
Mutations Can Cause Enamel-Renal Syndrome (ERS)
Enamel-renal syndrome (ERS) is an autosomal recessive disorder characterized by severe enamel hypoplasia, failed tooth eruption, intrapulpal calcifications, enlarged gingiva, and nephrocalcinosis. Recently, mutations in FAM20A were reported to cause amelogenesis imperfecta and gingival fibromatosis syndrome (AIGFS), which closely resembles ERS except for the renal calcifications. We characterized three families with AIGFS and identified, in each case, recessive FAM20A mutations: family 1 (c.992G>A; g.63853G>A; p.Gly331Asp), family 2 (c.720-2A>G; g.62232A>G; p.Gln241_Arg271del), and family 3 (c.406C>T; g.50213C>T; p.Arg136* and c.1432C>T; g.68284C>T; p.Arg478*). Significantly, a kidney ultrasound of the family 2 proband revealed nephrocalcinosis, revising the diagnosis from AIGFS to ERS. By characterizing teeth extracted from the family 3 proband, we demonstrated that FAM20A−/− molars lacked true enamel, showed extensive crown and root resorption, hypercementosis, and partial replacement of resorbed mineral with bone or coalesced mineral spheres. Supported by the observation of severe ectopic calcifications in the kidneys of Fam20a null mice, we conclude that FAM20A, which has a kinase homology domain and localizes to the Golgi, is a putative Golgi kinase that plays a significant role in the regulation of biomineralization processes, and that mutations in FAM20A cause both AIGFS and ERS.
Published in the journal:
. PLoS Genet 9(2): e32767. doi:10.1371/journal.pgen.1003302
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003302
Summary
Enamel-renal syndrome (ERS) is an autosomal recessive disorder characterized by severe enamel hypoplasia, failed tooth eruption, intrapulpal calcifications, enlarged gingiva, and nephrocalcinosis. Recently, mutations in FAM20A were reported to cause amelogenesis imperfecta and gingival fibromatosis syndrome (AIGFS), which closely resembles ERS except for the renal calcifications. We characterized three families with AIGFS and identified, in each case, recessive FAM20A mutations: family 1 (c.992G>A; g.63853G>A; p.Gly331Asp), family 2 (c.720-2A>G; g.62232A>G; p.Gln241_Arg271del), and family 3 (c.406C>T; g.50213C>T; p.Arg136* and c.1432C>T; g.68284C>T; p.Arg478*). Significantly, a kidney ultrasound of the family 2 proband revealed nephrocalcinosis, revising the diagnosis from AIGFS to ERS. By characterizing teeth extracted from the family 3 proband, we demonstrated that FAM20A−/− molars lacked true enamel, showed extensive crown and root resorption, hypercementosis, and partial replacement of resorbed mineral with bone or coalesced mineral spheres. Supported by the observation of severe ectopic calcifications in the kidneys of Fam20a null mice, we conclude that FAM20A, which has a kinase homology domain and localizes to the Golgi, is a putative Golgi kinase that plays a significant role in the regulation of biomineralization processes, and that mutations in FAM20A cause both AIGFS and ERS.
Introduction
Enamel-Renal Syndrome (ERS; OMIM #204690) is a recessive syndrome characterized by severely hypoplastic (thin) or aplastic enamel on both the primary and secondary dentitions, pulp stones, and failed or delayed eruption of much of the permanent dentition, particularly the posterior teeth. Coronal dentin is sometimes resorbed and replaced by lamellar bone and there is often hypercementosis on root surfaces. These dental symptoms are associated with nephrocalcinosis, although blood chemistry analyses are typically normal [1]–[3]. Gingival enlargement is sometimes noted [4], [5]. The initial patient complaint is the lack of enamel and failed eruption of many permanent teeth. Nephrocalcinosis is typically discovered by a renal ultrasound scan ordered because of the known association between this rare pattern of dental defects and renal dysfunction, rather than due to a patient complaint or history of renal problems [4]–[6]. In the original report of ERS one of the two affected individuals died of renal failure [1]. Another report described the results of a series of renal evaluations on an ERS patient that observed minimal renal calcifications at age 5 that became progressively denser in roentgenograms taken at ages 8, 11, and 14 years, and then stabilized [2]. Subsequent reports found the kidney calcifications in patients with ERS to be benign [3], [4], [7].
The literature describes patterns of recessive tooth defects similar to that observed in enamel renal syndrome (ERS), but without evidence of nephrocalcinosis [8]–[23]. As in ERS, the patient's initial chief complaint relates to the lack of enamel and failure of permanent tooth eruption. Dental radiographs reveal that most if not all of the teeth lack an enamel layer and have extensive pulp calcifications. The unerupted teeth show pericoronal radiolucencies delimited by a sclerotic margin. The teeth are usually smaller than normal, often with misshapened roots [12]. A common observation on radiographs is resorption of the occlusal surface (sometimes all the way to the pulp) of unerupted teeth [20]. When the malformed teeth are characterized histologically, they lack dental enamel, but show normal-looking dentin with well-formed dentinal tubules [13], [14]. The minimal “enamel” has no prismatic structure. On some teeth there is extensive localized root and/or crown resorption with partial replacement of the resorbed dentin by lamellar bone or in some places by globular structures comprised of incompletely coalesced concentric calcifications [12]. The thin roots are often covered by an abnormally thick layer of what appears to be cellular cementum [11], [19].
Recently, advanced genetic methods involving targeted exome capture, next generation DNA sequencing, and bioinformatics computer analyses implicated FAM20A (family with sequence similarity 20, member A) located on chromosome 17q24.2 as the defective gene in a recessive disorder manifesting the same oral features as described above [22], [24] and designated “Amelogenesis Imperfecta and Gingival Fibromatosis Syndrome” (AIGFS; OMIM #614253). The association between FAM20A and a syndrome that included severe enamel hypoplasia and gingival hypertrophy was confirmed by mutational analyses in four additional families that identified three homozygous FAM20A mutations (c.34_35delCT; c.813-2A>G; c.1175_1179delGGCTC) and compound heterozygous mutations (c.590-2A>G with c.826C>T) in four families [25]. In none of the families with FAM20A mutations were teeth available for microscopic examination or were renal ultrasounds performed.
We have characterized three families with a recessive syndrome caused by FAM20A mutations. All affected individuals in these families had mutations in both FAM20A alleles. Extracted molars were characterized histologically and shown to have hypercementosis and dentin replaced by lamellar bone. All findings were consistent with a diagnosis of AIGFS; however, we were intrigued by the similarity of AIGFS with ERS and inquired further about whether or not our probands had kidney problems.
Results
Family 1
The proband's parents were born in the Caribbean. Oral photos and a panoramic radiograph were obtained for the proband and DNA was collected from nine family members (Figure 1). The proband (III:1), his younger sister (III:4), and niece (IV:1) were affected. According to the proband, when his adult teeth finally came in they were the same size as his baby teeth and very short. As far as he remembers, his gums have always been large and bumpy. He reported that he was otherwise healthy with average height and weight. Intraoral photographs of the proband showed a mixed dentition, small dental crowns with generally thin enamel, and yellow discoloration. Over-retained primary molars in the mandibular arch and partially erupted maxillary premolars were observed. The proband had a deep anterior overbite, a posterior cross-bite, and a class III molar relationship. The vertical dimension appeared to be reduced. Radiographically, the proband had the full complement of permanent teeth, but eruption of the canines, mandibular premolars and molars was failed or delayed. No radiopaque enamel layer was apparent on any of the teeth. Even unerupted teeth with completed root formation lacked enamel. Pericoronal radiolucencies outlined by sclerotic borders were observed around unerupted teeth, symptomatic of a slow expansion of the dental follicle covering the crown. In some cases the dentin occlusal surface appeared concave and close to the pulp chamber, suggesting pre-eruptive crown resorption. Most teeth showed intrapulpal calcifications. The gingiva appeared to be hyperplastic.
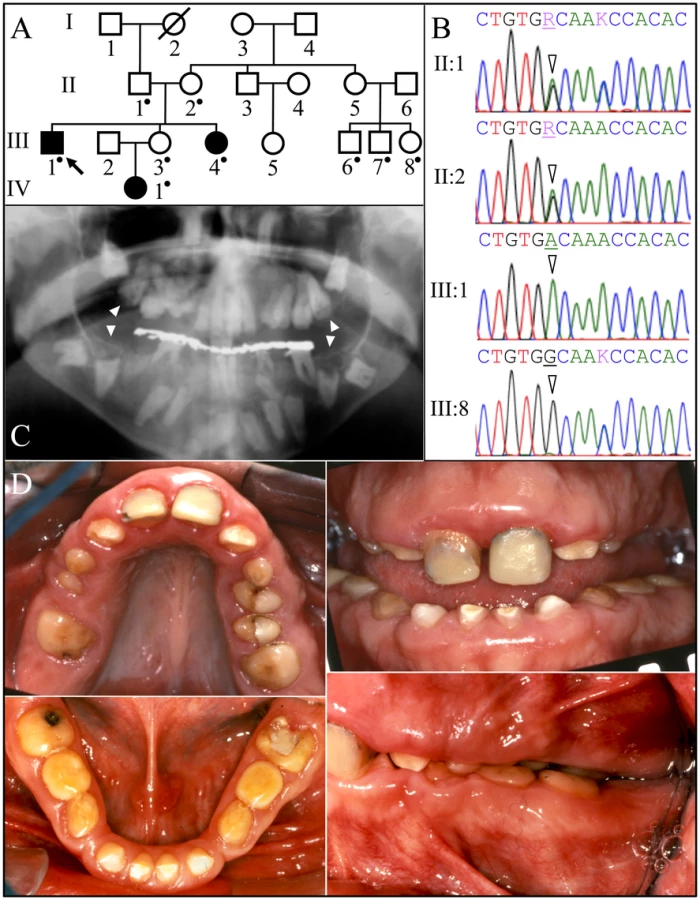
Family 1 was one of the original 24 AI families that we recruited for genetic studies [26]. No disease-causing mutations in the proband's DNA were identified during mutational analyses of the proven AI candidate genes (AMELX, ENAM, FAM83H, WDR72, KLK4, and MMP20) and AMBN [27]. FAM20A analyses however identified a G to A transition resulting in a missense mutation in exon 7 (c992G>A; g.63853G>A; p.Gly331Asp) that is homozygous in the proband (III:1), his sister (III:4) and niece (IV:1), heterozygous in proband's parents (II:1 and II:2) and unaffected sister (III:3), and absent from his three first cousins (III:6, III:7:, and III:8). This sequence variation has not been previously identified in the dbSNP database or in 1000 Genomes Project Pilot Data [28]. The glycine (G331) that is replaced by aspartic acid is conserved throughout vertebrate evolution (Figure S1) and the substitution was predicted to be probably damaging by PolyPhen-2 analyses [29]. This family was recruited almost 10 years ago and we have not been able to obtain any information concerning kidney calcifications.
Family 2
Family 2 was a consanguineous family from Jordan (Figure 2). A panorex radiograph of the proband showed the retention of primary teeth and delayed eruption of permanent cuspids, premolars, and second molars. No radiopaque enamel was detected and expanded peri-coronal radiolucencies were evident on all unerupted teeth. The pulp chambers were typically calcified and nearer the occlusal surface than expected. On some unerupted teeth the crown occlusal to the pulp chambers had disappeared, as if by resorption. An ultrasound of the proband's kidneys revealed bilateral medullary nephrosis with small calcifications in both kidneys causing acoustic shadowing. Otherwise both kidneys were normal in size and corticomedullary differentiation, each measuring about 11 cm in bipolar length.
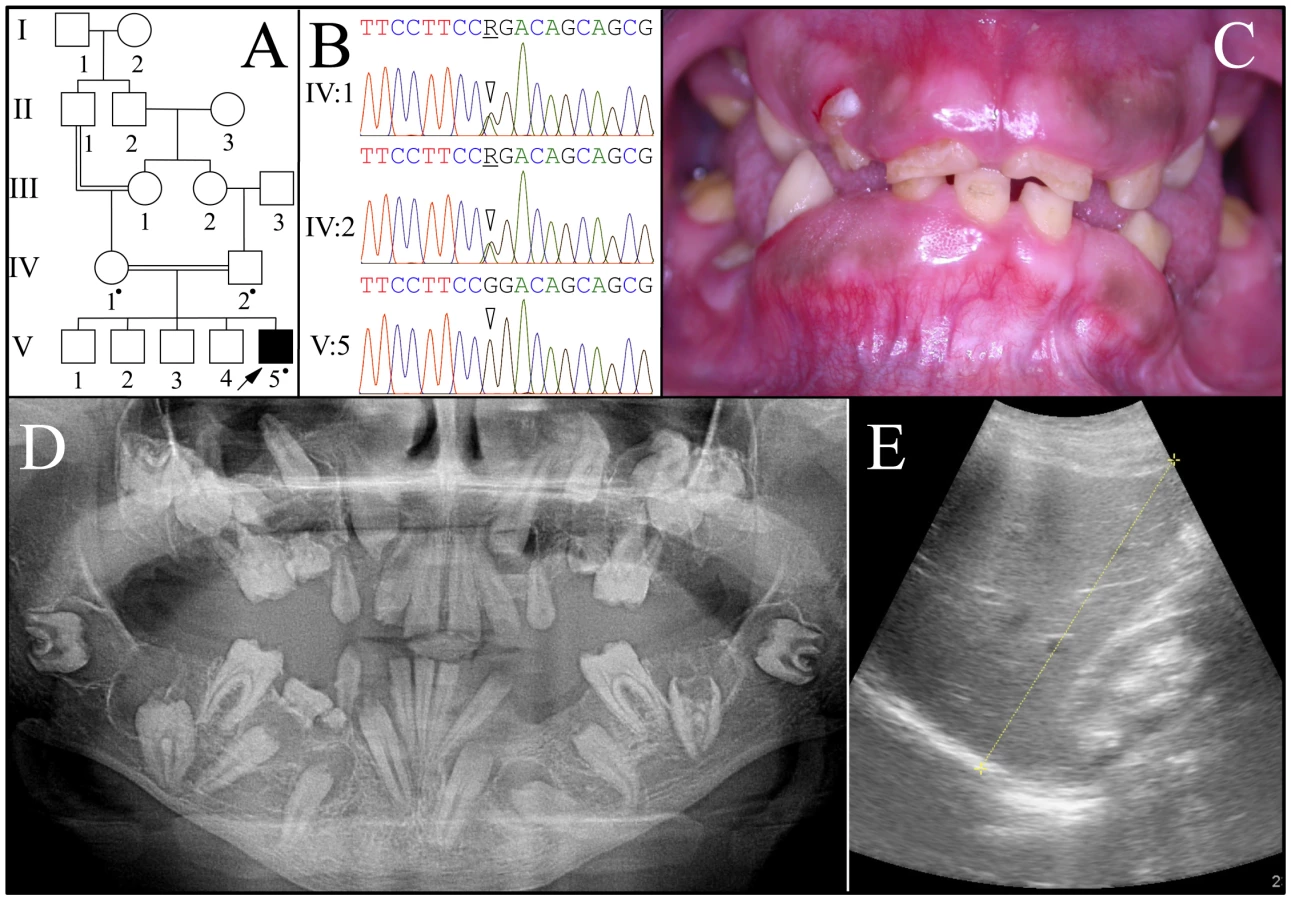
The proband (V:5) was the only affected person. FAM20A mutation analyses of the proband and his parents (IV:1 and IV:2) identified an A to G transition that altered the splice acceptor site at the end of intron 4 (c.720-2A>G; g.62232A>G) that is homozygous in the proband and heterozygous in both parents. This sequence variation has not been previously identified in the dbSNP database or in 1000 Genomes Project Pilot Data. Several possible aberrant RNA splicing outcomes could occur [30]. Skipping of exon 5 would delete 31 amino acids (p.Q241_R271del). No normal transcript variants skipping exon 5 are listed in GenBank. Retention of intron 4 would introduce a premature termination codon, and likely cause mutant FAM20A transcripts to be degraded by nonsense-mediated decay. Human Splice Finder version 2.4.1 [31] suggests there could be activation of a 5′ cryptic splice site that would add one nucleotide to exon 5 and lead to a frameshift and subsequent nonsense-mediated decay.
Family 3
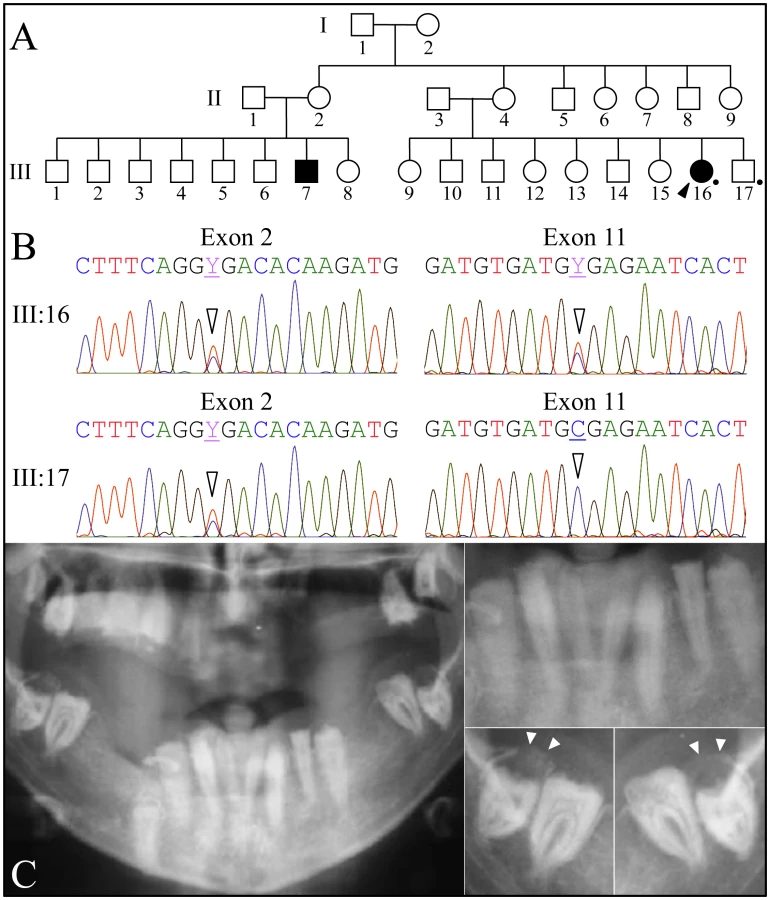
Family 3 was a large kindred from Iran with two affected cousins (Figure 3). All of the proband's teeth were extracted (and some saved) prior to recruitment. A pre-surgical panoramic radiograph showed no radiopaque enamel, delayed tooth eruption, intrapulpal calcifications, and pericoronal radiolucencies. DNA was obtained from the proband and her younger, unaffected brother. Only FAM20A was characterized by mutational analyses. The proband was a compound heterozygote for two C>T transitions that both resulted in premature translation termination (TGA) codons. The nonsense mutations were in exon 2 (c.406C>T; g.50213C>T; p.R136*) and in exon 11 (c.1432C>T; g.68284C>T; p.R478*). The unaffected younger brother was heterozygous for the nonsense mutation in exon 2, but his exon 11 sequence was normal on both alleles. The exon 2 nonsense mutation was previously reported to cause AIGFS when found on both FAM20A alleles [24].
Scanning Electron Microscopy
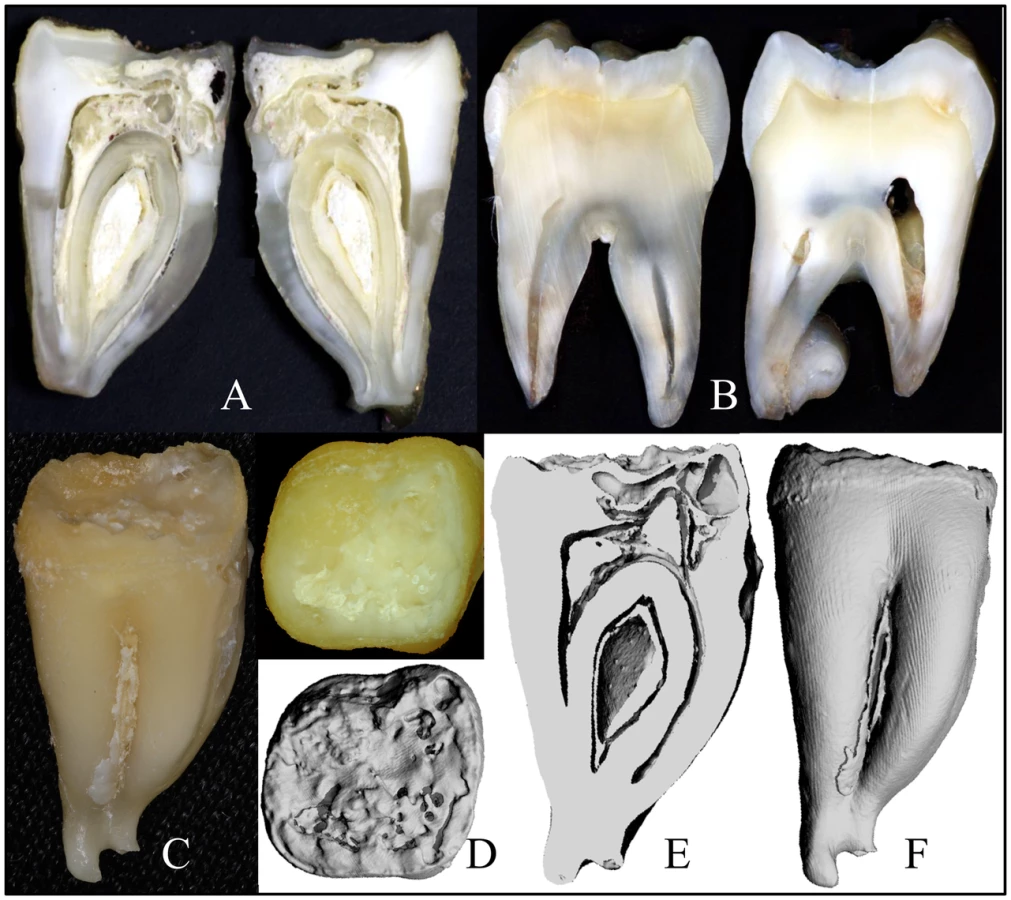
Four extracted secondary teeth from the proband of family 3 were provided to us for analyses (Figure S2). The “enamel” on the crowns was thin, soft and crusty, and only evident near the cervical margins. The teeth were smaller than normal and showed irregularities of root form. The area coronal to the root furcation was sometimes expanded, while the roots themselves were short, thin, and sometimes fused. Some parts of the roots showed pronounced concavities resembling a row of bites from an apple. A mesial-distal cut was made through #18, the mandibular left second molar (Figure 4A, 4C) and compared to the normal tooth (Figure 4B). Much of the mesial half of the crown had been resorbed and was only partially replaced by mineral, producing “hollow” areas on 3-D μCT images (Figure 4D–4F), while much of the pulp chamber was calcified.
SEM analysis of the occlusal surface of the FAM20A−/− mandibular second molar (tooth #18; Figure 5A) revealed a variety of surface features, including rough, knob-like calcifications (Figure 5B), dentin with exposed dentinal tubules (Figure 5C), some relatively smooth mineral near the dentin surface (Figure 5D), and pitted “enamel” mineral superficially resembling volcanic rock on the lateral aspect of the crown (Figure 5E–5F). SEMs of deliberately fractured areas showed no mineral organization characteristic of true enamel (Figure 6). SEMs of dentin looked the same as normal dentin (Figure 7). SEMs of the mandibular third molar (tooth #32; Figure 8A) showed a relatively smooth root surface (Figure 8B) perforated by small holes (dentinal tubules) and larger craters suggestive of resorption lacunae (Figure 8C–8E).



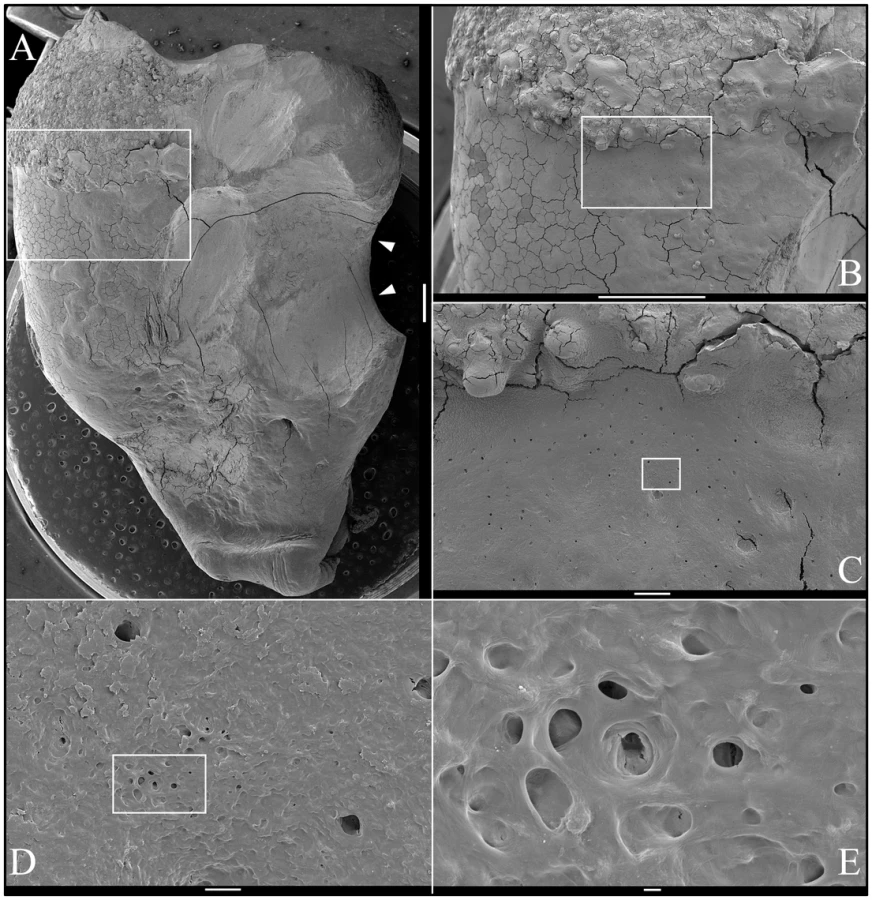
Backscatter Scanning Electron Microscopy
Backscatter SEMs of tooth #18 revealed that there was no enamel on the occlusal surface of the crown, although a thin, crusty material more highly mineralized than dentin covered part of the lateral coronal surfaces (Figure 9). Remarkably, only a small remnant of dentin, with apparent resorption lacunae at its edges, was evident in the mesial half of this FAM20A−/− molar crown. In its place was a laminated, bone-like material with osteocyte lacunae. Based upon the patterns of the growth lines, this lamellar bone had undergone repeated cycles of resorption and deposition. The calcifications in the pulp were unevenly mineralized. The most highly mineralized pulp material was comprised of incompletely coalesced spherical structures (calcospherites). Thus there appeared to be at least two types of pathological mineralization within the crown: lamellar-like bone and calcospherites that apparently formed by a different mechanism. Backscatter SEMs of the root region revealed thin roots, with normal-looking root dentin covered by a thick layer of laminated, cementum-like mineralized tissue similar to the lamellar mineralized tissue that had replaced dentin in the crown but with fewer osteocyte lacunae (Figure 10). The dentin was often separated from this thick cementum-like layer by a hypermineralized line that might represent the original cementum. This line was sometimes interupted at places where the root surface had been resorbed locally and replaced.
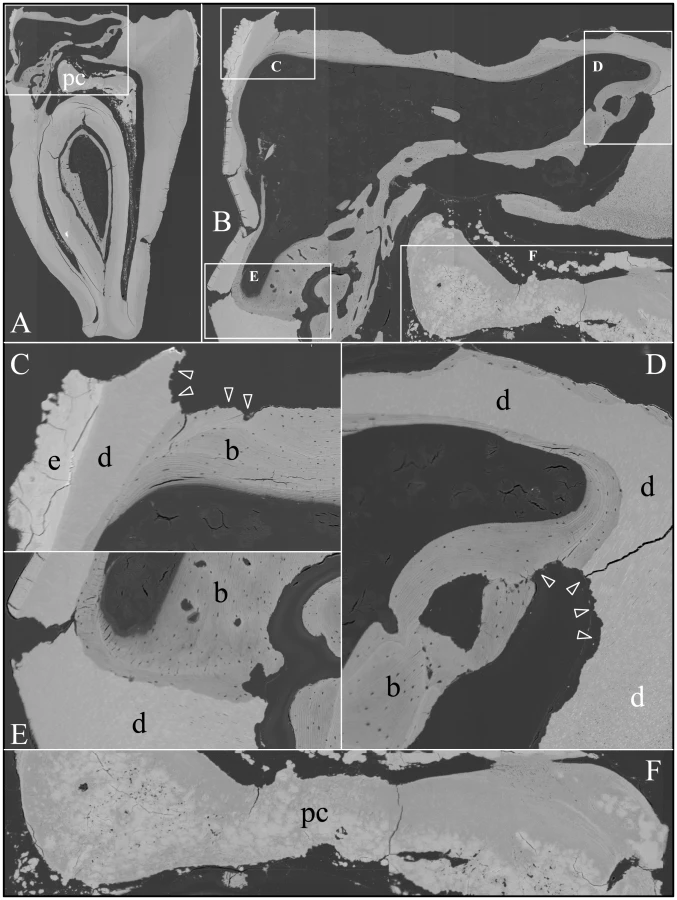

An unerupted third molar (#32) was also characterized by bSEM and showed a somewhat different pattern of pathological resorption and mineralization (Figure 11). The “enamel” layer was very thin, rough, and discontinuous. It was more highly mineralized than dentin in most places, and appeared to have coalesced from multiple mineral foci. The dentin had well-organized dentinal tubules. The pulp was partially calcified. Besides major resorption of dentin from part of the root surface, the bulk of the pathology was in the furcation area, which included a large hypermineralized region of coalesced calcospherites surrounded by lamellar bone that extended coronally to the pulp and apically beyond the furcation so that the bone-like material seems to have entirely replaced the dentin at the furcation but retained the original morphology of the furcation.

FAM20A Localization to the Golgi
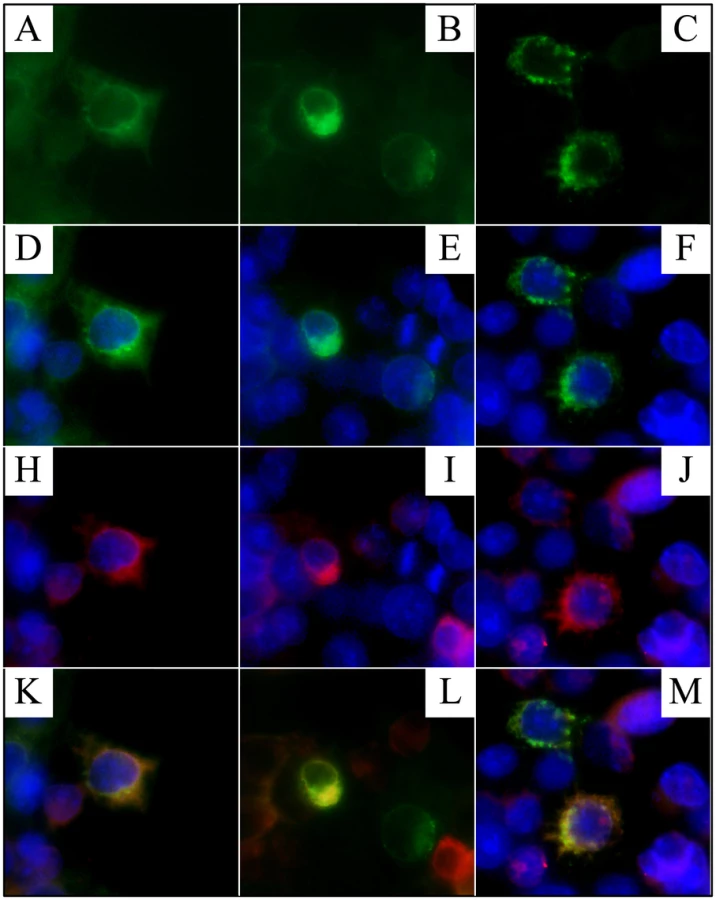
Human embryonic kidney (HEK) 293 cells were cotransfected with two plasmid constructs that expressed Flag-tagged FAM20A and a Golgi-GFP (green fluorescent protein) marker, respectively (Figure 12). In cells that received both constructs, the FAM20A signal superimposed upon that of the Golgi marker.
Discussion
Amelogenesis imperfecta (AI) generally refers to non-syndromic forms of inherited enamel malformations that may include other local defects such as delayed tooth eruption and misshapened roots [32]. It is also used to describe this phenotype in syndromes. In the case of Enamel-Renal Syndrome (ERS) the dental problems may appear prior to any evidence of kidney disease and kidney ultrasounds are rarely performed on AI patients, so the condition can easily be misdiagnosed as autosomal recessive enamel agenesis (AI Type IG) [32]. This was the diagnosis we made years ago for the proband of family 1 [26]. We noted the mild gingival enlargement but did not consider the possibility of a kidney phenotype.
With the breakthrough discovery that FAM20A mutations cause AI associated with a variety of oral manifestations including eruption failures and gingival enlargement [24], we performed mutation analyses in three families exhibiting this pattern of defects, and in each case we identified disease-causing mutations in both FAM20A alleles and made the diagnosis of Amelogenesis Imperfecta and Gingival Fibromatosis Syndrome” (AIGFS). However, we read several reports showing the AIGFS pattern of oral manifestations in a person with kidney calcifications (nephrocalcinosis) [2]–[5], [33], [34]. In every case the nephrocalcinosis was discovered by radiographic or ultrasound images of the kidneys and not through a history of kidney problems in the family. Among the reports of similar cases without nephrocalcinosis, we found no cases in which a renal ultrasound was performed and found to be negative. After finally appreciating the kidney association, we were able to contact family 2 to obtain a kidney ultrasound, which detected nephrocalcinosis. The appropriate diagnosis for this family then is Enamel-Renal Syndrome (ERS). Unfortunately, we have been unable to obtain renal ultrasounds from affected individuals in families 1 and 3.
Additional support for the association of nephrocalcinosis and the absence of FAM20A comes from recent analyses of Fam20a null mice [35], which discovered that “two-thirds of Fam20a−/− mice had small kidneys with pitted surfaces, which showed widespread calcification…”. Another interesting finding was detecting Fam20a gene expression in the parathyroid gland, given the failure of posterior tooth eruption in AIGFS and ERS and that primary failure of tooth eruption (PFE, OMIM #125350) can be caused by mutations in PTH1R (OMIM 168468; 3p21.31), the gene encoding a receptor for both parathyroid hormone and parathyroid hormone-related protein [36].
Scanning Electron Microscopy (SEM)
Our SEM analyses of FAM20A−/− molars detail an assortment of developmental malformations juxtaposed with secondary modifications. Developmentally, the enamel failed to form and the roots were small and misshapened. The crown and roots were susceptible to secondary resorption and turnover. Lamellar bone replaced parts of the resorbed crown, while a thick material resembling cellular cementum covered the roots. Highly mineralized, coalesced spherical calcifications were observed in the pulp and/or radicular area. We expect that the secondary root resorption and pathological mineralization occurred during the period of impaction. These unusual dental changes are rare in patients with non-syndromic amelogenesis imperfecta [27], [37], but are hallmark features of Enamel-Renal Syndrome (ERS) and were observed in the teeth from our probands with FAM20A mutations and nephrocalcinosis. These distinctive dental histological changes have previously been described in persons diagnosed as having AI without checking for nephrocalcinosis [8], [11], [12], [15]. The similarities between our histological observations and those reported for extracted teeth with a comparable dental phenotype are remarkable and increase our suspicion that ERS has been historically under-diagnosed.
FAM20A is part of a small gene family that in human and mouse has three members: FAM20A, FAM20B, and FAM20C. All three proteins have signal peptides of 21 amino acids and appear to be secreted [38]. The FAM20 genes encode proteins of similar size with a conserved C-terminal putative kinase domain (cd10469). FAM20A (17q24.2; cDNA reference sequence NM_017565.3) encodes a 541 amino acid protein. FAM20B (1q25; NM_014864.3) encodes 409 amino acids and FAM20C (7p22.3; NM_020223.2) 584 amino acids. Analysis of human expressed sequence tags (ESTs) suggests that the FAM20 family is expressed in many tissues. The National Center for Biotechnology Information (NCBI) human EST database currently has 5,779,625 entries for 45 healthy tissues. Among these are 103 EST entries for FAM20A (Hs.268874) from 22 tissues, with larynx (82/million), testes (60/million), and kidney (47/million) showing the highest proportion of FAM20A transcripts. Mice homozygous for a defined 58-kb deletion in the 5′ region of Fam20a showed growth-cessation and growth-delay [39]. Recently, Fam20a null mice were characterized with severe ectopic calcifications in the kidneys [35].
FAM20B is a xylose kinase in the Golgi required for the efficient addition of glycan attachments on secreted proteins. [40]. FAM20C has recently been identified as Golgi casein kinase [41], the enzyme that phosphorylates the secretory calcium binding phosphoproteins critical for biomineralization [42]. FAM20C mutations cause autosomal recessive lethal osteosclerotic bone dysplasia (Raine syndrome; OMIM #259775) [43], as well as non-lethal osteosclerotic bone dysplasia [44], [45]. FAM20A localizes to the Golgi, so perhaps FAM20A is a Golgi kinase like FAM20B and FAM20C, and its deficiency results in altered post-translational modifications of secreted proteins. In the absence of FAM20A, the dental follicle does not support tooth eruption, slowly expands, and generates psammomatous calcifications. The connective tissue of the gingiva also slowly expands and psammomatous calcifications are deposited within the hyperplastic gingiva [22]. Similar calcifications occur in the dental pulp and possibly in the kidneys, causing nephrocalcinosis. This pattern of ectopic mineralization might be explained by failure to catalyze appropriate post-translational modifications on extracellular matrix molecules that inhibit mineralization when FAM20A is absent.
The nine novel, disease-causing FAM20A mutations so far reported are obviously destructive of protein structure and function. Three are nonsense mutations (c.406C>T, p.Arg136*; c.826C>T, p.Arg276*; c.1432C>T, p.Arg478*). Three are splice junction mutations at the borders of exon 3 (c.590-2A>G, p.Asp197_Ile214delinsV), exon 5 (c.720-2A>G, p.Gln241_Arg271del) and exon 6 (c.813-2A>G, p.Arg271Serfs*70). Two are frameshifts (c.34_35delCT, pLeu12Alafs*67; c.1175_1179delGGCTC, p.Arg392Profs*22), and one is a missense mutation (c.992G>A, p.Gly331Asp) at a highly conserved site. Only one of the nine FAM20A disease-causing mutations was found in more than one family (c.406C>T). These data strongly implicate FAM20A in the etiology of this recessive disorder that combines enamel defects, retention of primary teeth, delayed and failed eruption of permanent teeth with pericoronal radiolucencies, pulp calcifications, small and misshapened teeth, gingival hyperplasia, and now, nephrocalcinosis. Only further studies can show if defects in other gene(s) can cause this pattern of malformations and if FAM20A defects were not previously associated with nephrocalcinosis due to a lack of penetrance, subclinical presentation, or delayed onset.
Materials and Methods
The human study protocol and patient consents were reviewed and approved by the Institution Review Board at the University of Michigan and appropriate local Ethics Committees where families were recruited.
DNA Isolation and PCR Amplification
Peripheral whole blood (5 cc) or buccal swabs were obtained from participating family members. Genomic DNA was isolated using the QIAamp DNA Blood Maxi Kit (Qiagen Inc, Valencia, CA) and (50 ng) from affected individuals was amplified using the Platinum PCR Supermix (Invitrogen, Carlsbad, CA), and the amplification products were purified using the QIAquick PCR Purification Kit (Invitrogen, Carlsbad, CA). The primer pairs and polymerase chain reaction conditions for the amplification of the coding regions were previously described for AMBN [26], AMELX [46], ENAM [47], FAM83H [48], WDR72 [49], KLK4 [50], and MMP20 [51]. Twelve primer pairs were synthesized to amplify the eleven FAM20A exons (Figure S3), which covered all coding sequences and intron/exon borders. These FAM20A reactions were annealed at 57°C for 60 s, extended at 72°C for 90 s, and run for 35 cycles.
Fabrication of Fam20a-Flag Expression Construct
A full-length mouse Fam20a cDNA clone (BC029169) in pCMV-SPORT6 was obtained from Thermo Scientific Open Biosystems (Lafayette, CO, USA). Restriction sites were introduced before the Fam20a translation initiation codon (NotI) and replacement of the translation termination codon (SalI) by PCR, which generated a 1645-bp amplicon (primer set: gcggCCGCTTGGGCCATGCCCG, agtcgacGCTCGTCAGATTAGCCTG). The amplification product was extracted from the gel and ligated into pCR2.1-TOPO (Invitrogen). The Fam20a coding region was excised by double digestion with NotI and SalI and ligated into pCMV-Tag 4 (Agilent), which had been restricted with NotI and SalI. Proper construction of the recombinant expression plasmid was verified by DNA sequencing.
Cell Culture, Plasmid Transfection, and Immunocytochemistry
HEK293 cells were cultured with 2 mL Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS) in a Lab-Tek chamber slide (1 chamber) with cover (70360-12, Electron Microscopy Sciences, Hatfield, PA, USA) to reach 60% confluence on the day prior to transfection. Four µg of pCMV-Tag 4-Fam20a plasmid in 10 µL of Lipofectamine2000 (Invitrogen) was diluted with 500 µL of Opti-MEM® I reduced serum media (Invitrogen), and incubated for 20 min at room temperature. The pCMV-Tag 4-Fam20a/Lipofectamine2000 complexes were then added to the culture media. After 6 h, the culture media with complexes were replaced with 2 mL fresh media containing 20 µL of Golgi marker, CellLight Golgi-GFP BacMan 2.0 (C10592, Invitrogen). After 18 h, the cells were fixed with 4% paraformaldehyde for 15 min at room temperature, washed with PBS buffer 3 times, and then permeabilized with PBST for 15 min at room temperature. Following blocking with 5% sheep serum in PBST for 30 min at room temperature, anti-Flag antibody (1∶200, F7425, Sigma-Aldrich) was applied. After over-night incubation of primary antibody at 4°C, the cells were washed with PBS buffer for 15 min and then incubated for 30 min at room temperature in solutions containing anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 594 (1∶500, A-11012, Invitrogen). The slides were then rinsed in PBS buffer for 15 min, mounted with ProLong Gold antifade reagent with DAPI (P-36931, Invitrogen), and examined under a Leica DM5000B fluorescence microscope.
Micro-CT
The teeth were secured on petri-dish containing 1% agarose, scanned and analyzed using a SCANCO μCT-100 series micro-computed tomography system at the University of Michigan School of Dentistry micro-CT core.
SEM
Tooth specimens were glued to a stub and sputter coated with gold for 75 s and then imaged using a Field Emission Gun Scanning Electron Microscope (FEG-SEM; Amray 1910 Field Emission Scanning Electron Microscope) at the Microscopy and Image Analysis Laboratory at the University of Michigan.
Backscatter SEM
Teeth were cut using a slow diamond saw (Model 650, South Bay Technology, Inc, San Clemente, CA, USA), infiltrated by 1∶1, 1∶2 and 1∶3 acetone∶Epon for 12 h, degassed twice, infiltrated overnight with pure Epon, polymerized at 60°C for 48 h with the cut surface placed face down in a 25 mm SeriForm mounting cup (Struers, Ballerup, DK). The blocks were sequentially polished with successively finer grades (400, 800 and 1200) of silicone carbide paper (South Bay Technology, Inc) followed by 4 h of polishing with 1.0 micro alumina abrasive with Multitex Polishing Cloth using a Buehler Supermet 2 Position Polisher (Lake Bluff, IL), sonication and rinsed with water. The finely polished tooth surface was coated with carbon and imaged using the Cameca SX-100 Electron Microprobe Analyzer (CAMECA, 92622 Gennevilliers Cedex, FR) at the University of Michigan Electron Microbeam Analysis Laboratory (EMAL) using the backscatter mode at a beam current of 15 kV and 10 nA.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. MacGibbonD (1972) Generalized enamel hypoplasia and renal dysfunction. Aust Dent J 17: 61–63.
2. LubinskyM, AngleC, MarshPW, WitkopCJJr (1985) Syndrome of amelogenesis imperfecta, nephrocalcinosis, impaired renal concentration, and possible abnormality of calcium metabolism. Am J Med Genet 20: 233–243.
3. HallRK, PhakeyP, PalamaraJ, McCredieDA (1995) Amelogenesis imperfecta and nephrocalcinosis syndrome. Case studies of clinical features and ultrastructure of tooth enamel in two siblings. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 79: 583–592.
4. Normand de la TranchadeI, BonarekH, MarteauJM, BoileauMJ, NancyJ (2003) Amelogenesis imperfecta and nephrocalcinosis: a new case of this rare syndrome. J Clin Pediatr Dent 27: 171–175.
5. PaulaLM, MeloNS, Silva GuerraEN, MestrinhoDH, AcevedoAC (2005) Case report of a rare syndrome associating amelogenesis imperfecta and nephrocalcinosis in a consanguineous family. Arch Oral Biol 50: 237–242.
6. HunterL, AddyLD, KnoxJ, DrageN (2007) Is amelogenesis imperfecta an indication for renal examination? Int J Paediatr Dent 17: 62–65.
7. DellowEL, HarleyKE, UnwinRJ, WrongO, WinterGB, et al. (1998) Amelogenesis imperfecta, nephrocalcinosis, and hypocalciuria syndrome in two siblings from a large family with consanguineous parents. Nephrol Dial Transplant 13: 3193–3196.
8. CatenaDL, MillerAS, LebermanOF, FreemanNC, BalickNL (1970) Consanguinity. Report of a case. Oral Surg Oral Med Oral Pathol 30: 207–212.
9. ChosackA, EidelmanE, WisotskiI, CohenT (1979) Amelogenesis imperfecta among Israeli Jews and the description of a new type of local hypoplastic autosomal recessive amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol 47: 148–156.
10. FritzGW (1981) Amelogenesis imperfecta and multiple impactions. Oral Surg Oral Med Oral Pathol 51: 460.
11. NakataM, KimuraO, BixlerD (1985) Interradicular dentin dysplasia associated with amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol 60: 182–187.
12. MockD, AidelbaumMR, ChapnickP (1986) Familial amelodentinal dysplasia. Oral Surg Oral Med Oral Pathol 61: 485–491.
13. van HeerdenWF, RaubenheimerEJ, DreyerAF, BennAM (1990) Amelogenesis imperfecta: multiple impactions associated with odontogenic fibromas (WHO) type. J Dent Assoc S Afr 45: 467–471.
14. OoyaK, NalbandianJ, NoikuraT (1988) Autosomal recessive rough hypoplastic amelogenesis imperfecta. A case report with clinical, light microscopic, radiographic, and electron microscopic observations. Oral Surg Oral Med Oral Pathol 65: 449–458.
15. PetersE, CohenM, AltiniM (1992) Rough hypoplastic amelogenesis imperfecta with follicular hyperplasia. Oral Surg Oral Med Oral Pathol 74: 87–92.
16. CollinsMA, MaurielloSM, TyndallDA, WrightJT (1999) Dental anomalies associated with amelogenesis imperfecta: a radiographic assessment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 88: 358–364.
17. CarnelioS, RaoN (2005) Amelogenesis imperfecta with gingival calcification: a rare presentation. Braz J Oral Sci 4: 932–935.
18. MacedoGO, TunesRS, MottaAC, Passador-SantosF, GrisiMM, et al. (2005) Amelogenesis imperfecta and unusual gingival hyperplasia. J Periodontol 76: 1563–1566.
19. FellerL, JadwatY, BouckaertM, BuskinA, RaubenheimerEJ (2006) Enamel dysplasia with odontogenic fibroma-like hamartomas: review of the literature and report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 101: 620–624.
20. KorbmacherHM, LemkeR, Kahl-NiekeB (2007) Progressive pre-eruptive crown resorption in autosomal recessive generalized hypoplastic amelogenesis imperfecta. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 104: 540–544.
21. FellerL, WoodNH, AnagnostopoulosC, BouckaertM, RaubenheimerEJ, et al. (2008) Enamel dysplasia with hamartomatous atypical follicular hyperplasia: review of the literature and report of a case. Sadj 63: 096–091, 096-097, 100-091.
22. Martelli-JuniorH, BonanPR, Dos SantosLA, SantosSM, CavalcantiMG, et al. (2008) Case reports of a new syndrome associating gingival fibromatosis and dental abnormalities in a consanguineous family. J Periodontol 79: 1287–1296.
23. ReddySS, NishaA, HarishBN (2010) Hypoplastic amelogenesis imperfecta with multiple impacted teeth–report of two cases. J Clin Exp Dent 2: e207–211.
24. O'SullivanJ, BituCC, DalySB, UrquhartJE, BarronMJ, et al. (2011) Whole-Exome Sequencing Identifies FAM20A Mutations as a Cause of Amelogenesis Imperfecta and Gingival Hyperplasia Syndrome. Am J Hum Genet 88: 616–620.
25. ChoSH, SeymenF, LeeKE, LeeSK, KweonYS, et al. (2012) Novel FAM20A mutations in hypoplastic amelogenesis imperfecta. Hum Mutat 33: 91–94.
26. KimJW, SimmerJP, LinBP, SeymenF, BartlettJD, et al. (2006) Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. Eur J Oral Sci 114 Suppl 1: 3–12.
27. ChanHC, EstrellaNM, MilkovichRN, KimJW, SimmerJP, et al. (2011) Target gene analyses of 39 amelogenesis imperfecta kindreds. Eur J Oral Sci 119: 311–323.
28. Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467: 1061–1073.
29. AdzhubeiIA, SchmidtS, PeshkinL, RamenskyVE, GerasimovaA, et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249.
30. SinghRK, CooperTA (2012) Pre-mRNA splicing in disease and therapeutics. Trends Mol Med 18: 472–482.
31. DesmetFO, HamrounD, LalandeM, Collod-BeroudG, ClaustresM, et al. (2009) Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37: e67.
32. WitkopCJJr (1989) Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol 17: 547–553.
33. KirziogluZ, UluKG, SezerMT, YukselS (2009) The relationship of amelogenesis imperfecta and nephrocalcinosis syndrome. Med Oral Patol Oral Cir Bucal 14: e579–582.
34. Martelli-JuniorH, dos Santos NetoPE, de AquinoSN, de Oliveira SantosCC, BorgesSP, et al. (2011) Amelogenesis imperfecta and nephrocalcinosis syndrome: a case report and review of the literature. Nephron Physiol 118: p62–65.
35. VogelP, HansenGM, ReadRW, VanceRB, ThielM, et al. (2012) Amelogenesis Imperfecta and Other Biomineralization Defects in Fam20a and Fam20c Null Mice. Vet Pathol 49: 998–1017.
36. DeckerE, Stellzig-EisenhauerA, FiebigBS, RauC, KressW, et al. (2008) PTHR1 loss-of-function mutations in familial, nonsyndromic primary failure of tooth eruption. Am J Hum Genet 83: 781–786.
37. WrightJT, TorainM, LongK, SeowK, CrawfordP, et al. (2011) Amelogenesis Imperfecta: Genotype-Phenotype Studies in 71 Families. Cells Tissues Organs 194: 279–283.
38. NalbantD, YounH, NalbantSI, SharmaS, CobosE, et al. (2005) FAM20: an evolutionarily conserved family of secreted proteins expressed in hematopoietic cells. BMC Genomics 6: 11.
39. AnC, IdeY, Nagano-FujiiM, KitazawaS, ShojiI, et al. (2009) A transgenic mouse line with a 58-kb fragment deletion in chromosome 11E1 that encompasses part of the Fam20a gene and its upstream region shows growth disorder. Kobe J Med Sci 55: E82–92.
40. KoikeT, IzumikawaT, TamuraJ, KitagawaH (2009) FAM20B is a kinase that phosphorylates xylose in the glycosaminoglycan-protein linkage region. Biochem J 421: 157–162.
41. TagliabracciVS, EngelJL, WenJ, WileySE, WorbyCA, et al. (2012) Secreted Kinase Phosphorylates Extracellular Proteins that Regulate Biomineralization. Science 336: 1150–1153.
42. KawasakiK, WeissKM (2003) Mineralized tissue and vertebrate evolution: the secretory calcium-binding phosphoprotein gene cluster. Proc Natl Acad Sci U S A 100: 4060–4065.
43. SimpsonMA, HsuR, KeirLS, HaoJ, SivapalanG, et al. (2007) Mutations in FAM20C are associated with lethal osteosclerotic bone dysplasia (Raine syndrome), highlighting a crucial molecule in bone development. Am J Hum Genet 81: 906–912.
44. SimpsonMA, ScheuerleA, HurstJ, PattonMA, StewartH, et al. (2009) Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet 75: 271–276.
45. FradinM, StoetzelC, MullerJ, KoobM, ChristmannD, et al. (2011) Osteosclerotic bone dysplasia in siblings with a Fam20C mutation. Clin Genet 80: 177–183.
46. KimJ-W, SimmerJP, HuYY, LinBP-L, BoydC, et al. (2004) Amelogenin p.M1T and p.W4S mutations underlying hypoplastic X-linked amelogenesis imperfecta. J Dent Res 83: 378–383.
47. KimJW, SeymenF, LinBP, KiziltanB, GencayK, et al. (2005) ENAM mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res 84: 278–282.
48. KimJW, LeeSK, LeeZH, ParkJC, LeeKE, et al. (2008) FAM83H mutations in families with autosomal-dominant hypocalcified amelogenesis imperfecta. Am J Hum Genet 82: 489–494.
49. LeeSK, SeymenF, LeeKE, KangHY, YildirimM, et al. (2010) Novel WDR72 Mutation and Cytoplasmic Localization. J Dent Res 89: 1378–1382.
50. HartPS, HartTC, MichalecMD, RyuOH, SimmonsD, et al. (2004) Mutation in kallikrein 4 causes autosomal recessive hypomaturation amelogenesis imperfecta. J Med Genet 41: 545–549.
51. KimJW, SimmerJP, HartTC, HartPS, RamaswamiMD, et al. (2005) MMP-20 mutation in autosomal recessive pigmented hypomaturation amelogenesis imperfecta. J Med Genet 42: 271–275.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 2
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- Complex Inheritance of Melanoma and Pigmentation of Coat and Skin in Grey Horses
- Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs
- Autophagy Induction Is a Tor- and Tp53-Independent Cell Survival Response in a Zebrafish Model of Disrupted Ribosome Biogenesis
- Assembly of the Auditory Circuitry by a Genetic Network in the Mouse Brainstem