
The νSaα Specific Lipoprotein Like Cluster () of . USA300 Contributes to Immune Stimulation and Invasion in Human Cells
Highly pathogenic and epidemic Staphylococcus aureus strains carry a pathogenicity island in their genome that contains a cluster of lipoprotein-encoding genes termed lpl. As the role lpl in virulence is still unclear, we deleted the entire lpl cluster in the community-acquired methicillin-resistant S. aureus (CA-MRSA) USA300 and found that the mutant was defective in stimulation of pro-inflammatory cytokines in human immune cells. Moreover, the major finding highlighted in this study is that the lpl cluster contributes to invasion into non-professional phagocytes such as epithelial cells and keratinocytes. Furthermore, the lpl-dependent increase in invasive activity, most likely, accounts for the enhanced bacterial burden observed in a murine kidney abscess model. In general, internalization of a pathogen into host epithelial cells shields the pathogen from immune defense and antibiotic treatment. However, further investigation is needed to clarify whether the increased ability to invade host cells is responsible for the potent disseminative activity and hypervirulent phenotype characterizing the νSaα type I island expressing S. aureus strains, including the USA300 CA-MRSA strain.
Published in the journal:
. PLoS Pathog 11(6): e32767. doi:10.1371/journal.ppat.1004984
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004984
Summary
Highly pathogenic and epidemic Staphylococcus aureus strains carry a pathogenicity island in their genome that contains a cluster of lipoprotein-encoding genes termed lpl. As the role lpl in virulence is still unclear, we deleted the entire lpl cluster in the community-acquired methicillin-resistant S. aureus (CA-MRSA) USA300 and found that the mutant was defective in stimulation of pro-inflammatory cytokines in human immune cells. Moreover, the major finding highlighted in this study is that the lpl cluster contributes to invasion into non-professional phagocytes such as epithelial cells and keratinocytes. Furthermore, the lpl-dependent increase in invasive activity, most likely, accounts for the enhanced bacterial burden observed in a murine kidney abscess model. In general, internalization of a pathogen into host epithelial cells shields the pathogen from immune defense and antibiotic treatment. However, further investigation is needed to clarify whether the increased ability to invade host cells is responsible for the potent disseminative activity and hypervirulent phenotype characterizing the νSaα type I island expressing S. aureus strains, including the USA300 CA-MRSA strain.
Introduction
In Staphylococcus aureus, the lipid moiety of lipoproteins represents the major signal activating innate immunity. This was confirmed by the fact that innate immune activation was significantly decreased in diacylglyceryl transferase enzyme encoding gene (lgt) deleted mutants [1]. The previously described stimulatory activity of lipoteichoic acid (LTA) was, thus, most likely due to contamination of the LTA fraction with highly active natural lipoproteins and/or lipopeptides [2,3].
One of the first lipoproteins to be analyzed and described in S. aureus was the membrane bound penicillinase [4]. The search of the S. aureus genomes for the lipobox motif revealed approximately 55–70 genes encoding putative lipoproteins [1,5]. Many of them exert crucial functions as transporters (iron, manganese, nickel, zinc, amino acid, oligopeptide, glycine betaine, sugar, teichoic acid transporter, or preprotein translocase subunit YidC) or chaperons, e.g. phage terminases, hem/copper-type cytochrome/quinol oxidase, pyruvate-formate-lyase-activating enzyme, protein-disulfide isomerase, or peptidyl-prolyl cis/trans isomerase (PrsA) [1]. It was, therefore, not surprising that the lgt mutation not only affected high-affinity metal ion uptake but mutants were also attenuated in virulence [6].
Highly pathogenic and, in particular, epidemic S. aureus strains bear various genomic islands in their genome that encode prophages, toxins and antibiotic resistance genes, e.g. SCCmec, or tandem paralogous genes such as those encoded in the νSaα island. The term νSaα refers to non-phage and non-SCC genomic islands that are exclusively present in S. aureus and inserted at specific loci in the chromosome [7]. The genomic island termed νSaα is present in all S. aureus genomes sequenced to date [8–10] but is not found in coagulase-negative species, including the non-pathogenic species S. carnosus [11]. The genetic organization of νSaα is highly conserved and is composed of two gene clusters: one cluster encodes a number of highly homologous exotoxin-encoding genes (set), the other one encodes lipoproteins, referred to as 'lipoprotein-like' (lpl), with a typical lipo-box containing signal sequence [12]. The genetic organization of νSaα of S. aureus USA300 is illustrated in Fig 1A. The set and lpl clusters are separated by the hsdM and hsdS genes that encode a restriction-modification system. While HsdS selects the genomic target sequence, HsdM is serves as a methylase. It has been proposed that this system contributes to stabilization and maintenance of the S. aureus genomic islands. It was further suggested that the νSaα genomic islands originate from mobile genetic elements that were acquired independently through intra-species genetic transfer between S. aureus strains [8]. The finding that some νSaα types contain a remnant transposase gene supports this hypothesis. As the hsdS alleles correlate with the structural similarity of the islands its sequence has been used for classification of νSaα islands into type I to IV.
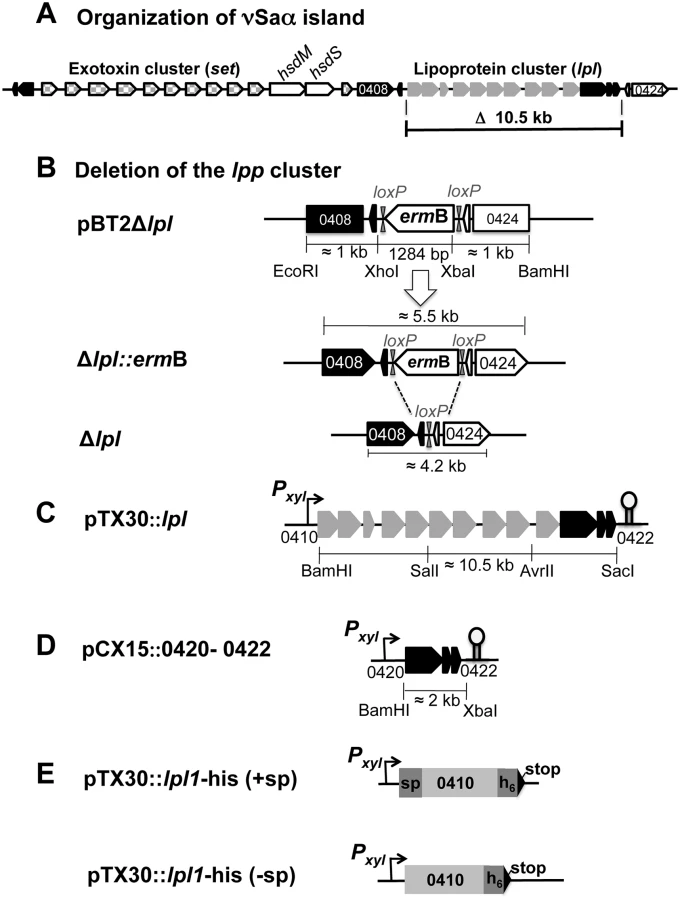
All lpl genes are sequentially lined up in the same orientation, thus comprising a paralog cluster. The lpl genes within one type are highly homologous (almost 99%), but are significantly different from those encoded in other types. The various lpl genes within one cluster are further distinguished by a 5′- and 3′-variable region and a highly conserved intragenic region, which might represent a structural prerequisite for gene shuffling, tandem duplications and diversification [7]. Homologs of these lpl genes were also found in loci other than the genomic island and represent the largest group of paralogous genes in various S. aureus strains [8,10].
The S. aureus strains USA300, N315, Mu50, NCTC8325, Newman, COL, JH1 and JH9 possess a type I νSaα island [8]. Interestingly, all of these strains belong to the clonal complex 5 and 8 (CC5 & CC8) whose representatives are characterized by high transmissibility and virulence. Although the genome of USA300 is known [9], the molecular genetic basis of the high transmissibility and hypervirulence of the CC8 strains and USA300, in particular, is unknown. However, the broad distribution and apparent persistence of the νSaα island in S. aureus suggests a potential function in virulence and, additionally, in the dissemination of epidemic strains. We, therefore, carried out our studies in S. aureus USA300, a community-associated MRSA (CA-MRSA) that is epidemic in the general population and has been associated with necrotizing fasciitis, pneumonia and other rapidly progressing and life-threatening infections [9,13]. According to multilocus sequence typing (MLST) USA300 belongs to the clonal complex CC8, which contains many strains with high clinical impact [14,15].
Since the role the lpl gene cluster of the νSaα islands in virulence is unclear to date, we investigated its function in S. aureus USA300 by deleting the entire lpl gene cluster. We found that the mutant was deficient in innate immune stimulation, host cell invasion and virulence.
Results
Genomic characterization of the νSaα specific lipoprotein like cluster (lpl) of S. aureus USA300
The lipoprotein-like cluster within the νSaα island of USA300 comprises ten lpl genes (lpl 0410–0419). All of them contain a classical lipo-box motif in the signal sequence (Fig 2A). Based on protein sequence similarity the corresponding Lpl proteins can be grouped into three clusters: Lpls encoded by the genes 0410, 0411, 0413, 0414, 0417 share > 80% similarity; the two Lpls encoded by 0416 and 0418 also share > 80% similarity but the similarity between 0410 and 0416 is only approximately 60% (Fig 2B). The similarity of the Lpls suggests that they might have redundant functions. The lpl cluster contains several potential promoters located upstream of the genes 0410, 0413, 0417 and 0419 (Fig 2A). By Northern blot analysis (S1 Fig) we could verify the transcripts starting from P2, P4 and P5 promoters. However, we assume that a run-through transcript of approximately 10.5 knt is generated because there is no conspicuous transcription terminator sequence in the lpl cluster (Fig 2A). There are two predicted rho-independent transcription terminators, Ω1 and Ω2, downstream of 0422 and 0424, respectively. Downstream of the lpl cluster, but upstream of the Ω2, there are three genes (0420–0422) with unknown function. These highly conserved genes are present in all presently sequenced νSaα islands. We, therefore, predict that they might be involved in Lpl biosynthesis/modification. The third transcription terminator, Ω3, is inserted in opposite direction, downstream of 0409 (Fig 2A).

To study the potential role of the νSaα-specific lpl cluster in USA300 we constructed a marker-less deletion mutant, USA300Δlpl, in which the entire lpl operon was deleted including the three downstream genes (0420–0422) of unknown function (Fig 1B). For complementation of the mutant two plasmids were constructed: pTX30::lpl contains all the genes (0410–0422) that had been deleted (Fig 1C), and pCX15::0420–0422 contains only the three non-lipoprotein genes (Fig 1D); in these two compatible plasmids the genes are expressed under the control of a xylose-inducible promoter.
The USA300 νSaα-specific lpl cluster enhances TNF-α and IL-6 production in monocytes
In USA300 the νSaα specific lpl genes represent roughly 16% of all annotated lipoproteins. To investigate whether the lpl cluster contributes to the stimulation of innate immune cells, the human monocytic cell line (Mono Mac 6) was infected with an MOI of 30:1 with living cells of the parental strain USA300 wt, the lpl deletion mutant Δlpl, and the complemented mutant Δlpl (pTX30::lpl). The production of pro-inflammatory cytokines such as TNF-α and IL-6 was determined after 4 and 24 h of stimulation. The stimulatory conditions for TNF-α and IL-6 were selected based on prior experiments (S2 and S3 Figs). The results showed that the production of TNF-α and IL-6 was significantly decreased when stimulating with the Δlpl mutant compared to the wt: TNF-α decreased by roughly 70% (Fig 3A) and IL-6 by approximately 40% (Fig 3C). In the complemented mutant the cytokine levels were restored to the parental levels, even in the absence of xylose.
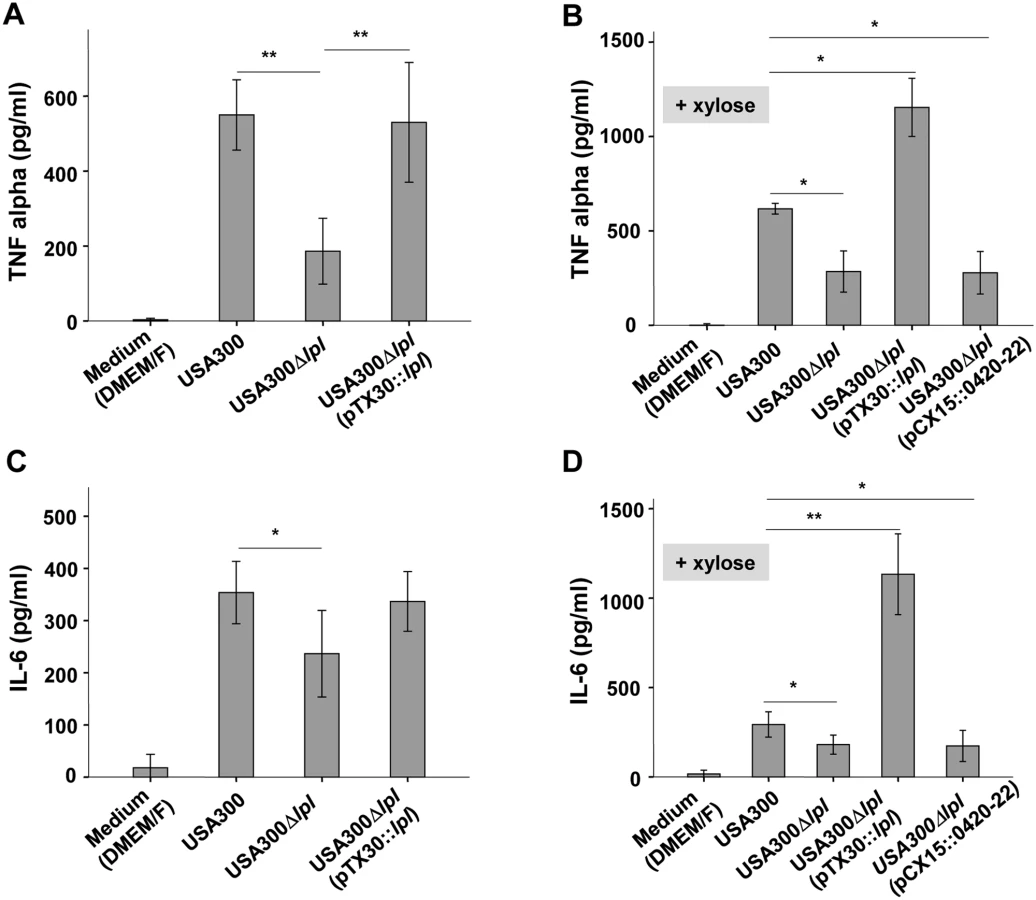
However, when the complemented mutant USA300lpl (pTX30::lpl) was cultivated in the presence of 0.8% xylose TNF-α and IL-6 production was increased 2- to 3-fold, respectively, when compared to the parental strain (Fig 3B and 3D). In order to investigate whether the three non-lipoprotein genes (0420–0422) downstream of the lpl cluster, which are part of the lpl operon, had an immune modulatory effect, USA300lpl (pCX15::0420–22) was constructed. However, the xylose-induced genes 0420–0422 showed no effect on TNF-α and IL-6 production (Fig 3B and 3D), suggesting that only the Lpl proteins contribute to TNF-α and IL-6 production. Similar results were obtained with different MOIs tested (S2 Fig).
We next investigated whether the lpl-cluster exerts a similar immune stimulatory effect in other staphylococcal strains. To this end we cloned pTX30::lpl into S. aureus HG003, which is a rsbU- and tcaR-repaired derivate of NCTC8325 [16] and into S. carnosus TM300, a non-pathogenic foodborne staphylococcal species [11,17]. The results showed that in HG003, as in USA300, xylose-induced expression of the lpl-cluster led to an approximately four-fold induction of TNF-α and IL-6 production (Fig 4A and 4B). In S. carnosus (pTX30::lpl) TNF-α production was only slightly increased and for IL-6 production no difference was observed (Fig 4A and 4B). However, one of the most remarkable differences between the pathogenic S. aureus strains and the non-pathogenic S. carnosus strain was the generally much higher activation of Mono Mac 6 by S. carnosus. While USA300 and HG003 triggered TNF-α production in a range of 500 pg/ml, S. carnosus TM300-derived TNF-α production was increased to > 8000 pg/ml, which is 16 times higher than that of the S. aureus strains. A similar difference was observed with IL-6 (Fig 4A and 4B). Thus, S. carnosus has a much higher TLR2-dependent immune stimulatory activity in Mono Mac 6 than USA300.

We hypothesize that the ability of S. carnosus to mount a strong TNF-α and IL-6 response is due to its non-pathogenicity. As a consequence it can rapidly be neutralized by a robust immune response. Alternatively, S. carnosus could be less toxic to Mono Mac 6 cells compared to S. aureus resulting in higher viable cell numbers that produce TNF-α/IL-6. We, therefore, checked the number of live Mono Mac 6 cells after stimulation with S. aureus and S. carnosus but observed no significant difference in cell viabilities (S4 Fig).
The lpl cluster enhances cytokine and antimicrobial peptide expression in differentiated primary human keratinocytes and macrophages
Differentiated primary human keratinocytes were infected with USA300, the Δlpl mutant, and the complemented mutant Δlpl (pTX30::lpl) with an MOI of 30 (± 10) for 8 h. mRNA expression of cytokines (TNF-α and IFN-γ) and the antimicrobial protein RNase7 was analyzed by RT-PCR. The relative expression of the target genes (TNF-α, IL-1β and RNase7) was normalized to β-actin as house keeping gene. In all cases the Δlpl mutant induced significantly lower levels of TNF-α, IL-1β and RNase7 transcripts than the parental strain or the complemented mutant, suggesting that the lpl cluster contributes to the expression of inflammatory cytokines and antimicrobial peptides in primary human keratinocytes (Fig 5A, 5B and 5C). To evaluate these findings in primary human leukocytes, human macrophages were generated in the presence of GM-CSF or M-CSF, respectively. Similarly, we observed significantly lower TNF-α production with the Δlpl mutant in both human macrophage subsets (Fig 5D).

Cytokine production induced by the lpl cluster is conserved in an lgt mutant
It is well known that the lipid-modification of lipoproteins is crucial for the activation of innate immune cells via TLR2 signaling. The S. aureus Δlgt mutants lacks lipidation of pre-prolipoproteins because the prolipoprotein diacylglyceryl transferase is absent. S. aureus Δlgt mutants were formerly not only completely deficient in lipidation of pre-prolipoproteins, but also induced significantly less proinflammatory cytokines and chemokines and were compromised in respect to virulence [1,2,6]. Due to the importance of the lipid-moiety of lipoproteins for immune stimulation we anticipated that expression of the lpl-cluster in SA113Δlgt mutant would have no effect on proinflammatory cytokine expression because the corresponding Lpls lack the lipid-moiety that is crucial for the initiation of TLR2/MyD88-dependent signaling. However, expression of the lpl-cluster in SA113Δlgt (pTX30::lpl) caused a nearly two-fold increase in TNF-α and IL-6 production, suggesting that, unexpectedly, not only the lipid-moiety but also the Lpl protein portion might possess stimulatory activity (Fig 4C and 4D). Subsequent experiments, however, revealed that the stimulatory activity is not exerted by the proteinaceous part of the Lpls but is rather due to cellular release of other PAMPs from the lgt mutant. The latter results from cellular stress induced by membrane jamming due to overexpression of the plasmid-encoded Lpls that cannot be correctly targeted to the membrane.
Purified lipidated Lpl1-his—but not unlipidated Lpl1-his—induces proinflammatory cytokines in human-derived Mono Mac 6 and TLR2-transfected HEK293 cells
We, next, investigated the effect of a purified Lpl protein on the induction of cytokine secretion. As a prototype we chose the first lpl-gene (lpl1; 0410) of the lpl cluster, which also displays a high degree of similarity with the other four lpl genes (Fig 2). The lpl1 was marked by a 3'- His-tag and was cloned into the pTX30::lpl1-his (Fig 1E). The Lpl1-his protein containing the lipid moiety (+sp) was purified from the membrane fraction of SA113 (pTX30::lpl1-his) by affinity chromatography and rabbits were immunized to generate anti-Lpl1-his rabbit antibodies. The purified Lpl1-his showed a high purity in Coomassie-stained SDS-PAGE (Fig 6A). In Western blot analysis of membrane fractions from USA300 wt, its Δlpl mutant and the complemented mutant, Lpl1-his was only detectable in the wt and complemented mutant (Fig 6B). Notably, Lpl1-his expression did not undergo relevant changes after 4, 8 and 14 h cultivation periods.
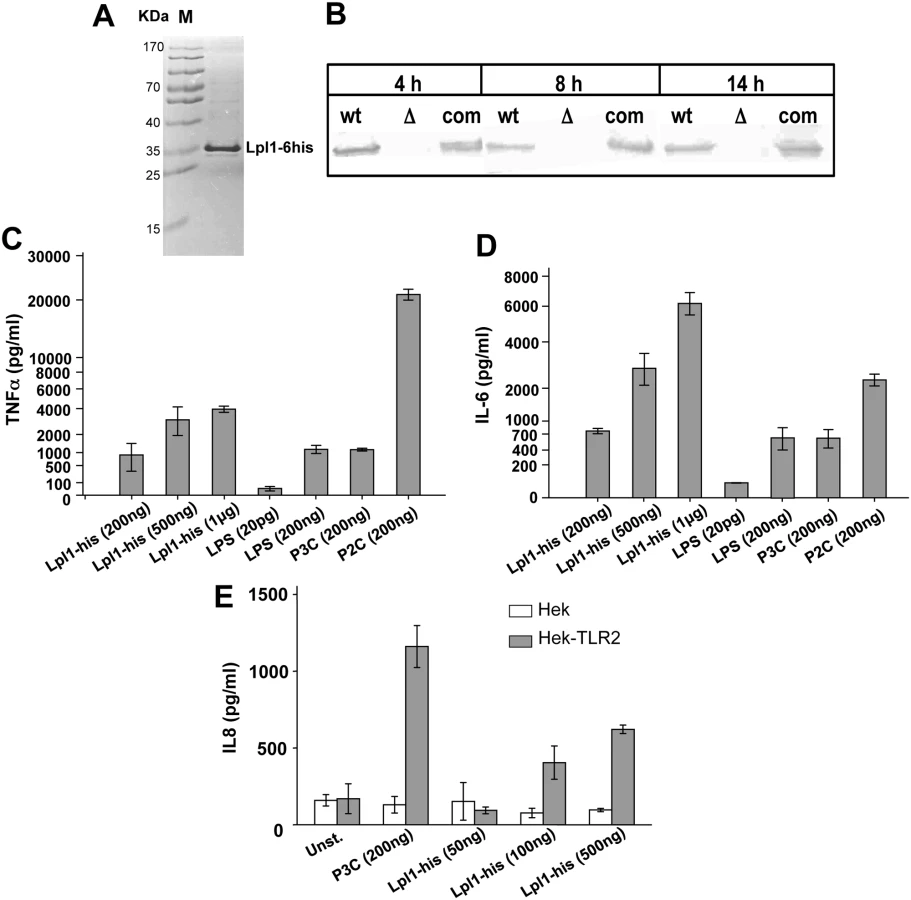
Subsequently, purified Lpl1-his was used in concentrations of 200 ng, 500 ng and 1 μg/ml to stimulate Mono Mac 6 cells. P3C (200 ng), P2C (200 ng) and LPS (200 ng) were used as controls. The results showed that Lpl1-his induced TNF-α and IL-6 production in a concentration-dependent manner (Fig 6C and 6D). 200 ng Lpl1-his induced comparable amounts of TNF-α and IL-6 as seen with 200 ng of LPS or P3C. Surprisingly, we observed an extremely high stimulation of TNF-α by P2C, which was approximately 20 fold higher than that induced with the same amount of P3C or LPS. To further assess its immunogenicity purified Lpl1 was tested for TLR2 activity. HEK293 cells transfected with or without TLR2-bearing plasmid were stimulated with Lpl1 or TLR2 agonist P3C. Evidently, Lpl1-his induced IL-8 via TLR2 activation as a dose-dependent manner only was seen in the presence of TLR2 (Fig 6E). Of note, purified lipidated Lpl1-his was essentially endotoxin-free. Specificity of the assay was previously demonstrated by comparing SA113 and the SA113Δlgt mutant that lacks TLR2-stimulating lipoproteins [1,18].
We also purified unlipidated Lpl1-his by expressing it in the absence of the lipo signal sequence (-sp) in S. aureus (Fig 1E). Purified unlipidated Lpl1 showed no stimulatory activity in Mono Mac 6 cells (S5 Fig), indicating that the lipidation of the Lpl proteins is essential for signaling, and the signaling observed in SA113Δlgt (pTX30::lpl) was most likely due to stress-induced secondary PAMPs.
The νSaα specific lpl cluster contributes to invasion into human keratinocytes and in mouse skin
For invasion studies differentiated primary human keratinocytes were infected with USA300 wt, its Δlpl mutant and the complemented mutant with a MOI of 30. In the Δlpl mutant the number of invaded bacteria was 2.5-fold lower compared to the wt; in the complemented mutant the invaded bacteria exceeded that of the wt by 1.5-fold (Fig 7A). This suggests that the lpl gene cluster significantly contributes to invasion of keratinocytes. Downstream of the lpl cluster there are three (black-labeled) genes 0420–0422 with unknown function (Fig 1A). To exclude that these genes affect invasion we transferred the genes into USA300Δlpl mutant by pCX15::0420–0422. The results confirmed that these genes do not enhance invasion (Fig 7A). We further investigated the invasion of S. carnosus and S. carnosus (pTX30::lpl). While S. carnosus and S. carnosus (pTX30) completely lacked invasion, S. carnosus (pTX30::lpl) showed a significant increase in invasive activity, which corresponds to approximately 5% of that seen with USA300 (Fig 7A). Thus, these results provided strong evidence that the Lpl proteins trigger host cell invasion.

To clarify whether invasion can also be observed in vivo, we, next, carried out invasion studies on murine skin after shaving and tape-stripping (Fig 7B). Here, too, we confirmed significantly lower invasion with the mutant compared to the wt; and in the complemented mutant invasion could be restored to the extent reached by the wt. In order to demonstrate that USA300 is internalized into human keratinocytes we monitored bacterial adherence and invasion with a mCherry-expressing USA300 strain. Using bright-field microscopy we could visualize adherence of USA300 to the host cell surface (Fig 7C). In the invasion studies adherent USA300 cells were lysed with lysostaphine. We could, therefore, not detect them in the bright-field microscopy but only by fluorescence microscopy (Fig 7D).
The νSaα-specific lpl cluster enhances the bacterial burden in a mouse kidney abscess model
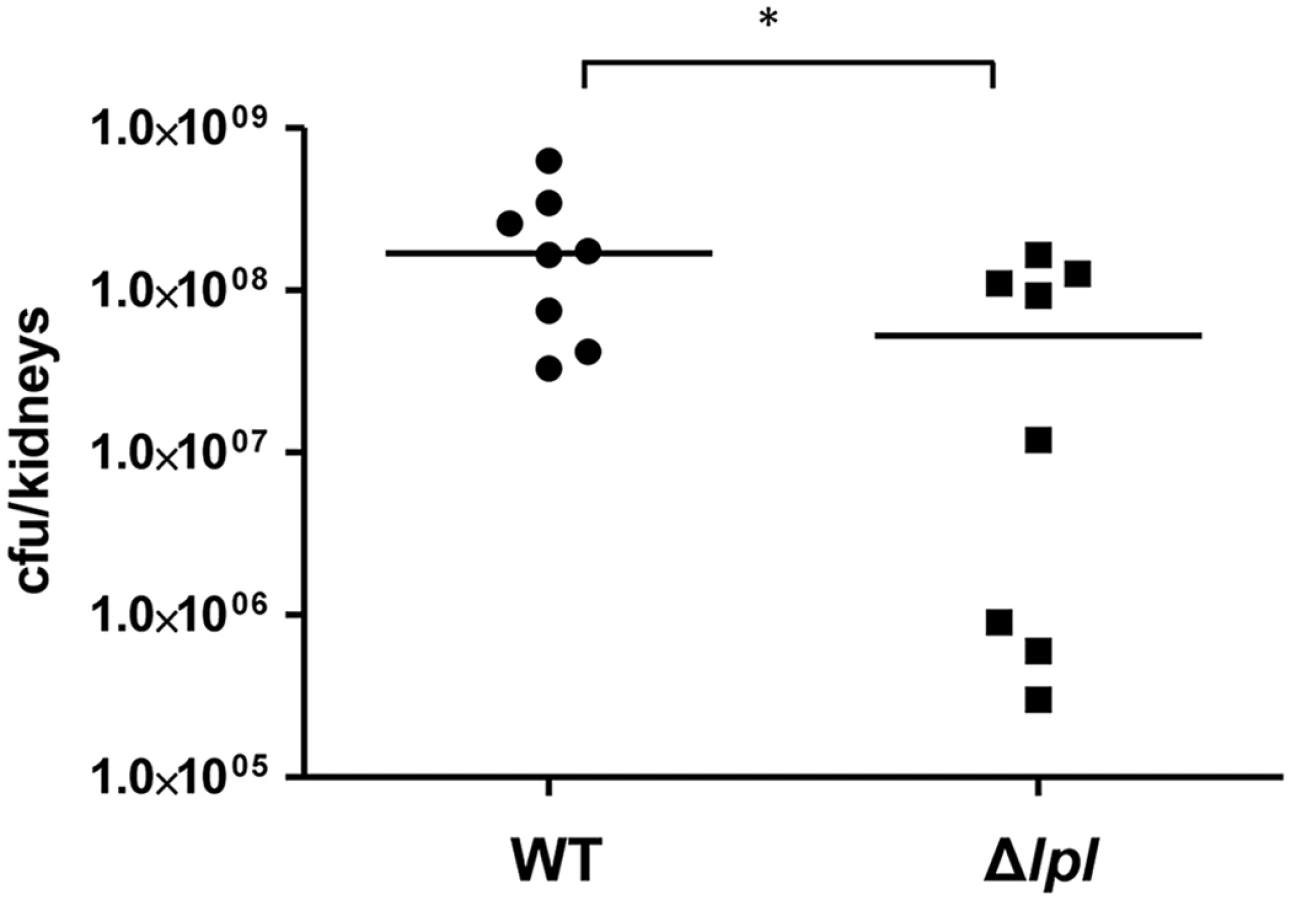
To see whether the lpl cluster contributes to pathogenicity we examined USA300 and the Δlpl mutant in a mouse kidney abscess model with Balb/c mice infected for 5 days. With the Δlpl mutant the bacterial burden of the kidneys was significantly lower when compared to the parental strain (Fig 8). The complemented mutant Δlpl (pTX30::lpl) was also tested in the kidney abscess model, however, the number of colony forming units (CFU) did not reach the level of the parental strain. We assume that the xylose-inducible expression of the lpl genes was instable over the 5-day infection period. Additionally, we observed a slight growth defect in TSB with clones containing the high-copy plasmid pTX30.
Discussion
Why the genomic island νSaα is so prevalent and highly conserved in S. aureus is a puzzling issue. For the genetic maintenance of the tandem lpl gene cluster it is currently believed that the highly conserved intragenic region flanked by the variable regions of the lpl genes is responsible for gene shuffling, tandem duplications and diversification [7]. On the other hand, if νSaα was not beneficial in infection we would expect it to get lost during evolution. Thus, the aim of this study was to investigate the potential benefit of the νSaα tandem lpl gene cluster in infection.
We chose S. aureus USA300 [9] as a model strain for several reasons: firstly it contains one of the most complex tandem lipoprotein clusters in the νSAα island, and, secondly, it is a member of the clonal complex (CC) 8 that encompasses several globally distributed epidemic lineages, including hospital-associated methicillin-resistant S. aureus (MRSA) and the highly prevalent community-associated MRSA clone USA300 [19]. However, the genetic basis for the high transmissibility and hypervirulence of the CC8 strains, and USA300 in particular, is unknown. Here, we reasoned that the νSaα island could represent a virulence determinant of S. aureus. After some difficulties we managed to construct a marker-less deletion mutant of the 10 kb lpl cluster. The mutant showed no apparent growth defect in both B-medium (rich medium) and DMEM/F (nutrient-poor medium) indicating that the tandem lipoproteins have no vital role in vitro, however, they might contribute to innate immune response.
It is well known that lipoproteins and lipopeptides are recognized by TLR2 [20,21] which can form heterodimers with TLR1 or TLR6 to trigger intracellular signaling by triacylated and diacylated lipopeptides, respectively [22–25]. In Staphylococcus lipoproteins do not only contribute to ion uptake and pathogenicity, they also represent the predominant immune stimulators [1,6,26–28]. Therefore, we first addressed whether and to which extent the lpl gene cluster contributes to innate immune stimulation. Searching the annotated proteins of USA300 by the DOLOP program there are approximately 63 proposed lipoproteins present including the 10 Lpl proteins of the νSaα island. The 10 Lpl proteins comprise 16% of all lipoproteins in USA300. Theoretically, one would expect only a minor effect on induction of pro-inflammatory cytokine expression in Mono Mac 6 cells. However, we observed in USA300Δlpl a 3-fold decrease in TNF-α and an almost 2-fold decrease in IL-6 expression (Fig 3A and 3C). The complemented strain USA300lpl (pTX30::lpl) largely reached parental expression levels even in the absence of xylose, while in the presence of xylose TNF-α and IL-6 expression exceeded that of the parental strain by a factor of 2 and 4, respectively (Fig 3A and 3C). These results show that despite the presence of other lipoproteins Lpl proteins have an unexpectedly high impact on innate immune stimulation. Notably, heterologous expression of pTX30::lpl in S. aureus HG003 caused a similarly high induction of TNF-α and IL-6 expression as with USA300 (Fig 4A and 4B).
Of note, in S. carnosus the induction of TNF-α and IL-6 expression was not as pronounced. However, the non-pathogenic S. carnosus was exceptional in so far, as it triggered an almost 20-fold higher TNF-α and IL-6 expression than the S. aureus strains. S. carnosus belongs to the non-pathogenic staphylococcal species and we assume that generally the non-pathogenic species are non-pathogenic not only because of low to absent toxin and adhesion production but also because of their better recognition by our immune system [17]. Interestingly, we also observed a comparable difference in stimulation with the synthetic lipopeptides: P2C was much more active than P3C (Fig 6C).
Since in S. aureus lipidation of the lipoproteins by diacylglyceryl transferase (encoded by the lgt gene) is crucial for innate immune activation [1], we wondered whether innate immune stimulation is preserved after cloning of the lpl gene cluster into an lgt deletion mutant. As shown in Fig 4C and 4D there was a roughly 2-fold increase in TNF-α and IL-6 expression in SA113lgt (pTX30::lpl) compared to SA113lgt. Although SA113lgt (pTX30::lpl) did not reach the SA113 parental level, we wondered why there was an increase at all. The most likely explanation is release of other PAMPs as a stress-response due to the lgt mutation. In this mutant all pro-lipoproteins are unlipidated but they are still transiently anchored in the cytoplasmic membrane by the unprocessed signal peptide. Taking into consideration that in SA113lgt (pTX30::lpl) ten additional lipoproteins are highly expressed and cannot be correctly targeted via their lipid moiety to the outer leaflet of the cytoplasmic membrane, the membrane translocation machinery might be crammed, which could result in release of peptidoglycan and/or DNA that could contribute to the observed innate immune stimulation. Membrane jamming has frequently been observed when proteins were overexpressed and not correctly targeted. For example, upon overproduction of β-galactosidase hybrid proteins, the export pathway is so severely jammed that other exported proteins accumulate in their precursor form in the cytoplasm [29]. The other thrilling but unlikely possibility would have been that unlipidated Lpls caused immune stimulation. To clarify the situation we purified his-tagged Lpl1, as a Lpl representative lipoprotein, with and without its lipid part by expressing it with and without its lipo signal sequence (Fig 1E) in S. aureus. Although applied in high concentration, the unlipidated protein Lpl1 showed no immune stimulating activity, suggesting that the increased immune stimulating activity in SA113lgt (pTX30::lpl) is probably due to a side effect, possibly, by higher release of peptidoglycan fragments, DNA or RNA that also contribute to innate immune stimulation [30]. Lastly, Lpl-mediated invasive activity might facilitate uptake and cytosolic sensing of these bacterial compounds.
The enhanced innate immune stimulation exerted by the lpl gene cluster in USA300 is not really beneficial in regards to virulence; on the contrary, these strains are better recognized by the innate immune system. Therefore, the key question was whether expression of the νSaα tandem lpl gene cluster could represent an advantage in infection. It turned out that the most beneficial function of the Lpl was increased invasiveness. Invasion studies in vitro and in vivo (Fig 7A and 7B) revealed that the mutant USA300Δlpl was significantly less invasive (>2-fold) than the parental strain, and that the complemented mutant showed an even higher invasiveness than the parental strain. The ability of S. aureus to be internalized by host cells is considered to be one of the most critical pathogenicity factors in persisting and relapsing infections because intracellular localization of bacteria evades antimicrobials and the host immune response [31]. It is now widely accepted that S. aureus is internalized by a variety of non-professional phagocytes, such as endothelial cells [32,33], epithelial cells [34–36], fibroblasts [37], or osteoblasts [38,39], and can persist inside the host cells for weeks, months or even years. The fibronectin binding proteins (FnBPA and FnBPB) are the major adhesins involved in S. aureus internalization by host cells. They use fibronectin as a bridging molecule and the α5β1 integrin as host cell receptor resulting in signal transduction, tyrosine kinase activity, and cytoskeletal rearrangements [34,36,40]. The FnBPs also bind to the heat shock protein 60 (Hsp60), which might act as a co-receptor in the FnBP-mediated uptake of S. aureus [41]. Furthermore, a role of the extracellular adherence protein (Eap) was described in internalization into fibroblasts or epithelial cells; the receptor is unknown to date [42]. Recently, the major autolysin of S. aureus and S. epidermidis Atl was found to contribute to host cell internalization by interacting with the heat shock cognate protein Hsc70 [43].
A very important confirmation of the invasive activity of the Lpls was the finding that the non-pathogenic and non-invasive S. carnosus strain becomes invasive when transformed with (pTX30::lpl) (Fig 7A). Although the frequency of invasion of S. carnosus (pTX30::lpl) was low, e.g. only approximately 10% of that of USA300, one has to consider that S. carnosus lacks FnBPs as the major players of S. aureus invasion and also Eap [44]. Furthermore, we do not know whether Atl plays a role in S. carnosus-mediated host cell invasion as its sequence and domain organization differs significantly from that of S. aureus [45]. Therefore, the invasive activity seen in S. carnosus probably reflects the impact of the Lpls.
In our studies we frequently used human keratinocytes because they represent the immune sentinels in the skin and as such they provide a first line of defense against microbial pathogens. They express almost all TLRs, which is crucial for promoting skin immune responses to adherent bacteria. Activation of these receptors on human keratinocytes leads to a predominant TH1-type immune response and to the production of type I interferons (IFNs) [46]. Moreover, keratinocytes also constitutively secrete, or are induced to release, numerous cytokines, including IL-1, IL-6, IL-10, IL-18 and TNF-α; and they process and release IL-1β through activation of the inflammasome.
Increased invasive activity is an important advantage for persistence. In our opinion, this overrules the disadvantage of enhanced innate immune stimulation. It is well known that if a pathogen invades new tissues the host responds by eliciting immune responses in an effort to eliminate infection. Internalization into non-professional antigen-presenting cells such as keratinocytes [46] most likely protects S. aureus from the immune response. It is, therefore, to expect that by contributing to host cell invasion the lpl gene cluster enhances S. aureus' virulence and particularly its persistence. Whether the enhanced host cell invasion also contributes to increased dissemination and epidemic spreading needs to be further investigated.
Lpl gene clusters are found in most S. aureus strains; however, these clusters differ with respect to the number of the tandem lpl genes and the type of νSaα. For example, most members of the CC8 (USA300, Newman, A5948, A9754) have 10 lpl genes, while NCTC8325 has only three. Members of the CC5 lineage also have an average of 10 lpl genes. Both CC5 and CC8 are distinguished by type I νSaα, while CC1, CC30 and CC151 possess type II, III and IV, respectively (S6 Fig). In this study we chose USA300 as a model strain for lpl gene cluster analysis. However, the lpl gene cluster in other S. aureus strains will most likely have a similar function in invasion. One of the questions which will be addressed in the future is whether the number of lpl repeats in νSaα is correlated with the invasion frequency. Probably, there is a fine-tuned balance between a stable maintenance of the repeats and their benefit in infection. We further performed electron microscopy to visualize potential differences in cell surface structures between USA300 and its Δlpl mutant. However, at first sight these images revealed no apparent differences in the morphology (S7 Fig). This was not unexpected because the mutant expresses 50 other lipoproteins.
Recently, it has been shown that S. aureus USA300 and HG001 induced autophagy in a human tumor cell line, while S. carnosus did not [47]. The S. aureus cells become entrapped in vesicles and dividing S. aureus cells were seen intracellularly suggesting that S. aureus cells are able to multiply intracellularly [47]. Whether the lpl gene cluster is involved in this process is currently unknown. However, the ability of S. aureus to invade different types of non-professional phagocytes, to escape from the host lysosomal degradation machinery and to persist within the intracellular location most likely represent essential steps in pathogenesis. After invasion there are several options: S. aureus cells may be killed by the host cell, they kill the host cell, or they survive and persist by switching to an SCV (small colony variant) phenotype, an essential step for establishing chronic infections [48].
What distinguishes an epidemic and disseminating strain from other strains is an important question. Not many cases are known where epidemic outbreaks could be traced to certain genes. One recent example is the Escherichia coli O104:H4 strain that caused the large German outbreak in 2011. This strain acquired genes from an entero-aggregative E. coli strain (EAEC) that increased intestinal colonization thus turning it into a highly virulent and disseminating hybrid strain [49,50]. In Listeria monocytogenes the interaction between the bacterial surface molecules, the internalins InlA and InlB, and their cellular receptors E-cadherin and the Hepatocyte Growth Factor Receptor (Met), respectively, triggers the recruitment of endocytic effectors, the subversion of the phosphoinositide metabolism, and the remodeling of the actin cytoskeleton that leads to bacterial engulfment [51]. In this pathogen the internalins are the basic virulence factors because they are a prerequisite for spreading and dissemination. It is, however, completely unknown how the Lpl proteins trigger invasion.
Conclusion
This is one of the first studies addressing the role of the νSaα-encoded tandem lpl gene cluster in a member of the high transmissible and hypervirulent clonal complex CC8. Firstly, the lpl gene cluster enhances the innate immune signaling suggesting that the lipoproteins carry the lipid moiety, which has been confirmed by testing purified lipidated and unlipidated Lpl1 lipoprotein in vitro. On the one hand, increased activation of the innate immune system is not really beneficial for a pathogen because it activates bacterial immune defense. On the other hand, however, it is highly beneficial for a first-class pathogen to acquire an enhanced ability to invade host cells, which may facilitate spreading in the host. This is illustrated by the enhanced bacterial burden in a murine kidney abscess model. The increased ability to invade host cells could further be responsible for the high disseminative activity of some S. aureus isolates. Investigation of the exact mechanism of host cell invasion is one of the expedient next steps. We postulate that the lpl gene cluster will have clinical, diagnostic, epidemiologic and evolutionary implications.
Materials and Methods
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in S1 Table. E. coli strains were cultivated aerobically in Luria Broth (LB) medium with shacking at 30°C. S. aureus strains aerobically grown in either basic medium, BM (1% soy peptone, 0.5% yeast extract, 0.5% NaCl, 0.1% glucose and 0.1% K2HPO4, pH 7.4), or Tryptic Soy Broth (TSB) at 37°C. When appropriate, the media were supplemented with ampicillin (100 μg/ml), erythromycin (10 μg/ml), tetracycline (25 μg/ml) or chloramphenicol (20 μg/ml).
Deletion of the tandem lipoprotein cluster by allelic replacement in S. aureus USA300
The deletion of entire lpl cluster in S. aureus USA300 was generated by homologous recombination. The basis for the construction of the knock-out plasmid was temperature-sensitive E. coli—Staphylococcus shuttle vector pBT2 [52] in which three DNA fragments were cloned between the EcoRI and BamHI sites: the approximately 1 kb upstream region comprised part of ORF 0408 and the entire ORF 0409, the 1 kb downstream region comprised ORF 0423 and part of ORF 0424. In between the up- and downstream region the 1.2 kb erythromycin gene flanked by loxP sites was integrated (Fig 1A). The 1 kb upstream and downstream regions were amplified by PCR from the genome of S. aureus USA300 using primer pairs Fr_up (EcoRI) and Re_up (Xhol) and primer pair Fr_down (XbaI) and Re_down (BamHI), respectively (S2 Table). The ermB gene together with the flanking loxP sites was amplified from pBT2-srtA [53] with primer pair Fr_erm (XbaI) and Re_erm (XhoI); the Cre-loxP sequence is in italics. The amplified upstream fragment was cut with EcoRI and XhoI, the amplified downstream fragment was cut with XbaI and BamHI, and the amplified ermB gene fragment was cut with XhoI and XbaI. These fragments were then ligated with vector pBT2 digested by BamHI and EcoRI, resulting pBT2Δlpl and transformed into E. coli DH5α. The positive clone containing plasmid pBT2Δlpl was isolated, purified and subsequently transformed by electroporation into S. aureus RN4220 as an intermediary host and then transformed into S. aureus USA300. The deletion of tandem lipoprotein cluster from genome was achieved by homologous recombination described previously [52]. The resultant deletion mutant was named USA300Δlpl::ermB (Fig 1A). This clone was transformed with thermo-sensitive replicon pRAB1 which encodes the cre-recombinase gene to remove the ermB cassette [53]. Positive clones (erythromycin and chloramphenicol sensitive) were selected and confirmed by PCR and sequencing (GATC-Biotech AG, Konstanz, Germany).
Construction of pTX30::lpl and pCX15::0420–0422
Plasmid pTX30::lpl contained all ten lpl genes under control of the xylose-inducible promoter. pTX30 is a derivative of pTX15 [54] with a strong transcription terminator inserted and a SalI site deleted by partial digestion. Into pTX30 the entire lpl gene cluster (10,529 bp) was inserted stepwise. The first fragment (4,146 bp) comprising the first five lipoprotein genes (USA300 0410–0414) was amplified by using forward primer F_0410 (BamHI) and reversed primer Re_0414 (Sal1). The second fragment (3,537 bp) comprising the 4 next lipoprotein genes (0415–0418) was amplified by using forward primer F_0415 (Sal1) and reversed primer Re 0418 (AvrII). The last fragment (2,892 bp) containing the last lipoprotein gene (USA300-0419) together with the three lpl flanking genes (USA300 0420–0422) were amplified by using forward primer F_0419 (AvrII) and reversed primer R_0422 (SacI). The amplified fragments and plasmid pTX30-mch-cw were cut by using the same digestion enzymes and subsequently ligated together. The ligation products were transformed first into S. carnosus TM300 by the protoplasts method [55] to yield the plasmid pTX30::lpl (17,343 bp), which was then transformed into S. aureus RN4220 and USA300Δlpl.
Plasmid pCX15::0420–0422 contained three genes (SAUSA 0420–0422) which are located at the end of the lpl cluster and are highly conserved. The corresponding DNA fragment (2,020 bp) was amplified by using two primers Fr_0420 and Re_0422 and were inserted between the BamHI and XbaI site of pCX15 [56]. The ligation products were first transformed into S. aureus RN4220 by electroporation; the resulting plasmid, pCX15::0420–0422 (5,912 bp), verified by DNA sequencing was subsequently transformed into S. aureus USA300Δlpl and S. carnosus TM300.
Construction of pTX30::lpl1-his with and without signal sequence
A fragment containing the first lpl gene SAUSA0410, named Lpl1-his, was amplified from the USA300 genome by using primers containing 6xHis-tag codons at the 3' end. We constructed two different lipoproteins Lpl1-his with and without signal sequence. The forward primers of both construction comprises an optimized SD-sequence AGGAGG, downstream a BamHI site, and upstream with the start codon and following the complemented sequence (S2 Table). Two constructed plasmid containing the same reversed primer, Re_Lpl1-his (SacI), comprises a 6-Histag coding sequence (sequence in italics), a stop codon and a SacI site. The amplified fragment was ligated into xylose inducible vector pTX30 after digestion with two BamHI and SacI enzymes to yield the plasmid pTX30::lpl1-his. This plasmid was transformed into S. aureus SA113 by electroporation.
Purification of Lpl1-his
Lpl1-his was isolated from the membrane fraction of S. aureus SA113 (pTX30::lpl1-his). The clone was first cultivated aerobically at 37°C in the absence of xylose (BO-medium) until OD578nm ≈ 0.5 was reached, then 0.5% xylose was added to induce Lpl1 expression and cultivation was continued for 4 h. The bacterial cells were harvested by centrifugation at 4,000 x g at 4°C. The cell pellets were washed two times with Tris buffer (20 mM Tris, 100 mM HCl, pH 8.0). Then the pellet was re-suspended with Tris buffer containing protease inhibitor table (Merck, Darmstadt, Germany) and lysostaphin (30 μg/ml) and incubated at 37°C for 2 h to disrupt the cell wall. Membrane protein extraction and purification were followed according to a previous study [27] with a small modification. After ultracentrifugation (235,000 x g for 45 min at 4°C), membrane proteins were dissolved overnight at 6°C with Tris buffer containing 2% Triton X100. After another ultracentrifugation step, the supernatant was incubated with Ni-NTA super flow beads (Qiagen, Germany) overnight at 6°C under mild rotation 20 rpm. One volume of Ni-NTA beads were washed twice with five volumes of extraction buffer (Tris buffer containing 0.25% TritonX-100 and 20 mM imidazole), subsequently the beads were washed four times with five volumes of the same buffer containing 40 mM imidazole and finally the Lpl1-his was eluted with the same buffer containing 500 mM imidazole. Lpl1-his was concentrated via centrifugal ultra-filter unit with a molecular mass cut-off of 10 kDa (Sartorius AG, Göttingen, Germany). Finally, the Lpl1-his purification was checked by SDS-PAGE and the total protein amount was determined by a Bradford assay kit. Purified Lpl1-his was used for generating polyclonal rabbit antibodies by Biogenes GmbH (Berlin, Germany).
The purification of Lpl1-his (-sp) was performed with the same procedures as Lpl1-his (+sp) except that the Lpl1-his (-sp) isolated from the cytoplasm of S. aureus SA113 by French press.
Determination of endotoxin contamination
The purified lipoprotein (Lpl1-his) stock used for stimulation of cytokine production was tested for endotoxin contamination as 0.19 EU (1 EU≅100 pg LPS) by the Endosafe-PTS system (Charles River, Charleston, USA). Positive reaction is usually due to LPS contamination but might also arise from lipoprotein content.
Localization of Lpl1-his
USA300 was cultivated aerobically in B-medium and cells were harvested after 4, 8 and 14 h. The cells were harvested by centrifugation (4000 x g, for 10 min at 4°C) and the supernatant was filtered with 0.2 μl Millipore filters to remove remaining cells. Two ml of supernatant were mixed with 10 μl StrataClean Resin (Agilent Technology, USA) and incubated for 5 min at room temperature. After centrifugation (12,000 x g, for 10 min, at 20°C), the pelleted resin with bound extracellular proteins was washed twice with 20 mM Tris buffer (pH 8). The membrane fraction was isolated as described above and membrane proteins were precipitated using chloroform and methanol method [57]. The extracellular and membrane proteins were dissolved in SDS running buffer, separated by SDS-PAGE and transferred onto nitrocellulose membrane by using semi-dry transfer apparatus in semidry buffer (25 mM Tris, 150 mM glycine, and 10% methanol) using 350 mA for 80 min. Lpl1-his was detected by Western blot using anti-Lpl1-his rabbit antibodies at a dilution of 1:1000.
Human cells and infection assay
Mono Mac 6 [58], a human monocytic leukemia cell line, was obtained from DSMZ (Braunschweig, Germany) and cultured in RPMI-1640 (Biochrom AG, Berlin, Germany) supplemented with 10% FBS superior (BiochromAG, Berlin, Germany), 1% OPI (O5003, Sigma, Taufkirchen, Germany) in NEA-non essential amino acids NEA (Biochrom AG, Berlin, Germany) and 1% Zell Shield (Minerva Biolabs GmbH, Berlin, Germany) at 37°C with 5% CO2. Prior to stimulation, 106 cells per 24-well microtiter plate were seeded out in 1ml of culture medium and incubated for 1 h at 37°C with 5% CO2 supplement. Mono Mac 6 cells were stimulated with staphylococcal strains with ratio MOI 30:1 or otherwise specifically indicated. Prior to stimulation, staphylococcal strains were cultured overnight in TSB or TSB with 0.8% xylose and washed twice with Dulbecco’s Modified Eagle Medium (DMEM/F-12) containing L-glutamine and 15mM HEPES (Gibco). The time period of stimulation was 4 h for TNF-α or 24 h for IL-6. The supernatants were collected and stored at -20°C until usage. In addition, different amounts of Lpl1-his (200 ng, 500 ng and 1 μg) were applied to 106 Mono Mac 6 cells and their supernatants were collected after 4 h and 24 h incubation for TNF-α and IL-6 measurement, respectively. LPS (Sigma Aldrich, Taufkirchen, Germany), Pam3Cys (P3C), and Pam2Cys (P2C) (EMC, Tuebingen, Germany) were used as controls.
Peripheral human blood mononuclear cells (PBMC) were isolated from buffy coats by Ficoll gradient centrifugation. Culture medium consisting of RPMI 1640 (PAA, Vienna, Austria) supplemented with 1% L-glutamine and 1% penicillin-streptomycin (both from Sigma-Aldrich, Munich, Germany) and 10% FCS (PAA Laboratories, Vienna, Austria) or 2% human antibody positive serum (Lonza, Cologne, Germany) for macrophage cultures. Medium supplements were controlled for endotoxin contamination by absence of TNF induction [59]. Human macrophages were obtained by culturing the adherent cell fraction from PBMC for five days in the presence of GM-CSF (10 ng/ml on day 0 and day 3, Miltenyi Biotec, Bergisch-Gladbach, Germany) or M-CSF (10 ng/ml on day 0 and day 3; Miltenyi Biotec). Cells were stimulated in 24-well plates for 24 hours with bacteria (MOI 10:1) or left unstimulated.
Transfection of adherent HEK293 cells 5x104 per well (200 μL) was performed with lipofectamine (Invitrogen, Karlsruhe, Germany) with or without 200 ng plasmid bearing TLR2 cDNA overnight. After washing, cells were stimulated with TLR2 ligands P3C (100 ng/ml), different amounts of Lpl1-his (50 ng, 100 ng and 500 μg) for 24 hours before harvest of cellular supernatants. TLR2 activity was assessed via quantification of secreted human IL-8 by ELISA (BD OptEIA, BD Biosciences, Heidelberg, Germany).
Cytokine and antimicrobial peptide (AMP) expression in differentiated primary human keratinocytes
With S. aureus it is difficult to determine cytokine concentration by ELISA because of IgG-binding protein A that affects Western and ELISA assays. Therefore, we used frequently transcript analysis as has been carried out previously [60]. Primary keratinocytes were isolated from human foreskin after routine circumcision. Cultivation of the cells was performed in collagen-coated flasks in keratinocytes growth medium (KGM) at 37°C, 5% CO2 as described previously [61]. Prior to infection, the cells were seeded into collagen-coated 24-well plates. For differentiation, medium was changed to keratinocyte base medium (KBM; KGM lacking BPE, supplements and antibiotics) and calcium concentration of KBM medium (0.15 mM) was altered to 1.7 mM. The cells were infected for 8 h with S. aureus USA300 strains with a MOI of 30 (±10). Before stimulation, S. aureus strains (USA300 wild type, USA300Δlpl and complemented mutant USA300Δlpl (pTX30::lpl) were cultured overnight in TSB and washed two times with PBS. Bacterial pellets were resuspended in KBM without any supplements for treatment of the cells. RNA extraction (NucleoSpin RNA, Macherey-Nagel, Düren, Germany) and reverse transcription (SuperScript II Reverse Transcriptase, Invitrogen, Carlsbad, USA) were performed according to the manufacturer's instructions. Quantitative RT-PCR for measurement of gene expression was performed using KAPA SYBR Fast (Peqlab, Erlangen, Germany) with the Roche LightCycler 480 Real-Time PCR system. Relative expression of target genes was calculated as ratio to β-actin as reference gene. Primer sequences are listed in S3 Table.
Detection of cytokines and immunoglobulin by ELISA
Human IL-8, TNF-α, IL-6 and IL-10 secretion was measured in cellular supernatants using the BD OptEIA ELISA kits according to the manufacturer´s instructions.
Murine kidney abscess model
Female BALB/c mice (18–20 g) were purchased from Charles River, Sulzfeld, Germany, housed in polypropylene cages and supplied with food and water ad libitum. S. aureus were cultured for 18 h in B-medium, washed 3 times with sterile PBS and suspended in sterile PBS to the desired concentration. To verify viable cell counts, appropriate dilutions were plated on B agar plates. Mice were inoculated with 100 μl of S. aureus via the tail vein. Eight mice were used for each strain of S. aureus tested. Five days after bacterial challenge, the kidneys were aseptically harvested and used for CFU enumeration. Kidneys were homogenized in 3 ml of sterile PBS using Dispomix (Bio-Budget Technologies GmbH, Krefeld, Germany) and serial dilutions of the organ homogenates were cultured on mannitol salt-phenol red agar plates for at least 48 h at 37°C. Colony-forming units were counted and the bacterial burden was calculated as CFU/kidneys. The statistical significance was determined using the Mann–Whitney test for bacterial burden.
Adherence and invasion of S. aureus USA300 in differentiated primary human keratinocytes and mouse skin
Differentiated primary human keratinocytes in 24-well plates were cultured as described above and were infected with USA300, its Δlpl mutant and the complemented mutant Δlpl (pTX30::lpl) with a MOI of 30. Before stimulation, the strains were cultured overnight in TSB and washed two times with PBS. Bacterial pellets were resuspended in KBM without any supplements and incubated with host cells for 1.5 h. For determination of the number of adhered bacteria, the cells were washed three times and lysed 1.5 h after infection and several dilutions were plated on TSB agar. For detection of invaded bacteria, cells were treated with lysostaphin (Sigma-Aldrich, Taufkirchen, Germany) for additional 1.5 h to remove extracellular bacteria. Keratinocytes were lysed with 0.1% Triton X-100 and 0.5% Trypsin in PBS. Agar plates were incubated overnight at 37°C and the obtained numbers of CFU were normalized to keratinocyte numbers.
For analysis of invasion in mouse skin, epicutaneous infection with S. aureus USA300 was performed. S. aureus strains (USA300, USA300Δtlpp and USA300Δtlpp (pTX30::tlpp) were cultured overnight in TSB. Afterwards they were transferred into pre-warmed TSB and grown to mid-logarithmic phase. Tape-stripping method is an established mouse model to follow the penetration of e.g. S. aureus into epidermal keratinocyte layers and the subsequent dissemination; the experimental infections do produce significant dermal damage, but the latter develops after dissemination has already taken place [62]. Here, C57BL/6 mice (Charles River Laboratories, Sulzfeld, Germany) were shaved and tape-stripping was performed 7 times (strong tape-stripping, STS). An inoculum of 3.5x106 (±1.5x106) bacteria was added to filter paper discs, placed onto the skin and covered by Finn Chambers on Scanpor (Smart Practice, Phoenix, USA). Overnight fixation occurred via Fixomull stretch plaster (BSN medical, Hamburg, Germany). 24 h after infection Finn Chambers and plasters were removed and two skin samples per mouse from the application site were taken. For determination of CFU skin samples were first washed in PBS to remove loosely attached bacteria and then scraped in 1 ml PBS to obtain the number of invaded bacteria. Several dilutions were plated on TSB agar plates and incubated overnight at 37°C. The number of invaded bacteria was calculated as percentage of initially applied CFU onto the skin sample.
Fluorescent microscopy
Staphylococcal strains were transformed with pCtuf-pp-mch [47] prior to incubation with 105 differentiated primary human keratinocyte cells in four-well chamber slides (Omnilab, Bremen, Germany). Adhered and invaded bacteria were microscopically analyzed. The cells were washed three times by PBS before fixing with paraformaldehyde 4% for 10 min. Subsequently, the nuclei of keratinocytes were stained with DAPI (4′,6-diamidino-2-phenylindole; Invitrogen, Karlsruhe, Germany) for 5 min and washed three times with PBS to remove the unspecific DAPI stain. Fluorescent microscopy was performed with Leica DM5500 B Upright microscope. Images were captured with Leica DFC360 FX high-sensitivity monochrome digital camera.
Electron microscopy
S. aureus strains USA300 (wild type) and USA300Δlpl were fixed with 2,5% glutaraldehyde and 1% Osmium tetroxide for 45 minutes each, dehydrated and embedded in Epon 812 according to standard procedures [63]. For better visualization of membranes cells were treated with 0,1% Tannin according to the protocol of Gelderblom et al. [64] prior to dehydration. 70 nm ultrathin sections were cut and stained with 2% uranyl acetate and 1% lead citrate. Preparations were examined in a CEM 902 electron microscope (Zeiss) and micrographs taken with TRS camera using Olympus iTEM 5.1 software.
Transcriptional analysis of lpl cluster
USA300 cultured in BM medium was harvested at 3 and 6 h. Total RNA was isolated following our previous study [65]. The qualification and quantity of RNA were determined by agarose gel electrophoresis and photometric measurements via NanoDrop apparatus. Total RNA was subsequently transferred onto a nylon membrane by using a vacuum blotter for 4 h. The RNA was cross-linked to the membrane by UV for 1 min. As control 16S and 23S rRNA bands were visualized by staining with methylene blue. Digoxigenin-labeled RNA probes of three genes in lpl cluster (USA300 0410, 0417 and 0420) were used to detect gene-specific hybridization by using ChemiDoc apparatus (Bio-Rad). RNA probes were prepared by using PCR with T7 RNA polymerase. The forward primers and reversed primers, the latter containing a T7 RNA polymerase recognition site at the 5’ end, are listed in S4 Table.
Statistical analysis
Student's t-tests or analysis of variance (ANOVA) and Mann-Whitney test were employed when appropriate to compare the difference of means. All the statistical analysis was performed by using SPSS v.19 or GraphPad Prism and the significant level was set at P value of less than 0.05.
Ethics statement
Buffy coats were obtained from the transfusion medicine department of the University of Bonn. Informed written consent for the blood donation is obtained at the transfusion unit. Minors are not allowed to donate blood. We received no personal information nor had access to any other type of information on the donors. The buffy coats remained strictly anonymous. The use of peripheral blood leukocytes isolated from buffy coats for this project was approved by the local ethics committee of the medical faculty of the University of Bonn (approval number 36/12).
The use of human skin tissue was approved by the medical ethical committee of the University of Tübingen and was performed in accordance with the Declaration of Helsinki principles. The use of human skin tissue was approved by the medical ethical committee of the University of Tübingen and was performed in accordance with the Declaration of Helsinki principles. The identification number of this approval is 331/2010BO2. All samples were anonymized and written consent was given to the physician in charge (the routine circumcisions were performed at the Loretto Clinic by Dr. med. Frunder).
The mouse skin belongs to the animal studies that were approved by the Regierungspräsidium Tübingen. During the tape-stripping procedure to prime the mouse skin for S. aureus infection the animals were anesthetized during the procedure. The identification number of this approval is HT1/12. For murine kidney abscess model, all the animal studies were approved by the local government of Franconia, Germany (approval number 55.2–2531.01-06/12) and performed in strict accordance with the guidelines for animal care and experimentation of German Animal Protection Law and the DIRECTIVE 2010/63/EU of the EU.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Stoll H, Dengjel J, Nerz C, Götz F (2005) Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect Immun 73: 2411–2423. 15784587
2. Hashimoto M, Tawaratsumida K, Kariya H, Aoyama K, Tamura T, et al. (2006) Lipoprotein is a predominant Toll-like receptor 2 ligand in Staphylococcus aureus cell wall components. IntImmunol 18: 355–362. 16373361
3. Parcina M, Wendt C, Götz F, Zawatzky R, Zähringer U, et al. (2008) Staphylococcus aureus-induced plasmacytoid dendritic cell activation is based on an IgG-mediated memory response. J Immunol 181: 3823–3833. 18768836
4. Nielsen JB, Lampen JO (1982) Membrane-bound penicillinases in Gram-positive bacteria. J Biol Chem 257: 4490–4495. 6802832
5. Sibbald MJ, Ziebandt AK, Engelmann S, Hecker M, de Jong A, et al. (2006) Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiol Mol Biol Rev 70: 755–788. 16959968
6. Schmaler M, Jann NJ, Ferracin F, Landolt LZ, Biswas L, et al. (2009) Lipoproteins in Staphylococcus aureus mediate inflammation by TLR2 and iron-dependent growth in vivo. J Immunol 182: 7110–7118. doi: 10.4049/jimmunol.0804292 19454708
7. Tsuru T, Kobayashi I (2008) Multiple genome comparison within a bacterial species reveals a unit of evolution spanning two adjacent genes in a tandem paralog cluster. MolBiolEvol 25: 2457–2473.
8. Baba T, Bae T, Schneewind O, Takeuchi F, Hiramatsu K (2008) Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J Bacteriol 190: 300–310. 17951380
9. Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, et al. (2006) Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367: 731–739. 16517273
10. Kuroda M, Ohta T, Uchiyama I, Baba T, Yuzawa H, et al. (2001) Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 357: 1225–1240. 11418146
11. Biswas L, Biswas R, Nerz C, Ohlsen K, Schlag M, et al. (2009) Role of the twin-arginine translocation pathway in Staphylococcus. J Bacteriol 191: 5921–5929. doi: 10.1128/JB.00642-09 19633084
12. Babu MM, Priya ML, Selvan AT, Madera M, Gough J, et al. (2006) A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J Bacteriol 188: 2761–2773. 16585737
13. Enright MC, Robinson DA, Randle G, Feil EJ, Grundmann H, et al. (2002) The evolutionary history of methicillin-resistant Staphylococcus aureus (MRSA). Proc Natl Acad Sci U S A 99: 7687–7692. 12032344
14. Cockfield JD, Pathak S, Edgeworth JD, Lindsay JA (2007) Rapid determination of hospital-acquired meticillin-resistant Staphylococcus aureus lineages. J Med Microbiol 56: 614–619. 17446283
15. Robinson DA, Kearns AM, Holmes A, Morrison D, Grundmann H, et al. (2005) Re-emergence of early pandemic Staphylococcus aureus as a community-acquired meticillin-resistant clone. Lancet 365: 1256–1258. 15811459
16. Herbert S, Ziebandt AK, Ohlsen K, Schafer T, Hecker M, et al. (2010) Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect Immun 78: 2877–2889. doi: 10.1128/IAI.00088-10 20212089
17. Rosenstein R, Götz F (2013) What distinguishes highly pathogenic staphylococci from medium- and non-pathogenic? Curr Top Microbiol Immunol 358: 33–89. doi: 10.1007/82_2012_286 23224647
18. Hilmi D, Parcina M, Bode K, Ostrop J, Schuett S, et al. (2013) Functional variation reflects intra-strain diversity of Staphylococcus aureus small colony variants in the host-pathogen interaction. Int J Med Microbiol 303: 61–69. doi: 10.1016/j.ijmm.2012.12.008 23375466
19. Strommenger B, Bartels MD, Kurt K, Layer F, Rohde SM, et al. (2014) Evolution of methicillin-resistant Staphylococcus aureus towards increasing resistance. J Antimicrob Chemother 69: 616–622. doi: 10.1093/jac/dkt413 24150844
20. Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, et al. (1999) Cell activation and apoptosis by bacterial lipoproteins through toll-like receptor-2. Science 285: 736–739. 10426996
21. Brightbill HD, Libraty DH, Krutzik SR, Yang RB, Belisle JT, et al. (1999) Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 285: 732–736. 10426995
22. Buwitt-Beckmann U, Heine H, Wiesmüller KH, Jung G, Brock R, et al. (2006) TLR1- and TLR6-independent recognition of bacterial lipopeptides. J Biol Chem 281: 9049–9057. 16455646
23. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, et al. (2007) Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 130: 1071–1082. 17889651
24. Takeda K, Takeuchi O, Akira S (2002) Recognition of lipopeptides by Toll-like receptors. J Endotoxin Res 8: 459–463. 12697090
25. Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, et al. (2001) Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol 13: 933–940. 11431423
26. Hashimoto M, Tawaratsumida K, Kariya H, Kiyohara A, Suda Y, et al. (2006) Not lipoteichoic acid but lipoproteins appear to be the dominant immunobiologically active compounds in Staphylococcus aureus. J Immunol 177: 3162–3169. 16920954
27. Müller P, Müller-Anstett M, Wagener J, Gao Q, Kaesler S, et al. (2010) The Staphylococcus aureus lipoprotein SitC colocalizes with Toll-like receptor 2 (TLR2) in murine keratinocytes and elicits intracellular TLR2 accumulation. InfectImmun 78: 4243–4250. doi: 10.1128/IAI.00538-10 20679445
28. Schäffler H, Demircioglu DD, Kuhner D, Menz S, Bender A, et al. (2014) NOD2 stimulation by Staphylococcus aureus-derived peptidoglycan is boosted by Toll-Like Receptor 2 costimulation with lipoproteins in Dendritic Cells. Infect Immun 82: 4681–4688. doi: 10.1128/IAI.02043-14 25156723
29. Bassford PJ Jr., Silhavy TJ, Beckwith JR (1979) Use of gene fusion to study secretion of maltose-binding protein into Escherichia coli periplasm. J Bacteriol 139: 19–31. 110778
30. Müller-Anstett MA, Müller P, Albrecht T, Nega M, Wagener J, et al. (2010) Staphylococcal peptidoglycan co-localizes with Nod2 and TLR2 and activates innate immune response via both receptors in primary murine keratinocytes. PLoS One 5: e13153. doi: 10.1371/journal.pone.0013153 20949035
31. Wright JA, Nair SP (2010) Interaction of staphylococci with bone. Int J Med Microbiol 300: 193–204. doi: 10.1016/j.ijmm.2009.10.003 19889575
32. Menzies BE, Kourteva I (1998) Internalization of Staphylococcus aureus by endothelial cells induces apoptosis. Infect Immun 66: 5994–5998. 9826383
33. Ogawa SK, Yurberg ER, Hatcher VB, Levitt MA, Lowy FD (1985) Bacterial adherence to human endothelial cells in vitro. Infect Immun 50: 218–224. 4044035
34. Sinha B, Francois PP, Nusse O, Foti M, Hartford OM, et al. (1999) Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell Microbiol 1: 101–117. 11207545
35. Bayles KW, Wesson CA, Liou LE, Fox LK, Bohach GA, et al. (1998) Intracellular Staphylococcus aureus escapes the endosome and induces apoptosis in epithelial cells. Infect Immun 66: 336–342. 9423876
36. Dziewanowska K, Patti JM, Deobald CF, Bayles KW, Trumble WR, et al. (1999) Fibronectin binding protein and host cell tyrosine kinase are required for internalization of Staphylococcus aureus by epithelial cells. Infect Immun 67: 4673–4678. 10456915
37. Usui A, Murai M, Seki K, Sakurada J, Masuda S (1992) Conspicuous ingestion of Staphylococcus aureus organisms by murine fibroblasts in vitro. Microbiol Immunol 36: 545–550. 1513269
38. Ellington JK, Reilly SS, Ramp WK, Smeltzer MS, Kellam JF, et al. (1999) Mechanisms of Staphylococcus aureus invasion of cultured osteoblasts. Microb Pathog 26: 317–323. 10343060
39. Jevon M, Guo C, Ma B, Mordan N, Nair SP, et al. (1999) Mechanisms of internalization of Staphylococcus aureus by cultured human osteoblasts. Infect Immun 67: 2677–2681. 10225942
40. Fowler T, Wann ER, Joh D, Johansson S, Foster TJ, et al. (2000) Cellular invasion by Staphylococcus aureus involves a fibronectin bridge between the bacterial fibronectin-binding MSCRAMMs and host cell beta1 integrins. Eur J Cell Biol 79: 672–679. 11089915
41. Dziewanowska K, Carson AR, Patti JM, Deobald CF, Bayles KW, et al. (2000) Staphylococcal fibronectin binding protein interacts with heat shock protein 60 and integrins: role in internalization by epithelial cells. Infect Immun 68: 6321–6328. 11035741
42. Haggar A, Hussain M, Lonnies H, Herrmann M, Norrby-Teglund A, et al. (2003) Extracellular adherence protein from Staphylococcus aureus enhances internalization into eukaryotic cells. Infect Immun 71: 2310–2317. 12704099
43. Hirschhausen N, Schlesier T, Schmidt MA, Götz F, Peters G, et al. (2010) A novel staphylococcal internalization mechanism involves the major autolysin Atl and heat shock cognate protein Hsc70 as host cell receptor. Cell Microbiol.
44. Rosenstein R, Nerz C, Biswas L, Resch A, Raddatz G, et al. (2009) Genome analysis of the meat starter culture bacterium Staphylococcus carnosus TM300. Appl Environ Microbiol 75: 811–822. doi: 10.1128/AEM.01982-08 19060169
45. Albrecht T, Raue S, Rosenstein R, Nieselt K, Götz F (2012) Phylogeny of the staphylococcal major autolysin and its use in genus and species typing. J Bacteriol 194: 2630–2636. doi: 10.1128/JB.06609-11 22427631
46. Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ (2009) Skin immune sentinels in health and disease. Nat Rev Immunol 9: 679–691. doi: 10.1038/nri2622 19763149
47. Mauthe M, Yu W, Krut O, Krönke M, Götz F, et al. (2012) WIPI-1 Positive Autophagosome-Like Vesicles Entrap Pathogenic Staphylococcus aureus for Lysosomal Degradation. Int J Cell Biol 2012: 179207. doi: 10.1155/2012/179207 22829830
48. Löffler B, Tuchscherr L, Niemann S, Peters G (2014) Staphylococcus aureus persistence in non-professional phagocytes. Int J Med Microbiol 304: 170–176. doi: 10.1016/j.ijmm.2013.11.011 24365645
49. Zhang W, Bielaszewska M, Kunsmann L, Mellmann A, Bauwens A, et al. (2013) Lability of the pAA Virulence Plasmid in O104:H4: Implications for Virulence in Humans. PLoS One 8: e66717. 23805269
50. Karch H, Denamur E, Dobrindt U, Finlay BB, Hengge R, et al. (2012) The enemy within us: lessons from the 2011 European Escherichia coli O104:H4 outbreak. EMBO Mol Med 4: 841–848. doi: 10.1002/emmm.201201662 22927122
51. Pizarro-Cerda J, Kuhbacher A, Cossart P (2012) Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb Perspect Med 2.
52. Brückner R (1997) Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS MicrobiolLett 151: 1–8.
53. Leibig M, Krismer B, Kolb M, Friede A, Götz F, et al. (2008) Marker removal in staphylococci via Cre recombinase and different lox sites. ApplEnvironMicrobiol 74: 1316–1323. doi: 10.1055/s-2008-1081293 18622904
54. Peschel A, Götz F (1996) Analysis of the Staphylococcus epidermidis genes epiF,-E, and-G involved in epidermin immunity. J Bacteriol 178: 531–536. 8550476
55. Götz F, Schumacher B (1987) Improvements of protoplast transformation in Staphylococcus carnosus. Fems Microbiology Letters 40: 285–288.
56. Wieland KP, Wieland B, Götz F (1995) A promoter-screening plasmid and xylose-inducible, glucose-repressible expression vectors for Staphylococcus carnosus. Gene 158: 91–96. 7789818
57. Fic E, Kedracka-Krok S, Jankowska U, Pirog A, Dziedzicka-Wasylewska M (2010) Comparison of protein precipitation methods for various rat brain structures prior to proteomic analysis. Electrophoresis 31: 3573–3579. doi: 10.1002/elps.201000197 20967768
58. Ziegler-Heitbrock HW, Schraut W, Wendelgass P, Strobel M, Sternsdorf T, et al. (1994) Distinct patterns of differentiation induced in the monocytic cell line Mono Mac 6. J Leukoc Biol 55: 73–80. 8283142
59. Bekeredjian-Ding I, Roth SI, Gilles S, Giese T, Ablasser A, et al. (2006) T cell-independent, TLR-induced IL-12p70 production in primary human monocytes. J Immunol 176: 7438–7446. 16751389
60. Wanke I, Skabytska Y, Kraft B, Peschel A, Biedermann T, et al. (2013) Staphylococcus aureus skin colonization is promoted by barrier disruption and leads to local inflammation. Exp Dermatol 22: 153–155. doi: 10.1111/exd.12083 23362876
61. Meier F, Nesbit M, Hsu MY, Martin B, Van Belle P, et al. (2000) Human melanoma progression in skin reconstructs: biological significance of bFGF. Am J Pathol 156: 193–200. 10623667
62. Hahn BL, Onunkwo CC, Watts CJ, Sohnle PG (2009) Systemic dissemination and cutaneous damage in a mouse model of staphylococcal skin infections. Microb Pathog 47: 16–23. doi: 10.1016/j.micpath.2009.04.007 19397991
63. Kraus B, Boller K, Reuter A, Schnierle BS (2011) Characterization of the human endogenous retrovirus K Gag protein: identification of protease cleavage sites. Retrovirology 8: 21. doi: 10.1186/1742-4690-8-21 21429186
64. Gelderblom HR, Hausmann EH, Ozel M, Pauli G, Koch MA (1987) Fine structure of human immunodeficiency virus (HIV) and immunolocalization of structural proteins. Virology 156: 171–176. 3643678
65. Saising J, Dube L, Ziebandt AK, Voravuthikunchai SP, Nega M, et al. (2012) Activity of gallidermin on Staphylococcus aureus and Staphylococcus epidermidis biofilms. Antimicrob Agents Chemother 56: 5804–5810. doi: 10.1128/AAC.01296-12 22926575
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 6
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- HIV Latency Is Established Directly and Early in Both Resting and Activated Primary CD4 T Cells
- A 21st Century Perspective of Poliovirus Replication
- Battling Phages: How Bacteria Defend against Viral Attack
- Adenovirus Tales: From the Cell Surface to the Nuclear Pore Complex