while all cells within our “latent” cell population were exposed to virus, only a proportion were actually infected (median of 3%). It is possible that some of the observed differences in gene expression may be due to uninfected cells that were exposed to HIV but not infected. This may include the genes downstream of type I interferons as our in vitro experiments have shown that pDC, the major producers of type I interferons, were not involved in the induction of T cell latency. Therefore, it is possible that type I interferons are necessary but alone are not sufficient to induce HIV latency. We have demonstrated significant differential expression of genes involved in cell cycle, in particular those associated with cell cycle arrest (Fig. 7 C). During DC-T cell interactions in the presence of HIV, differential expression of co-stimulatory and negative regulatory factors determines the fate of the interacting CD4+ T cell [43]. These interactions can result in active suppression of T cell cycle and as a result may inhibit post-integration steps in viral replication and promote the establishment of latency. Indeed, in HIV-infected patients on ART, HIV DNA is found at higher frequencies in CD4+ T cells expressing the negative regulator PD-1 [44].
Latency has also been shown to be triggered by the absence of certain transcriptional machinery in resting CD4+ T cells, such as NF-κB [45] and nuclear factor of activated T (NFAT) [11], [46]. In DC-induced latently infected CD4+ T cells we observed the suppression of multiple genes associated with the activation of NF-κB (Table S1), including protein kinase C alpha, PRKCA, which also plays a role in the activation of NFAT [47], [48]. Therefore, it is possible that the global suppression of genes associated with the activation of NF-κB and/or NFAT may also contribute to the maintenance of latency in DC- T cell co-cultures by preventing progression to productive infection in cells that contain integrated HIV. However, while this data provides insights into genes that may potentially be important for both the establishment and maintenance of latency, it will be important to conduct gene knockdown experiments within our model in order to determine the specific role of individual genes in establishing and maintaining mDC-induced T cell latency.
In summary, this study has demonstrated a novel pathway for the establishment of latency in resting memory CD4+ T cells that was dependent on close proximity to mDC. Efficient infection of resting CD4+ T cells in close contact with mDC and HIV could explain the rapid early establishment of the latent HIV reservoir. Additionally, if infectious virus persists in tissues such as lymph node in patients on ART, mDC may facilitate ongoing infection of resting T cells leading to replenishment of the reservoir.
Materials and Methods
Isolation of cellular subsets
PBMC were isolated from buffy coats obtained from the Australian Red Cross Blood Service (Melbourne, Australia). Resting CD4+ T cells were negatively selected using magnetic cell sorting and a cocktail of antibodies to CD8, CD11b, CD16, HLA-DR, CD19 and CD69, as previously described [17], [49]. Sorted cells were routinely negative for CD69, CD25 and HLA-DR (Fig. S1 A). In some experiments bulk resting CD4+ T cells were further sorted into CD45RA+ naïve and CD45RA− memory CD4+ T cells using phycoerythrin (PE)-labelled antibody to CD45RA and a FACSAria (BD Biosciences). DC were isolated from blood as previously described [50]. Briefly, DC were enriched using magnetic bead depletion and antibodies to CD3, CD11b and CD19. Enriched cells were then sorted using a FACSAria (BD Biosciences) to obtain a bulk cocktail− HLA-DR+ DC population, HLA-DR+CD11c+ mDC or HLA-DR+CD123+ pDC. The purity of sorted cells was always >98% (Fig. S1 B).
Plasmids, virus production and infection
In all experiments except where noted we used an NL4-3 virus with EGFP inserted into the nef open reading frame at amino acid position 75 at the aKpnI (Acc651) site with a CCR5-tropic (AD8) envelope (NL(AD8)-nef/EGFP), alternatively we used this virus with a CXCR4-tropic (NL4-3) envelope (NL4-3-nef/EGFP; both kindly provided by Damian Purcell, University of Melbourne, Melbourne, Australia). In one set of experiments we used a Nef-competent EGFP reporter virus, kindly provided by Yasuko Tsunetsugu-Yokota (National Institute of Infectious Diseases, Tokyo, Japan) [51]. HIV stocks were generated by FuGene (Promega, Madison, WI) transfection of 293T cells as previously described [49], [50]. Cells were infected at 37°C for 2 hours at an MOI of 0.5 or 5, as determined by limiting dilution using the Reed and Muench method [52], followed by a wash step to remove unbound virus.
In vitro HIV latency model
Resting CD4+ T cells were labelled with proliferation dye, either SNARF (10 µM; Invitrogen) or eFluor®670 (5 µM; eBiosciences, San Diego, CA), according to the manufacturer's instructions. SNARF/eFluor670-labelled resting CD4+ T cells were cultured in media supplemented with IL-2 (2 U/mL; Roche Diagnostics) for 24 hours, with or without syngeneic bulk DC or sorted DC subsets (DC: T cell ratio of 1∶10), in the presence or absence of SEB (10 ng/mL; Sigma). Cells were then infected using an EGFP-reporter virus and cultured for a further 5 days (Fig. 1 A). In some experiments, cells were cultured with and without the protease inhibitor Indinavir (0.1 µM final) for 30 minutes at 37°C prior to infection. At day 5 post-infection, cells were analysed by flow cytometry for productive infection by detecting EGFP+ cells. Subsequently, the non-proliferating (SNARFhi/eFluor670hi) CD4+ T cells that were not productively infected (EGFP−) were sorted using a FACSAria (BD Biosciences). In order to amplify any latent infection, the sorted CD4+ T cells were stimulated with PHA (10 ug/mL)/IL-2 (10 U/mL) in the presence of PBMC and cultured for a further 5 days. The number of EGFP+ cells following re-stimulation was used as a surrogate measure for the number of latently infected, non-proliferating CD4+ T cells in the original cultures (Fig. 1 B). In some experiments, we also stimulated the sorted SNARFhi/eFluor670hiEGFP− CD4+ T cells directly with plate bound anti-CD3 (Beckman Coulter; 5 µg/mL) and soluble anti-CD28 (BD; 5 µg/mL). Flat bottomed 96 well plates were coated with anti-CD3 (5 µg/mL) for 3 hours at 37°C. Unbound antibody was then removed and 1×105 SNARFhi/eFluor670hiEGFP− CD4+ T cells were plated per well in 200 µL of media containing soluble anti-CD28 (5 µg/mL), IL-7 (50 ng/mL) and the integrase inhibitor, L8 (1 µM final). The number of EGFP+ cells was determined following 72 hours of culture. As a control for the activity of the integrase inhibitor L8, we cultured SEB-stimulated PBMC with and without L8 for 30 minutes prior to infection, and productive infection was determined at day 5 post-infection (Fig. S2).
Phenotyping
For phenotypic analysis of the CD4+ T cells before culture, we stained sorted resting CD4+ T cells with CD69-FITC, CD25-PE and HLA-DR-perCP (BD Bioscience) on ice for 25 minutes. To determine whether co-culture with DC had altered the activation state of the resting CD4+ T cells, in some experiments the sorted SNARFhiEGFP− CD4+ T cells were labelled with either CD69-FITC or HLA-DR-FITC (BD Biosciences). Intracellular staining was also performed on the sorted SNARFhiEGFP− CD4+ T cells to detect expression of the cell cycle marker Ki67. Cells were permeabilised with 500 µL of 1× FACS Permeabilising Buffer (BD Biosciences) in the dark at room temperature for 10 minutes, washed once with FACS wash and incubated with Ki67-FITC (5 µL/105 sorted CD4+ T cells; Dako) for 45 minutes on ice. Following incubation, cells were washed twice with FACS wash and resuspended in 1% FACS fix. We performed analyses on a FACSCalibur (BD Biosciences) and results were analysed using Weasel software (Walter and Elisa Hall Institute, Melbourne, Australia).
Bead arrays
Cytokine bead arrays (eBioscience) were used according to the manufacturer's directions to determine the concentration of IL-1-beta, IL-6, IL-10, IL-12p70, TNF-alpha, CXCL9 and CXCL10 in the cell cultures.
Inhibition of soluble factors/surface receptors
In some experiments, nAbs to CD18 (10 µg/mL (prior to infection) and 20 µg/mL (post-infection); clone 7E4; Beckman Coulter), CCL19 (25 µg/mL), CXCR3 (20 µg/ml), IFN-alpha (5 µg/mL) or control IgG (R&D Systems, Minneapolis, MN); IL-6 or IgG1 (10 ug/mL; BioLegend, San Diego, CA); IL-10R or IgG2 (10 ug/mL; Biolegend) were used. In these experiments, both the DC and the resting CD4+ T cells were pre-incubated with nAbs for 15 minutes on ice prior to culture. The nAbs were added again to the co-cultures following infection. Neutralising activity of anti-CCL19 (25 µg/mL) and anti-CXCR3 (20 µg/ml) was confirmed using a chemokine-induced migration assay. Resting CD4+ T cells were added to the top chamber of a 3 µM pore transwell migration plate (Sigma) and either CCL19 (100 nM) or CXCL10 (300 nM) was added to the bottom chamber. In experiments using anti-CXCR3, cells were treated with nAb for 15 minutes at 37°C and washed off prior to chemokine treatment. In comparison, in experiments using anti-CCL19, nAb was added together with chemokine to the bottom chamber. Migrated cells in the bottom chamber were then counted in duplicate at 20 hours post addition of chemokine. Anti-IL-6 and anti-IL-10R were used at neutralising concentrations previously described [53], [54]. As positive controls for these nAbs we demonstrated that 10 µg/mL of anti-IL-6 and anti-IL10R or 5 µg/mL of anti-IFN-alpha efficiently blocked IL-6 (100 ng/mL), IL-10 (50 ng/mL) or IFN-alpha (50 ng/mL) mediated STAT3 phosphorylation respectively (Fig. S2).
ICAM-fc stimulation
In order to determine the role of ICAM-1 and LFA-1 interactions in mDC-induced latency, resting CD4+ T cells were cultured alone or with 10 ug/mL of ICAM-1fc together with 6 µg/mL of anti-IgG-fc (both from R&D Systems) for 24 hours prior to infection and maintained post-infection.
Transwell/transfer experiments
DC were cultured with resting CD4+ T cells in the presence and absence of 0.4 µm cell culture inserts (BD, Franklin Lakes, NJ) with DC in the top chamber and resting CD4+ T cells in the lower chamber. Following 24 hours of culture, both the DC and the CD4+ T cells were infected as described above. In other experiments, we added supernatant from infected mDC-T cell co-cultures to uninfected resting CD4+ T cells and then infected these cells. Media changes were performed daily using supernatant from infected mDC-T cell co-cultures. To determine the role of DC-T cell transfer, resting CD4+ T cells were infected in the absence of mDC and uninfected mDC were added back to the T cells only after virus had been washed off.
Microarrays
SNARFhiEGFP− CD4+ T cells cultured with DC, in the presence (latently infected) or absence (mock-infected) of HIV, were sorted 5 days following infection with NL(AD8)-nef/EGFP. In these experiments, all culture media was supplemented with 10 ng/mL of IL-7 (Sigma) instead of IL-2, in order to increase cell survival of resting cells, and infections were performed at an MOI of 5 to ensure high numbers of latently infected cells.
Microarrays were performed as previously described [55]. Briefly, cells were lysed and RNA extracted (Qiagen, Valencia, CA), amplified (Ambion Applied Biosystems, Austin, TX) and hybridised to an Illumina Human-Ref8 (v3) BeadChip (Illumina, San Diego, CA). Beadchips were scanned using an Illumina BeadStation 500GX scanner and Illumina BeadStudio (version 3) software (Ilumina). Illumina probe data was exported from BeadStudio as raw data and screened for quality. Samples failing chip visual inspection and control examination were removed. Gene expression data was analysed using Bioconductor (http://bioconductor.org/) [56], an open-source software library for the analyses of genomic data based on R, a language and environment for statistical computing and graphics (www.r-project.org). The R software package was used for pre-processing, first to filter out genes with intensities below background in all samples, then to minimum-replace (a surrogate-replacement policy) values below background using the mean background value of the built-in Illumina probe controls as an alternative to background subtraction (which may introduce negative values) to reduce “over inflated” expression ratios determined in subsequent steps, and finally quantile-normalise the gene probes intensities. Genes were then filtered by intensity and by variance filters to allow a reduction in the number of tests and a corresponding increase in power of the differential gene expression analysis. The resulting matrix showing filtered genes as rows and samples as columns was log2 transformed and used as input for linear modelling using Bioconductor's limma package, which estimates the fold-change between two predefined groups by fitting a linear model and using an empirical Bayes method to moderate standard errors of the estimated log-fold changes for expression values from each gene. P values from the resulting comparison were adjusted for multiple testing according to the method of Benjamini and Hochberg [57]. This method controls the false discovery rate, which was set to 0.05 in this analysis. Microarray data is available through the National Center for Biotechnology Information Gene Expression Omnibus (GEO), series accession number pending.
Functional microarray analysis and network generation
Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, www.ingenuity.com) was used to identify canonical signalling pathways and networks associated with the expression profiles of the non-proliferating CD4+ T cells cultured with DC in the presence (HIV T (+DC)) or absence (Mock T (+DC)) of HIV. Differentially expressed Illumina Probe IDs were imported into the Ingenuity software and mapped to the Gene Symbol from Ingenuity knowledge database. The significance of the association between the dataset and the canonical pathway was measured in two ways: 1) A ratio of the number of genes from the dataset that map to the pathway divided by the total number of genes that map to the canonical pathway; 2) Over-representation Fisher's exact test was used to calculate a p-value determining the probability that the association between the genes in the dataset and the canonical pathway is explained by chance alone. The pathways were ranked by −log p-value. This score was used as the cut-off for identifying significant canonical pathways (p value<0.05). IPA's networks are built from direct or indirect physical, transcriptional, and enzymatic interactions between the mapped genes (focus genes). Two genes are considered to be connected if there is a path in the network between them. Ingenuity's approach is based on a multi-stage, heuristic algorithm that iteratively constructs networks that greedily optimize for both interconnectivity and number of Focus Genes under the constraint of a maximal network size. Each individual IPA network has a maximum of 35 focus genes and is assigned a significance score (based on P value) representing the likelihood that the focus genes within the network are found there by random chance.
Real-time PCR analysis
As previously described, full length viral DNA was quantified using primers specific for the HIV-1 long terminal repeat (LTR) and Gag [58], and integrated HIV-1 DNA was quantified using a nested Alu-LTR real-time PCR [15], [59], [60]. Results were normalised for total input DNA as determined by real-time PCR for the CCR5 gene [61]. The correlation between gene arrays and real-time PCR was performed using a Spearman correlation test.
Microarray expression data were validated in two donors by reverse transcriptase real-time PCR (RT-qPCR), as previously described [62]. Briefly, SNARFhiEGFP− CD4+ T cells were lysed for RNA extraction and DNAse treatment (Qiagen, RNAeasy mini kit). cDNA was generated using CellsDirect qRT-PCR mix (Invitrogen). After reverse transcription all target genes were pre-amplified (18 cycles) using Taqman primers (Roche Probe library) specific for the transcripts of interest, which were also used for quantification. qPCR were performed on a Roche Light Cycler 348II and analysed according to the ΔΔct method.
Statistical analysis
In all experiments, Wilcoxon signed-rank or student paired t tests (for n<5) were performed for comparisons between populations using Graphpad Prism 5.0 software. P values of less than 0.05 were considered significant. Statistical analyses for microarray data were performed with program R, according to the method of Benjamini and Hochberg [57].
Supporting Information
Zdroje
1. ChurchillMJ, WesselinghSL, CowleyD, PardoCA, McArthurJC, et al. (2009) Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol 66: 253–258.
2. ElleryPJ, TippettE, ChiuYL, PaukovicsG, CameronPU, et al. (2007) The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol 178: 6581–6589.
3. LambotteO, TaoufikY, de GoerMG, WallonC, GoujardC, et al. (2000) Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. J Acquir Immune Defic Syndr 23: 114–119.
4. WightmanF, SolomonA, KhouryG, GreenJA, GrayL, et al. (2010) Both CD31(+) and CD31 naive CD4(+) T cells are persistent HIV type 1-infected reservoirs in individuals receiving antiretroviral therapy. The Journal of infectious diseases 202: 1738–1748.
5. ChunTW, FinziD, MargolickJ, ChadwickK, SchwartzD, et al. (1995) In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med 1: 1284–1290.
6. FinziD, HermankovaM, PiersonT, CarruthLM, BuckC, et al. (1997) Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278: 1295–1300.
7. ChunTW, CarruthL, FinziD, ShenX, DiGiuseppeJA, et al. (1997) Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387: 183–188.
8. NorthTW, HigginsJ, DeereJD, HayesTL, VillalobosA, et al. (2010) Viral sanctuaries during highly active antiretroviral therapy in a nonhuman primate model for AIDS. J Virol 84: 2913–2922.
9. ChunTW, NickleDC, JustementJS, MeyersJH, RobyG, et al. (2008) Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J Infect Dis 197: 714–720.
10. YuklSA, GianellaS, SinclairE, EplingL, LiQ, et al. (2010) Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. The Journal of infectious diseases 202: 1553–1561.
11. BosqueA, PlanellesV (2009) Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113: 58–65.
12. MariniA, HarperJM, RomerioF (2008) An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol 181: 7713–7720.
13. YangHC, XingS, ShanL, O'ConnellK, DinosoJ, et al. (2009) Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J Clin Invest 119: 3473–3486.
14. SalehS, SolomonA, WightmanF, XhilagaM, CameronPU, et al. (2007) CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 110: 4161–4164.
15. SwiggardWJ, BaytopC, YuJJ, DaiJ, LiC, et al. (2005) Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J Virol 79: 14179–14188.
16. YoderA, YuD, DongL, IyerSR, XuX, et al. (2008) HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell 134: 782–792.
17. CameronPU, SalehS, SallmannG, SolomonA, WightmanF, et al. (2010) Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc Natl Acad Sci U S A 107: 16934–16939.
18. LassenKG, HebbelerAM, BhattacharyyaD, LobritzMA, GreeneWC (2012) A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. Plos One 7: e30176.
19. YoderA, GuoJ, YuD, CuiZ, ZhangXE, et al. (2011) Effects of microtubule modulators on HIV-1 infection of transformed and resting CD4 T cells. Journal of virology 85: 3020–3024.
20. BaratC, GilbertC, TremblayMJ (2009) Efficient replication of human immunodeficiency virus type 1 in resting CD4+ T lymphocytes is induced by coculture with autologous dendritic cells in the absence of foreign antigens. J Virol 83: 2778–2782.
21. CameronPU, PopeM, GezelterS, SteinmanRM (1994) Infection and apoptotic cell death of CD4+ T cells during an immune response to HIV-1-pulsed dendritic cells. AIDS Res Hum Retroviruses 10: 61–71.
22. ScheerenRA, KoopmanG, Van der BaanS, MeijerCJ, PalsST (1991) Adhesion receptors involved in clustering of blood dendritic cells and T lymphocytes. Eur J Immunol 21: 1101–1105.
23. WangJH, KwasC, WuL (2009) Intercellular adhesion molecule 1 (ICAM-1), but not ICAM-2 and -3, is important for dendritic cell-mediated human immunodeficiency virus type 1 transmission. Journal of virology 83: 4195–4204.
24. HungCM, Garcia-HaroL, SparksCA, GuertinDA (2012) mTOR-dependent cell survival mechanisms. Cold Spring Harb Perspect Biol 4: a008771.
25. SlavinDA, KoritschonerNP, PrietoCC, Lopez-DiazFJ, ChattonB, et al. (2004) A new role for the Kruppel-like transcription factor KLF6 as an inhibitor of c-Jun proto-oncoprotein function. Oncogene 23: 8196–8205.
26. BenzenoS, NarlaG, AllinaJ, ChengGZ, ReevesHL, et al. (2004) Cyclin-dependent kinase inhibition by the KLF6 tumor suppressor protein through interaction with cyclin D1. Cancer research 64: 3885–3891.
27. YuKY, KwonB, NiJ, ZhaiY, EbnerR, et al. (1999) A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. The Journal of biological chemistry 274: 13733–13736.
28. GrootF, van CapelTM, KapsenbergML, BerkhoutB, de JongEC (2006) Opposing roles of blood myeloid and plasmacytoid dendritic cells in HIV-1 infection of T cells: transmission facilitation versus replication inhibition. Blood 108(6): 1957–64.
29. ThackerTC, ZhouX, EstesJD, JiangY, KeeleBF, et al. (2009) Follicular dendritic cells and human immunodeficiency virus type 1 transcription in CD4+ T cells. J Virol 83: 150–158.
30. CameronPU, FreudenthalPS, BarkerJM, GezelterS, InabaK, et al. (1992) Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science 257: 383–387.
31. PopeM, BetjesMG, RomaniN, HirmandH, CameronPU, et al. (1994) Conjugates of dendritic cells and memory T lymphocytes from skin facilitate productive infection with HIV-1. Cell 78: 389–398.
32. AgostoLM, YuJJ, DaiJ, KaletskyR, MonieD, et al. (2007) HIV-1 integrates into resting CD4+ T cells even at low inoculums as demonstrated with an improved assay for HIV-1 integration. Virology 368: 60–72.
33. WangW, GuoJ, YuD, VorsterPJ, ChenW, et al. (2012) A dichotomy in cortical actin and chemotactic actin activity between human memory and naive T cells contributes to their differential susceptibility to HIV-1 infection. The Journal of biological chemistry 287: 35455–35469.
34. DaiJ, AgostoLM, BaytopC, YuJJ, PaceMJ, et al. (2009) Human immunodeficiency virus integrates directly into naive resting CD4+ T cells but enters naive cells less efficiently than memory cells. Journal of virology 83: 4528–4537.
35. van der SluisRM, van MontfortT, PollakisG, SandersRW, SpeijerD, et al. (2013) Dendritic cell-induced activation of latent HIV-1 provirus in actively proliferating primary T lymphocytes. PLoS Pathog 9: e1003259.
36. HarmanAN, ByeCR, NasrN, SandgrenKJ, KimM, et al. (2013) Identification of lineage relationships and novel markers of blood and skin human dendritic cells. J Immunol 190: 66–79.
37. Ziegler-HeitbrockL, AncutaP, CroweS, DalodM, GrauV, et al. (2010) Nomenclature of monocytes and dendritic cells in blood. Blood 116: e74–80.
38. MacDonaldKP, MunsterDJ, ClarkGJ, DzionekA, SchmitzJ, et al. (2002) Characterization of human blood dendritic cell subsets. Blood 100: 4512–4520.
39. JongbloedSL, KassianosAJ, McDonaldKJ, ClarkGJ, JuX, et al. (2010) Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med 207: 1247–1260.
40. GuntherC, StarkeJ, ZimmermannN, SchakelK (2012) Human 6-sulfo LacNAc (slan) dendritic cells are a major population of dermal dendritic cells in steady state and inflammation. Clin Exp Dermatol 37: 169–176.
41. van KooykY, van de Wiel-van KemenadeP, WederP, KuijpersTW, FigdorCG (1989) Enhancement of LFA-1-mediated cell adhesion by triggering through CD2 or CD3 on T lymphocytes. Nature 342: 811–813.
42. Iglesias-UsselM, VandergeetenC, MarchionniL, ChomontN, RomerioF (2013) High Levels of CD2 Expression Identify HIV-1 Latently Infected Resting Memory CD4+ T Cells in Virally Suppressed Subjects. J Virol 87: 9148–9158.
43. HochwellerK, AndertonSM (2005) Kinetics of costimulatory molecule expression by T cells and dendritic cells during the induction of tolerance versus immunity in vivo. Eur J Immunol 35: 1086–1096.
44. ChomontN, El-FarM, AncutaP, TrautmannL, ProcopioFA, et al. (2009) HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15: 893–900.
45. CoirasM, Lopez-HuertasMR, RullasJ, MittelbrunnM, AlcamiJ (2007) Basal shuttle of NF-kappaB/I kappaB alpha in resting T lymphocytes regulates HIV-1 LTR dependent expression. Retrovirology 4: 56.
46. MouzakiA, DoucetA, MavroidisE, MusterL, RunggerD (2000) A repression-derepression mechanism regulating the transcription of human immunodeficiency virus type 1 in primary T cells. Molecular medicine 6: 377–390.
47. HaoS, KurosakiT, AugustA (2003) Differential regulation of NFAT and SRF by the B cell receptor via a PLCgamma-Ca(2+)-dependent pathway. Embo J 22: 4166–4177.
48. LiY, SedwickCE, HuJ, AltmanA (2005) Role for protein kinase Ctheta (PKCtheta) in TCR/CD28-mediated signaling through the canonical but not the non-canonical pathway for NF-kappaB activation. J Biol Chem 280: 1217–1223.
49. SalehS, WightmanF, RamanayakeS, AlexanderM, KumarN, et al. (2011) Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology 8: 80.
50. EvansVA, LalL, AkkinaR, SolomonA, WrightE, et al. (2011) Thymic plasmacytoid dendritic cells are susceptible to productive HIV-1 infection and efficiently transfer R5 HIV-1 to thymocytes in vitro. Retrovirology 8: 43.
51. YamamotoT, Tsunetsugu-YokotaY, MitsukiYY, MizukoshiF, TsuchiyaT, et al. (2009) Selective transmission of R5 HIV-1 over X4 HIV-1 at the dendritic cell-T cell infectious synapse is determined by the T cell activation state. PLoS Pathog 5: e1000279.
52. ReedLJ, MuenchH (1938) A simple method of estimating fifty per cent endpoints. American journal of hygiene 27: 493–497.
53. SaidEA, DupuyFP, TrautmannL, ZhangY, ShiY, et al. (2010) Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med 16: 452–459.
54. SagiY, LandriganA, LevyR, LevyS (2012) Complementary costimulation of human T-cell subpopulations by cluster of differentiation 28 (CD28) and CD81. Proc Natl Acad Sci U S A 109: 1613–1618.
55. GaucherD, TherrienR, KettafN, AngermannBR, BoucherG, et al. (2008) Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. The Journal of experimental medicine 205: 3119–3131.
56. GentlemanRC, CareyVJ, BatesDM, BolstadB, DettlingM, et al. (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80.
57. BenjaminiY, HochbergY (1995) Controlling the False Discovery Rate : A Practical and Powerful Approach to Multiple Testing. JR Statist Soc B 57: 289–300.
58. ZackJA, HaislipAM, KrogstadP, ChenIS (1992) Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J Virol 66: 1717–1725.
59. ZhouY, ZhangH, SilicianoJD, SilicianoRF (2005) Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J Virol 79: 2199–2210.
60. LewinSR, MurrayJM, SolomonA, WightmanF, CameronPU, et al. (2008) Virologic determinants of success after structured treatment interruptions of antiretrovirals in acute HIV-1 infection. J Acquir Immune Defic Syndr 47: 140–147.
61. EcksteinDA, PennML, KorinYD, Scripture-AdamsDD, ZackJA, et al. (2001) HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 15: 671–682.
62. FlatzL, RoychoudhuriR, HondaM, Filali-MouhimA, GouletJP, et al. (2011) Single-cell gene-expression profiling reveals qualitatively distinct CD8 T cells elicited by different gene-based vaccines. Proceedings of the National Academy of Sciences of the United States of America 108: 5724–5729.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2013 Číslo 12
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- Influence of Mast Cells on Dengue Protective Immunity and Immune Pathology
- Host Defense via Symbiosis in
- Coronaviruses as DNA Wannabes: A New Model for the Regulation of RNA Virus Replication Fidelity
- Myeloid Dendritic Cells Induce HIV-1 Latency in Non-proliferating CD4 T Cells