
Cdk2 Is Required for p53-Independent G/M Checkpoint Control
The activation of phase-specific cyclin-dependent kinases (Cdks) is associated with ordered cell cycle transitions. Among the mammalian Cdks, only Cdk1 is essential for somatic cell proliferation. Cdk1 can apparently substitute for Cdk2, Cdk4, and Cdk6, which are individually dispensable in mice. It is unclear if all functions of non-essential Cdks are fully redundant with Cdk1. Using a genetic approach, we show that Cdk2, the S-phase Cdk, uniquely controls the G2/M checkpoint that prevents cells with damaged DNA from initiating mitosis. CDK2-nullizygous human cells exposed to ionizing radiation failed to exclude Cdk1 from the nucleus and exhibited a marked defect in G2/M arrest that was unmasked by the disruption of P53. The DNA replication licensing protein Cdc6, which is normally stabilized by Cdk2, was physically associated with the checkpoint regulator ATR and was required for efficient ATR-Chk1-Cdc25A signaling. These findings demonstrate that Cdk2 maintains a balance of S-phase regulatory proteins and thereby coordinates subsequent p53-independent G2/M checkpoint activation.
Published in the journal:
. PLoS Genet 6(2): e32767. doi:10.1371/journal.pgen.1000863
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000863
Summary
The activation of phase-specific cyclin-dependent kinases (Cdks) is associated with ordered cell cycle transitions. Among the mammalian Cdks, only Cdk1 is essential for somatic cell proliferation. Cdk1 can apparently substitute for Cdk2, Cdk4, and Cdk6, which are individually dispensable in mice. It is unclear if all functions of non-essential Cdks are fully redundant with Cdk1. Using a genetic approach, we show that Cdk2, the S-phase Cdk, uniquely controls the G2/M checkpoint that prevents cells with damaged DNA from initiating mitosis. CDK2-nullizygous human cells exposed to ionizing radiation failed to exclude Cdk1 from the nucleus and exhibited a marked defect in G2/M arrest that was unmasked by the disruption of P53. The DNA replication licensing protein Cdc6, which is normally stabilized by Cdk2, was physically associated with the checkpoint regulator ATR and was required for efficient ATR-Chk1-Cdc25A signaling. These findings demonstrate that Cdk2 maintains a balance of S-phase regulatory proteins and thereby coordinates subsequent p53-independent G2/M checkpoint activation.
Introduction
Cdks associate with cyclins to form heterodimers that are sequentially activated during the cell cycle. Metazoan cells have multiple Cdks and cyclins that are temporally regulated [1],[2]. In normal cell cycles, Cdk4 and Cdk6 pair with D-type cyclins during G1, Cdk2 pairs with E- and A-type cyclins during S and G2, and Cdk1 pairs with A- and B-type cyclins during G2 and M. The importance of Cdks in cell cycle transitions was suggested by studies in which expression of dominant negative mutants or introduction of inhibitory antibodies or small molecule inhibitors caused phase-specific cell cycle arrest [3]. However recent genetic studies have called into question the requirement for multiple Cdks [3]–[5]. RNAi-mediated depletion of Cdks in human cells [6] and gene knockouts in mice [7]–[10] showed that Cdk2, Cdk4 and Cdk6 are dispensable for cell cycle progression. Cdk1 can bind D-, E-, and A-type cyclins and functionally substitute for the non-essential Cdks [11]. While it is clear that Cdk1 alone can drive unperturbed cell cycle progression, it remains unclear whether the non-essential Cdks have non-redundant functions in cell cycle responses to stress.
Cdks are targeted by checkpoints that halt the cell cycle in response to DNA damage. Cdk2 is primarily considered a downstream target of the S-phase checkpoint [12],[13]. However Cdk2 can also signal upstream via the phosphorylation of ATRIP, a binding partner of the ATR kinase [14]. Despite this data suggesting a role for Cdk2 in the regulation of checkpoint signaling, conflicting genetic evidence challenges the functional requirement for Cdk2 in DNA damage responses. Both the G1/S and G2/M checkpoints appear to remain fully functional in CDK2−/− mouse embryonic fibroblasts (MEFs) [15],[16]. As Cdk2 and Cdk1 are functionally redundant in supporting DNA replication, it would seem plausible that Cdk1 could similarly substitute for Cdk2 in checkpoint pathways. Such redundancy would account for the lack of apparent checkpoint defects in the mouse CDK2-knockout.
Here we show that Cdk2 uniquely activates the G2/M checkpoint and that this function is masked by the presence of p53, which functions independently to arrest cells in G2 after DNA damage. Unlike the functions of Cdk2 during unperturbed S-phase, the role of Cdk2 in the G2/M checkpoint is non-redundant and cannot be performed by Cdk1.
Results
Stabilization of Cdc25A in Cdk2-deficient cells
Does Cdk2 contribute to human checkpoints? We first tested whether Cdk2 is required for the regulation of Cdc25A, a common target of checkpoint kinases and a critical mediator of cell cycle transitions. Depletion of Cdk2 with siRNA resulted in increased Cdc25A protein levels in human colorectal cancer cells (Figure 1A).
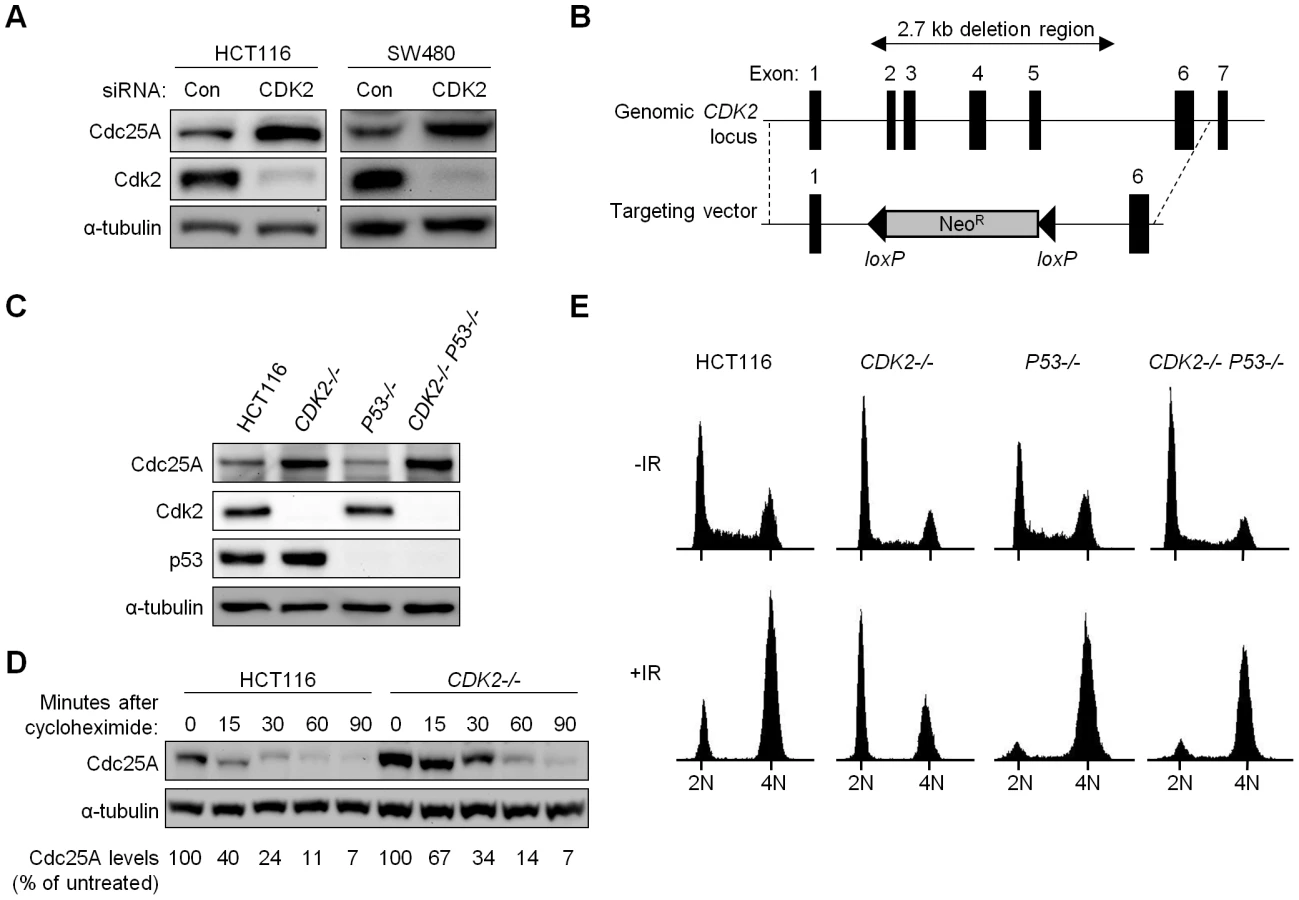
To unambiguously evaluate the role of Cdk2 in checkpoint responses, we disrupted both CDK2 alleles in the human colorectal cancer cell line HCT116 (Figure 1B). HCT116 cells have intact DNA damage-responsive checkpoints [17]–[19] and detailed analysis of these cells has revealed that p53 is required for maintaining stable arrest at G1/S and at G2/M after ionizing radiation (IR) [17]. To compare the contributions of p53 and Cdk2, we disrupted P53 and CDK2 individually to generate P53−/− and CDK2−/− cells, respectively, and together to generate double knockout cells (CDK2−/− P53−/−). Two double knockout clones were obtained in independent experiments. As expected, homozygous disruption of P53 and CDK2 led to loss of protein expression in a genotype-specific manner (Figure 1C). Consistent with the established role of Cdk2 in promoting the G1-S transition, asynchronous CDK2−/− cells exhibited an elevated G1 fraction with fewer cells in S-phase (Figure 1E). Following IR treatment, 60% of CDK2−/− cells arrested at G1/S (Figure 1E), consistent with previous observations of an intact G1/S checkpoint in CDK2−/− MEFs [15],[16]. P53 disruption caused a characteristic loss of the G1/S checkpoint, irrespective of CDK2 genotype (Figure 1E). Stabilization of p53 and the induction of its downstream target p21 after IR were not affected by CDK2 disruption (Figure S1C).
In P53+/+ and P53−/− backgrounds, Cdk2 deficiency resulted in increased Cdc25A (Figure 1C). Cdc25A protein levels are known to be tightly controlled by phosphorylation, in both stressed and unstressed cells [20],[21]. To determine if increased Cdc25A protein following loss of Cdk2 was due to changes in stability, we assessed Cdc25A turnover by treating HCT116 and CDK2−/− cells with the protein synthesis inhibitor cycloheximide. While Cdc25A was degraded by 90 min in CDK2−/− cells, the rate of degradation was decreased (Figure 1D) indicating that Cdk2 contributes to normal Cdc25A protein turnover.
Cdk2 and p53 cooperatively mediate G2/M checkpoint arrest
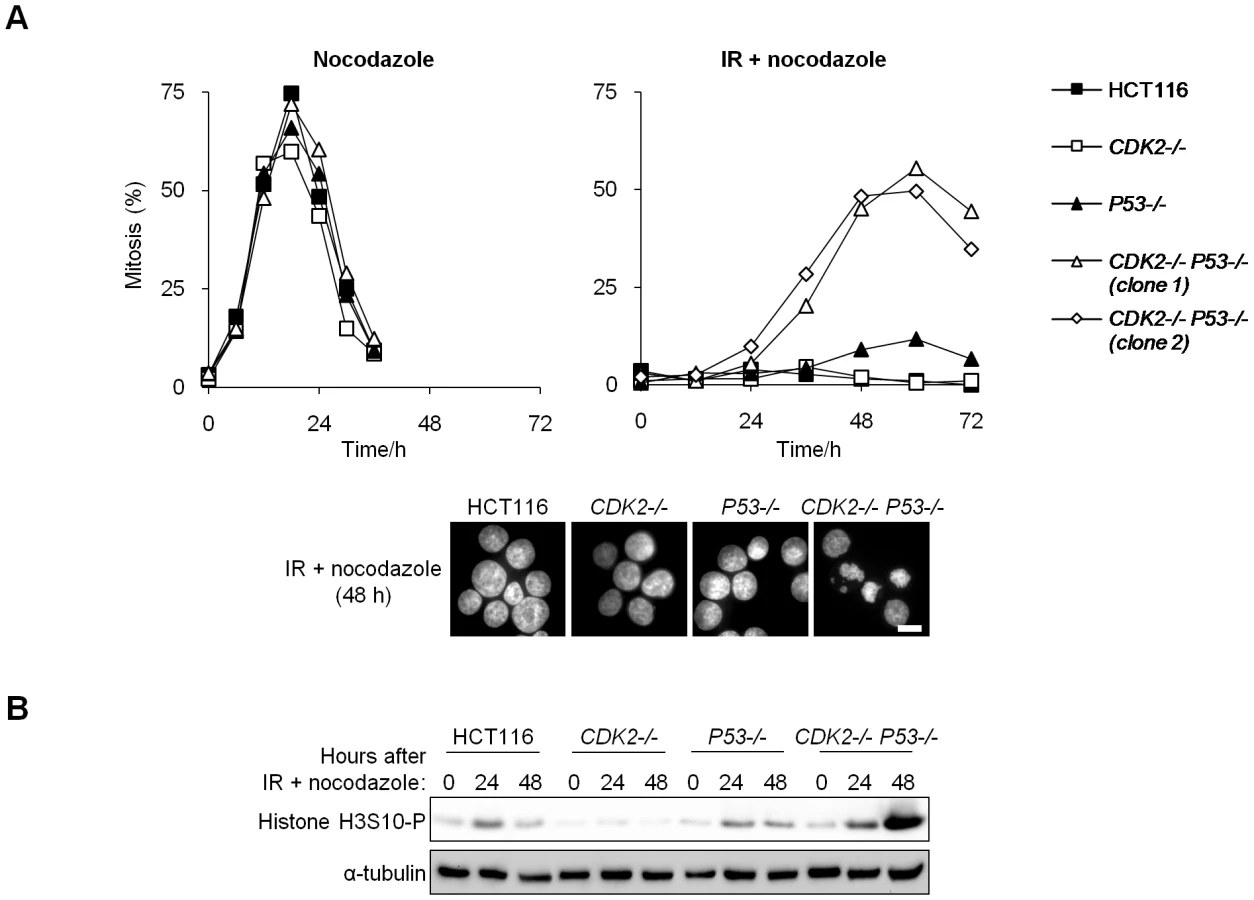
To assess the integrity of the G2/M checkpoint response to DNA double strand breaks, we treated isogenic cultures with IR and trapped the cells that subsequently entered mitosis with the microtubule-destabilizing drug nocodazole. Cells of all genotypes arrested normally in mitosis when treated with nocodazole alone (Figure 2A). p53-deficient cells do not stably arrest at G2/M following IR [17], and therefore exhibited a modest increase in mitotic entry after 48–60 h, compared with wild type cells in which the mitotic index remained below 4% (Figure 2A). The extent of mitotic entry was greatly elevated in double knockout cells (CDK2−/− P53−/−; Figure 2A). Accordingly, the mitotic marker phospho-histone H3 S10 (H3S10-P) was strongly expressed in CDK2−/− P53−/− cells 48 h following IR/nocodazole treatment (Figure 2B). Unirradiated cells entered mitosis within 24 h of the addition of nocodazole (Figure 2A). The temporal delay in the mitotic entry of irradiated double knockout cells compared with unirradiated controls suggests that checkpoint pathways were activated in the absence of Cdk2 and p53, but were apparently insufficient to facilitate stable arrest. This G2/M checkpoint defect was apparent over a range of IR doses (Figure S1A) and could be detected as early as 24 h after IR/nocodazole treatment (Figure 2 and Figure S1A). In contrast, the majority of CDK2 knockout-P53 wild type cells (CDK2−/−) arrested at G1/S after IR treatment, and the remaining subpopulation (about 40%) of these cells arrested at G2/M with 4N DNA content (Figure 1E). A very small number of these 4N cells entered mitosis over the course of the experiment (Figure 2 and Figure S1A). We conclude that Cdk2 plays an important role in G2/M arrest after DNA damage and that the requirement for Cdk2 was masked by the function of p53 at both the G1/S and G2/M checkpoints.
Cdk2-null cells fail to sequester Cdk1 in the cytoplasm after IR
The G2-M transition is controlled in part by Cdk1 localization. In unperturbed cells, Cdk1 is cytoplasmic during interphase and enters the nucleus in prophase to trigger mitosis. After DNA damage, Cdk1 is excluded from the nucleus, thus contributing to arrest in G2 [22],[23].
CDK2−/− MEFs have been reported to exhibit altered Cdk1 localization [15]. In these cells, deregulated Cdk1 localization has been attributed to the redistribution of cyclin E, normally associated with Cdk2, to Cdk1 [15]. Several studies demonstrate a similar redistribution of the nuclear protein cyclin A to Cdk1 in the absence of Cdk2 in both mouse and human cells [11],[15],[24],[25], and such complexes have been shown to promote mitotic entry [26]–[30].
Consistent with these previous studies, the amount of cyclin E and cyclin A associated with Cdk1 was increased in CDK2-knockout human cells (Figure 3A). As Cdk localization is dependent on its partner cyclin [31],[32], we asked whether the changes in Cdk1-cyclin complexes observed in CDK2−/− cells might affect Cdk1 localization.
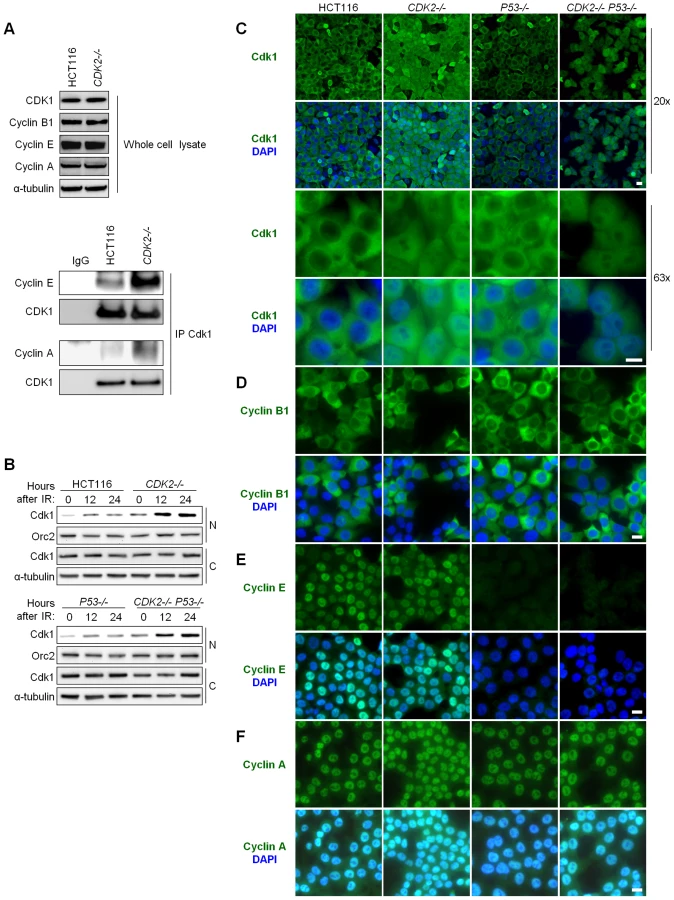
Total Cdk1 protein levels were unaffected by CDK2 genotype or IR (Figure 3A and Figure S1C). After IR treatment, the amount of Cdk1 in the nucleus was increased in CDK2−/− cells compared to wild type cells (Figure 3B). The increase in nuclear Cdk1 was independent of P53 genotype, and temporally preceded entry of double knockout cells into mitosis (Figure 3B). Together, these data suggest that aberrant nuclear Cdk1 was a cause rather than a consequence of defective G2/M checkpoint function in CDK2−/− P53−/− cells. The failure of CDK2-knockout cells to exclude Cdk1 from the nucleus in response to IR was confirmed by immunofluorescence. The localization of Cdk1 in untreated cells was similar in HCT116 and all isogenic derivatives (data not shown). By 24 h after IR, virtually all cells with wild type CDK2 had sequestered Cdk1 in the cytoplasm, while CDK2−/− cells exhibited Cdk1 staining in both the nuclear and cytoplasmic compartments (Figure 3C).
To determine which cyclin partners might contribute to the altered Cdk1 localization in CDK2−/− cells, we examined the localization of cyclin B1, cyclin A and cyclin E after IR. Cyclin B1 was cytoplasmic in all cell lines (Figure 3D), suggesting that aberrant nuclear localization of Cdk1 was not caused by deregulated cyclin B1 localization. In contrast, cyclins E and A were nuclear in checkpoint-proficient P53-wild type cells (Figure 3E and 3F). Cyclin E was barely detectable in P53−/− cells after IR (Figure 3E), presumably because these cells bypass the G1/S checkpoint and progress to G2/M wherein cyclin E is not normally expressed; cyclin A, which is normally expressed from interphase until prometaphase, was located in the nucleus in these cells (Figure 3F). In agreement with studies of CDK2−/− MEFs [15], these findings suggest that the redistribution of cyclin E and cyclin A to Cdk1 results in its aberrant localization to the nucleus after DNA damage.
Deregulation of Cdc25A is a cause of G2/M checkpoint failure
In addition to localization, Cdk1 is also controlled by inhibitory phosphorylation. The Cdc25A phosphatase promotes mitotic entry by removing the inhibitory Y15 phosphate moiety on Cdk1 (Cdk1Y15-P) [33]. This mode of activation is turned off following IR, when Cdc25A is degraded in a Chk1-dependent manner [20]. Cdk1 Y15 phosphorylation is a p53-independent checkpoint mechanism [33].
The increased Cdc25A protein levels in untreated, Cdk2-deficient cells (Figure 1C and 1D) prompted us to examine whether Cdc25A was also aberrantly regulated in response to IR. While Cdc25A was rapidly degraded after IR in wild type cells, Cdc25A levels remained high in cells lacking Cdk2 (Figure 4A). Next, we asked whether the failure to degrade Cdc25A after IR affected the ability of CDK2-knockout cells to induce Cdk1Y15-P and arrest at G2/M. As expected, p53-deficient cells normally induced Cdk1Y15-P after IR (Figure 4B); in contrast, the levels of Cdk1Y15-P did not increase after IR in cells that were also Cdk2-deficient. Interestingly, the phosphorylation of Cdk1 Y15 was increased in untreated Cdk2-deficient cells compared to untreated cells with wild type Cdk2 (Figure 4B). This increase in basal Cdk1Y15-P could be a consequence of the redistribution of Cdk1 to alternative Cdk1-cyclin heterodimers and the consequent expansion of the role of Cdk1 to multiple phases of the cell cycle, in Cdk2-deficient cells (Figure 3A) [11],[15],[24],[25]. It is unknown if non-canonical Cdk1-cyclin heterodimers are efficient substrates for activating phosphatases and inhibitory kinases.
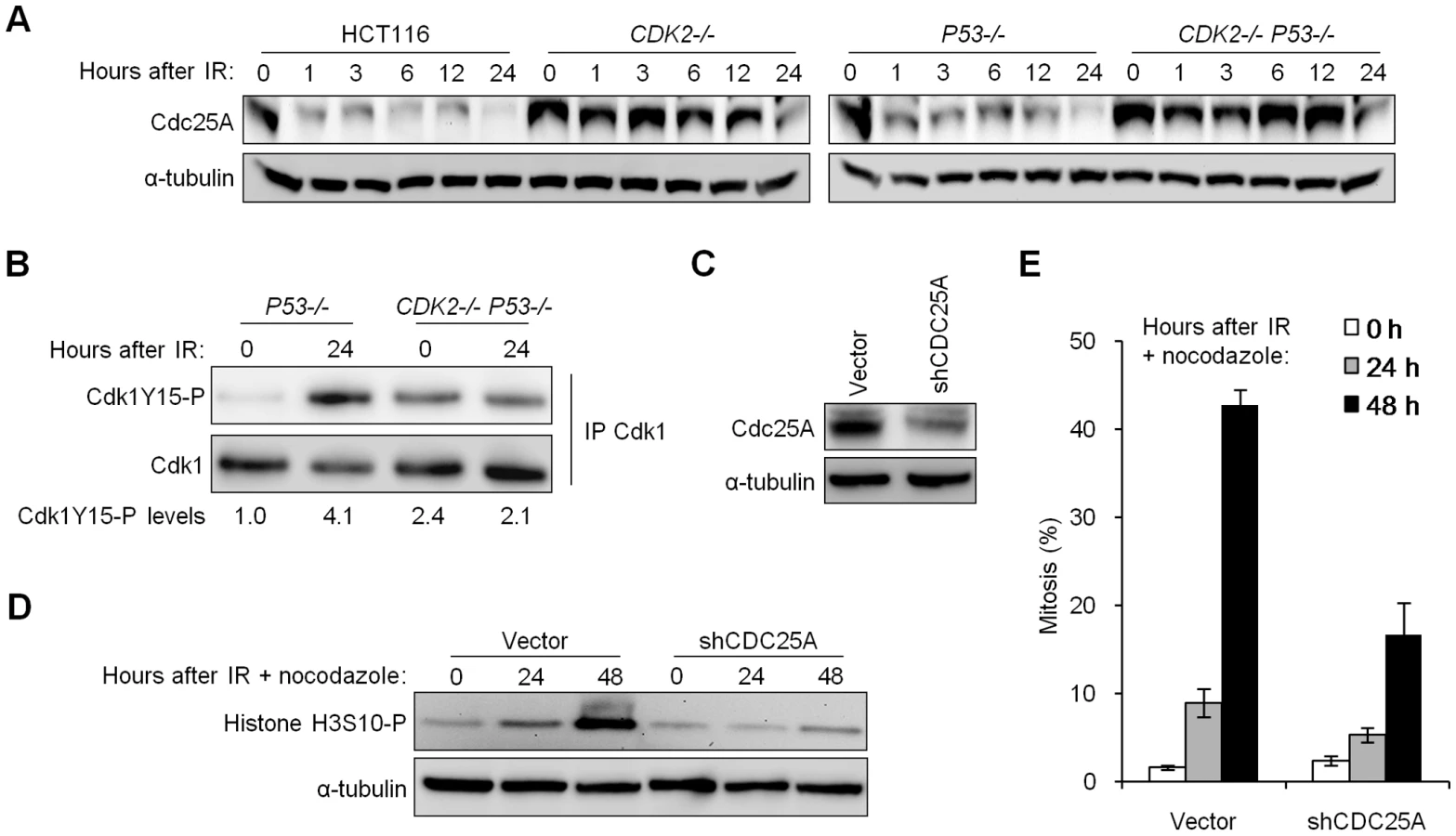
To assess the relevance of increased Cdc25A protein levels to the observed checkpoint defect, we tested whether stable depletion of Cdc25A could restore checkpoint function. Depletion of Cdc25A in CDK2−/− P53−/− cells using short hairpin RNAs (Figure 4C) suppressed histone H3S10-P induction (Figure 4D) and mitotic chromosome condensation (Figure 4E) after IR/nocodazole treatment. Mitotic entry of unirradiated cells was not affected by Cdc25A knockdown (Figure S1B). These results show that elevated Cdc25A contributes significantly to the checkpoint defect.
Cdk2 facilitates ATR-Chk1-Cdc25A pathway activation in part by stabilizing Cdc6
Cdc25A protein stability is regulated by two pathways in response to IR. ATM phosphorylates the checkpoint kinase Chk2 [34], which then triggers Cdc25A degradation [12]. Cdc25A is also targeted for IR-dependent degradation by Chk1 [21], which is activated after phosphorylation by ATR on residues S317 and S345 [35],[36]. The ATR-Chk1 signaling pathway is active at a reduced physiological level during unperturbed cell growth, and regulates basal Cdc25A protein turnover during S-phase [21].
IR-induced phosphorylation of Chk1 S317 and S345 was reduced in CDK2-knockout cells, irrespective of P53 genotype (Figure 5). Levels of IR-induced Chk1 S345 and S317 phosphorylation (Chk1S345-P and Chk1S317-P) were stably high in wild type cells but increased over time in p53-deficient cells. This effect is likely due to the entry of G1/S checkpoint-defective P53−/− cells into S-phase (Figure 1C), when ATR activity is known to increase [37]. RNAi-mediated knockdown of Cdk2 in a diverse panel of human cell lines consistently reduced IR-dependent Chk1 phosphorylation (Figure S2A). In contrast, Chk2 phosphorylation by ATM and the formation of DNA damage foci containing phosphorylated ATM were unaffected by CDK2 genotype (Figure S1C and S1D). These observations suggest that Cdk2 is required for efficient ATR- but not ATM-mediated checkpoint signaling.
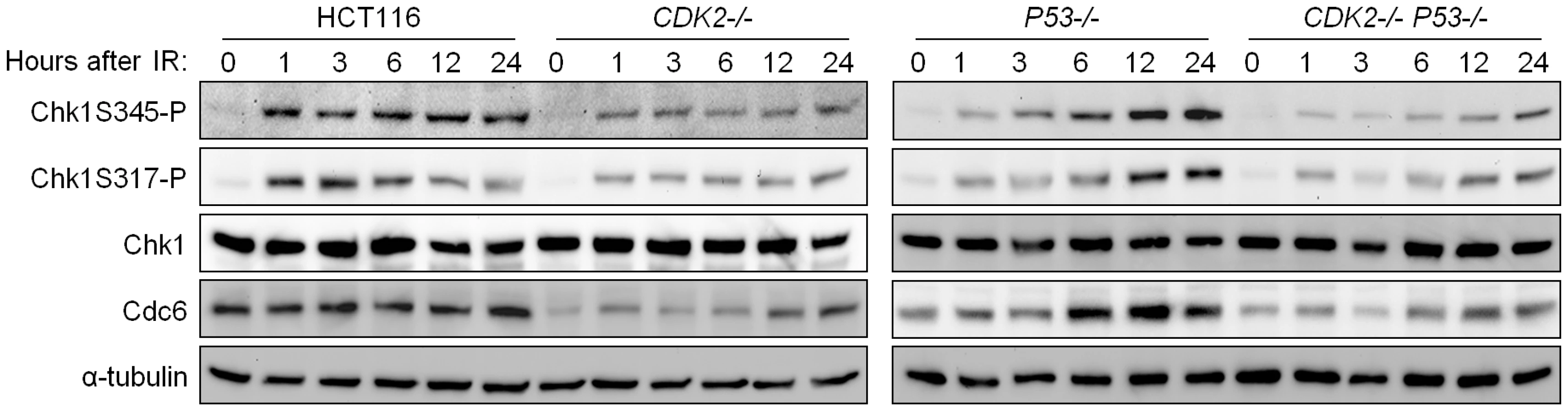
To determine how Cdk2 might control ATR signaling, we first examined the status of ATR localization and known interacting proteins. The ability of ATR to localize to DNA damage foci was unaffected by disruption of CDK2 (Figure S2C), as was the interaction between ATR and ATRIP, the requisite ATR binding partner [38] (Figure S2D). ATRIP is phosphorylated on residue S224 by Cdk2 [14] and this phosphorylation event has been shown to contribute to G2/M checkpoint arrest. We observed that cells lacking Cdk2 exhibited somewhat reduced ATRIP S224 phosphorylation (Figure S2B). Given the modest deficiency in ATRIP phosphorylation, we investigated additional mediators that might also contribute to checkpoint signaling by Cdk2.
One compelling candidate is the Cdk2 substrate Cdc6. Cdc6 is a loading factor for the DNA replicative helicase complex required for replication origin licensing [39]. In addition to its role in DNA replication, Cdc6 has also been implicated as a regulator of checkpoint function and mitotic entry [39]–[44]. In human cells, depletion of Cdc6 causes cells with actively replicating DNA to aberrantly enter mitosis [42], while overexpression of Cdc6 causes Chk1 phosphorylation and G2/M arrest [40]. Cdc6 has also been implicated in ATR-Chk1 signaling in fission yeast [41]and Xenopus [44].
Inherently unstable, Cdc6 can be stabilized as a direct result of phosphorylation by Cdk2 [45]. Although Cdc6 is an essential DNA replication protein, cells lacking functional Cdk2 are able to progress through S-phase despite significantly reduced Cdc6 levels [6],[45]. Therefore the relatively small amount of Cdc6 remaining in Cdk2-deficient cells is clearly sufficient to support DNA replication and cell cycle progression. In concordance with previous studies, Cdc6 protein levels were decreased (Figure 5 and Figure S3A), and turnover was increased (Figure 6A), in Cdk2-deficient cells. In checkpoint-deficient P53−/− cells, Cdc6 levels increased after IR in tandem with Chk1 phosphoprotein (Figure 5). These results are consistent with a potential role for Cdc6 in the regulation of the upstream kinase of Chk1, ATR.
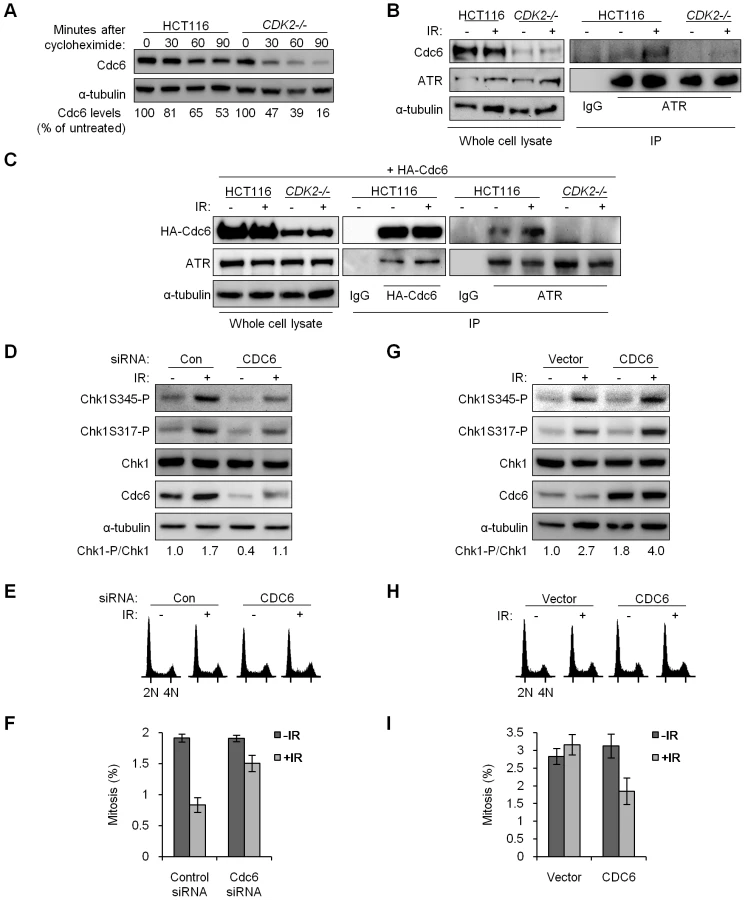
In fission yeast, a direct interaction between the Cdc6 homologue Cdc18 and the ATR homologue Rad3 is induced in response to replication stress; this complex then activates checkpoint signaling [41]. ATR and Cdc6 also interact following replication stress in human cells [43]. We asked whether Cdc6 and ATR might similarly interact after IR. Complexes of endogenous ATR and Cdc6 were detected after IR in HCT116 cells (Figure 6B). To confirm this interaction we exogenously expressed HA-tagged Cdc6 (HA-Cdc6; [45]) in HCT116 and CDK2-knockout cells and probed for an interaction with ATR (Figure 6C). Exogenous expression of HA-Cdc6 was higher in wild type cells (Figure 6C), which mirrored the relative abundance of endogenous Cdc6 and further illustrated the stabilizing effect of Cdk2 (Figure 6B and Figure S3A). ATR co-precipitated with HA-Cdc6, and the amount of ATR bound increased after IR treatment (Figure 6C, Figure S3C and S3D). In the reciprocal experiment, ATR was able to pull down increased HA-Cdc6 after IR treatment in both HCT116 and U2OS cell lines (Figure 6C and Figure S3B). The coimmunoprecipitation of Cdc6 and ATR was not disrupted by 50 µg/ml ethidium bromide, suggesting that this interaction is specific and not simply mediated by DNA (data not shown).
We could not reliably detect ATR-Cdc6 complexes in CDK2−/− cells, before or after IR (Figure 6B and 6C). As Cdc6 stability and protein levels were markedly decreased in CDK2-knockout cells (Figure 6A–6C and Figure S3A), we asked whether the lack of detectable interaction was simply due to reduced Cdc6 protein levels or, alternatively, if loss of Cdk2-dependent phosphorylation on Cdc6 directly disrupted its interaction with ATR. Mutant Cdc6 proteins, wherein the Cdk2-phosphorylated serine residues (S54, S74, S106) were replaced either with non-phosphorylatable alanine residues (HA-Cdc6AAA) or with phosphomimetic aspartic acid residues (HA-Cdc6DDD; [45]), were expressed and pulled down. ATR co-precipitated with HA-Cdc6AAA in wild type HCT116 cells and with HA-Cdc6DDD in CDK2−/− cells (Figure S3C and S3D). These results indicate that the ATR-Cdc6 interaction is independent of Cdc6 phosphorylation by Cdk2 per se, and that the differences in complex formation observed were most likely the result of decreased Cdc6 levels caused by Cdk2 deficiency.
To examine whether Cdk2-mediated stabilization of Cdc6 could functionally contribute to ATR-Chk1 signaling, we experimentally manipulated Cdc6 levels. First, we knocked down Cdc6 by siRNA. Cdc6 protein levels could be transiently lowered in U2OS cells, which express wild type p53, by siRNA transfection (Figure 6D).
The effects of Cdc6 levels on cell growth have been intensively studied. Depending on the extent and timing of Cdc6 depletion and the type of target cell, Cdc6 knockdown has been shown to result in variable changes to cell cycle distribution as well as cell death [42], [46]–[51]. In many cases, transient depletion of Cdc6 in various cell types, including HCT116 [46],[48],[51], has been reported to have minimal effects. Normal cells and cancer cells have been observed to respond differently to Cdc6 knockdown [49],[52], but the genetic alterations in cancer cells that might underlie such differences have not been conclusively identified. We observed that changes to the cell cycle distribution 48 h after partial Cdc6 knockdown were minimal, with a similar proportion of cells in S-phase and a small decrease of cells in G1 (Figure 6E). Knockdown of Cdc6 caused a reduction in IR-induced Chk1 phosphorylation (Figure 6D) that was reminiscent of the observed changes in checkpoint signaling after CDK2 knockdown (Figure S2A) or knockout (Figure 5). While knockdown of Cdc6 was less efficient in HCT116 cells, decreased Cdc6 also led to reduced IR-induced Chk1 phosphorylation, irrespective of P53 genotype (Figure S3E). Knockdown of Cdc6 also led to increased levels of the Chk1 target Cdc25A, before and after IR treatment (Figure S3F and S3G). To determine if Cdc6 protein levels could functionally impact G2/M checkpoint arrest, we assessed mitotic index after CDC6 knockdown followed by sequential treatment with IR and nocodazole. Twenty-four hours after IR/nocodazole treatment, cells pretreated with CDC6 siRNA entered mitosis in higher numbers as compared with control siRNA (Figure 6F).
We next increased Cdc6 levels by transient transfection of an untagged Cdc6 expression construct. While partial restoration of Cdc6 expression in CDK2−/− P53−/− knockout cells did not appreciably alter the cell cycle profile (Figure 6H), it did result in increased IR-induced Chk1 phosphorylation (Figure 6G) and reduced mitotic entry after IR/nocodazole treatment (Figure 6I). Together, the results of these overexpression and knockdown experiments suggest that stabilization of Cdc6 by Cdk2 contributes to efficient IR-induced Chk1 phosphorylation by ATR and p53-independent G2/M checkpoint function. The Cdk2-Cdc6 pathway appears to have a direct affect on ATR-Chk1 signaling, as cell cycle profiles were only minimally changed by Cdc6 manipulation under these conditions (Figure 6E and 6H).
Discussion
Studies of knockout mice have now unequivocally shown that the essential S-phase functions previously attributed to Cdk2 can also be conducted by Cdk1 in somatic cells [3]–[5]. These seminal observations raised the question of why mammalian cells express multiple Cdks that appear to be non-essential. In this report, we demonstrate that loss of Cdk2 alters the regulation of several proteins that are known to regulate S-phase progression, but also control mitotic entry, including Cdc25A, Chk1, Cdc6 and ATRIP [33],[39],[53]. The altered balance of these bifunctional proteins did not affect the transition of CDK2−/− cells through the phases of the unperturbed cell cycle, but did compromise their ability to mount effective checkpoint signaling through the ATR-Chk1 pathway. Cdk2 and Cdk1 are therefore redundant with respect to essential cell cycle functions, but have distinct, non-redundant roles in a key DNA damage response. IR-induced ATR activation is restricted to the S- and G2-phases [37], when Cdk2 is normally active. We show that Cdk2 plays a unique role in facilitating robust DNA damage checkpoint control by the ATR-Chk1-Cdc25A pathway. Cdk2 appears to promote the formation of active ATR complexes in at least two ways: via the phosphorylation of ATRIP and by the stabilization of Cdc6 (Figure 7). It is possible that Cdk2 also controls checkpoint signaling through additional mechanisms such as the recently described Cdk2 interacting protein (CINP) which facilitates robust ATR signaling [54].
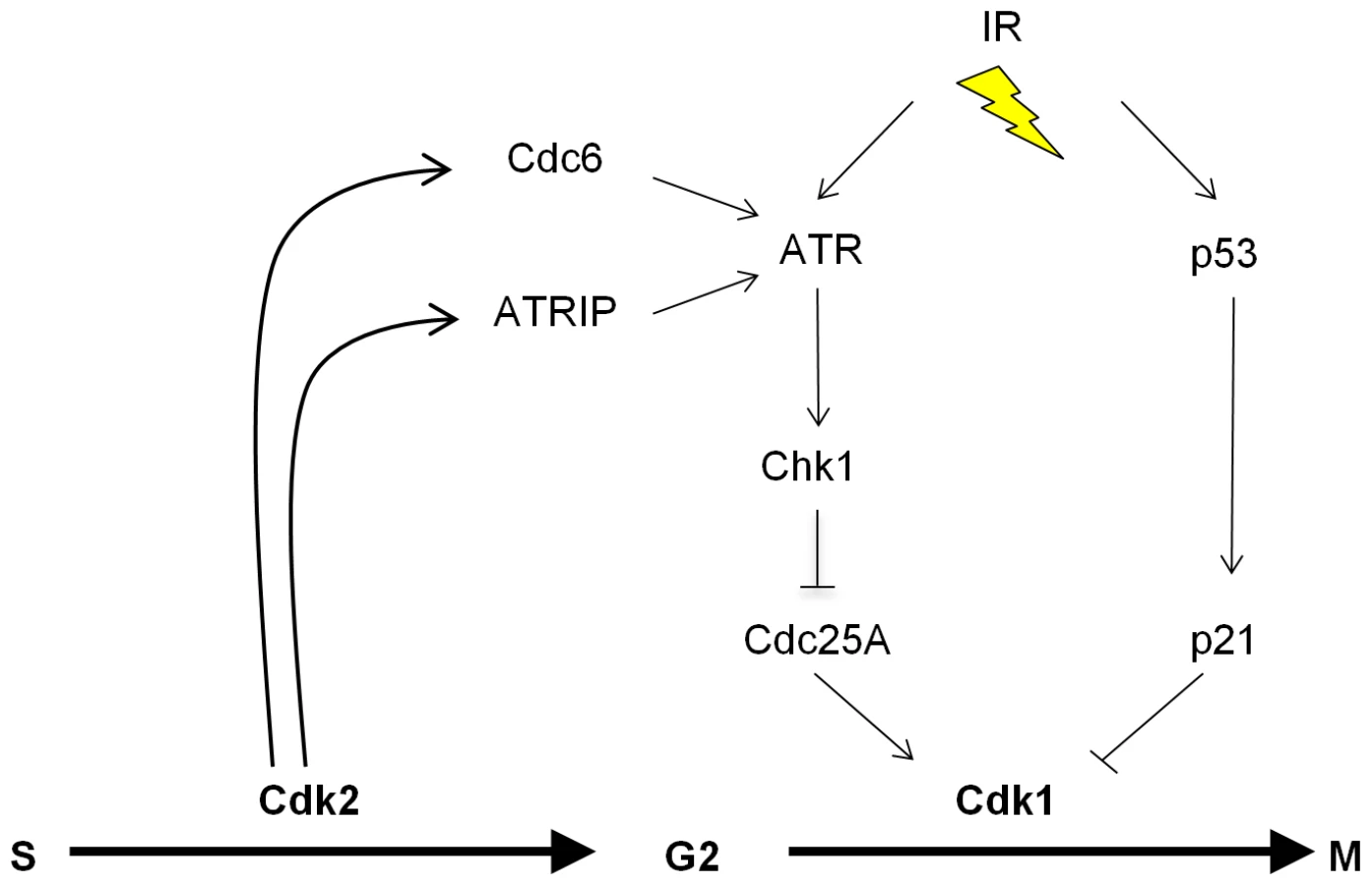
At present the precise mechanism by which Cdc6 affects ATR signaling remains unclear. Our data suggest that Cdk2-mediated phosphorylation of Cdc6 merely regulates Cdc6 levels but is not otherwise required for ATR-Cdc6 complex formation (Figure S3C). In agreement with our results, a paper published during the preparation of this manuscript has reported an interaction between ATR and Cdc6 in human and Xenopus cells [51] and showed that the non-phosphorylatable Cdc6-AAA mutant could interact with ATR, albeit with somewhat lower efficiency. The fission yeast Cdc6 homologue Cdc18 is required to anchor the ATR homologue Rad3 to chromatin [41], but our results suggest that Cdc6 may not perform an analogous function in human cells (Figure S2B). Further study is required to determine if Cdc6 might directly affect ATR catalytic activity, or if Cdc6 might promote the assembly of higher order complexes required for full ATR activation.
In conjunction with checkpoint pathways that target Cdk1 via Cdc25 phosphatases, the exclusion of Cdk1 from the nucleus is an important G2/M checkpoint mechanism [23]. In mouse [15] and human (Figure 3B and 3C) cells lacking Cdk2, Cdk1 becomes aberrantly localized to the nucleus. It would therefore appear that the formation of non-canonical Cdk1-cyclin heterodimers that allow Cdk1 to compensate for Cdk2 in the completion of S phase in unperturbed cells [11],[24],[25] also impairs the ability of damaged cells to arrest at G2/M. We propose that the temporal division of respective S-phase and G2-phase functions between Cdk2 and Cdk1 is a critical feature of the metazoan cell cycle that allows its progress to be efficiently halted after DNA damage.
Defective checkpoints are a feature of the majority of human cancers [55]–[57]. In many cancers, checkpoint deficiencies are caused by loss-of-function mutations in P53. The genetic interaction between P53 and CDK2 described here demonstrates a novel, non-redundant requirement for Cdk2 in the p53-independent G2/M checkpoint pathways that remain intact in cancer cells.
Materials and Methods
Gene targeting
Endogenous CDK2 and P53 loci were disrupted in HCT116 cells using recombinant adeno-associated virus (rAAV)-based gene targeting methods [58],[59]. Briefly, the targeting constructs pAAV-CDK2 and pSEPT-p53 [58] were packaged into infectious rAAV subsequently used to generate transgenic clones. Identification and expansion of homologous recombinant cell lines was performed as described [59]. At least two independent clones were isolated and analyzed for each cell line.
Cell culture, siRNA, and cell cycle analysis
HCT116, SW480 and derivatives were cultured in McCoys 5A supplemented with 6% FCS. U2OS cells were cultured in DMEM supplemented with 10% FCS. CDK2 and CDC6 siRNA pools and non-targeting control pools were purchased from Dharmacon. Transfections were performed with 100 nM siRNA and Lipofectamine 2000 (Invitrogen). In HCT116, optimal Cdk2 knockdown was achieved by two transfections 48 h apart and cells were analyzed 96 h after initial transfection. In SW480, optimal Cdk2 knockdown was achieved by a single transfection and cells were analyzed after 72 h. Cdc6 knockdown was achieved by a single transfection and cells were analyzed after 48 h. IR and nocodazole treatment were performed as described [17]. For cell cycle analysis cells were fixed, stained with Hoechst 33258 and analyzed by flow cytometry or microscopy for mitotic chromosome condensation as described [17]. Mitotic index for U2OS cells were determined by immunofluorescence for histone H3S10-P staining.
Protein analysis
Total cell lysates were prepared using NuPAGE sample buffer (Invitrogen). Non-denatured cell lysates for immunoprecipitation were collected in Cell Lysis Buffer (Cell Signaling). Immunoprecipitations were performed by incubation of lysates with antibody and Protein A/G PLUS-Agarose beads (Santa Cruz) overnight at 4°C. Beads were washed, resuspended and boiled in NuPAGE lysis buffer. Proteins were separated on NuPAGE gels (Invitrogen), transferred to PVDF membranes, probed with antibodies and developed using Enhanced Chemiluminescence (Amersham). Primary antibodies were directed against α-tubulin, ATR, Cdc6, Cdk1, Cdk2, Chk1, cyclin A, cyclin E, cyclin B1, HA, p53 (Santa Cruz), Cdc25A (Neomarkers), ATMS1981-P, Cdk1Y15-P, Chk1S317-P, Chk1S345-P, Chk2T68-P (Cell Signaling), ATRIP, histone H3S10-P (Millipore), Orc2 (BD Biosciences), and ATRIPS224-P (a gift from D Cortez), as indicated. The Quantity One 4.6.1 software package (Bio-Rad) was used for quantitation. For analysis of protein stability, cells were incubated in 100 µg/ml cycloheximide prior to lysis; band intensities were measured and normalized to α-tubulin abundance. Protein levels were expressed as a percentage of untreated control cells.
Cell fractionation and immunofluorescence microscopy
Subcellular fractionation was performed as described [60]. For immunofluorescence, cells were grown on chamber slides and fixed with 3.75% paraformaldehyde/2% sucrose. Fixed cells were permeabilized in 0.2–0.5% Triton X-100 and blocked in BSA. Immunofluorescence staining was performed using Cdk1, cyclin A, cyclin B1, cyclin E or histone H3S10-P antibodies followed by biotin-conjugated secondary antibody (Santa Cruz) and Alexa-488 conjugated avidin (Molecular Probes). Cells were counterstained with 4′-6′-diamidino-2-phenylindole (DAPI) and mounted with Fluoromount-G (Southern Biotech). Images were captured at room temperature using an AxioImager Z1 microscope equipped with an AxioCam HRm camera, Axiovision 4.6.3 software, and a Plan Neofluar 20x/0.25NA, 40x/1.3NA or 63x/1.25NA lens (Zeiss), as indicated. Images were processed for brightness and contrast using Adobe Photoshop.
Retroviral gene transfer
CDC25A shRNA was cloned from pSUPER-Cdc25A [61] into the retroviral plasmid pBabe to generate pBabe-shCDC25A. Retroviral production using Amphopack293 cells (Clontech) and subsequent gene transfer was performed according to protocols supplied by the manufacturer.
Plasmids
Plasmids encoding HA-Cdc6, HA-Cdc6AAA and HA-Cdc6DDD were previously described [45]. To generate the untagged full length Cdc6 construct, human Cdc6 cDNA was cloned into pCDNA3.1/Hygro (Invitrogen). Cells were transfected using Lipofectamine 2000 (Invitrogen) and analyzed after 24–48 h.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MalumbresM
BarbacidM
2005 Mammalian cyclin-dependent kinases. Trends Biochem Sci 30(11) 630 641
2. SherrCJ
RobertsJM
2004 Living with or without cyclins and cyclin-dependent kinases. Genes Dev 18(22) 2699 2711
3. HocheggerH
TakedaS
HuntT
2008 Cyclin-dependent kinases and cell-cycle transitions: Does one fit all? Nat Rev Mol Cell Biol 9(11) 910 916
4. MalumbresM
BarbacidM
2009 Cell cycle, CDKs and cancer: A changing paradigm. Nat Rev Cancer 9(3) 153 166
5. BerthetC
KaldisP
2007 Cell-specific responses to loss of cyclin-dependent kinases. Oncogene 26(31) 4469 4477
6. TetsuO
McCormickF
2003 Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 3(3) 233 245
7. BerthetC
AleemE
CoppolaV
TessarolloL
KaldisP
2003 Cdk2 knockout mice are viable. Curr Biol 13(20) 1775 1785
8. BerthetC
KlarmannKD
HiltonMB
SuhHC
KellerJR
2006 Combined loss of Cdk2 and Cdk4 results in embryonic lethality and rb hypophosphorylation. Dev Cell 10(5) 563 573
9. OrtegaS
PrietoI
OdajimaJ
MartinA
DubusP
2003 Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat Genet 35(1) 25 31
10. MalumbresM
SotilloR
SantamariaD
GalanJ
CerezoA
2004 Mammalian cells cycle without the D-type cyclin-dependent kinases Cdk4 and Cdk6. Cell 118(4) 493 504
11. SantamariaD
BarriereC
CerqueiraA
HuntS
TardyC
2007 Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448(7155) 811 815
12. FalckJ
MailandN
SyljuasenRG
BartekJ
LukasJ
2001 The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410(6830) 842 847
13. FalckJ
PetriniJH
WilliamsBR
LukasJ
BartekJ
2002 The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 30(3) 290 294
14. MyersJS
ZhaoR
XuX
HamAJ
CortezD
2007 Cyclin-dependent kinase 2 dependent phosphorylation of ATRIP regulates the G2-M checkpoint response to DNA damage. Cancer Res 67(14) 6685 6690
15. SatyanarayanaA
HiltonMB
KaldisP
2008 p21 inhibits Cdk1 in the absence of Cdk2 to maintain the G1/S phase DNA damage checkpoint. Mol Biol Cell 19(1) 65 77
16. MartinA
OdajimaJ
HuntSL
DubusP
OrtegaS
2005 Cdk2 is dispensable for cell cycle inhibition and tumor suppression mediated by p27(Kip1) and p21(Cip1). Cancer Cell 7(6) 591 598
17. BunzF
DutriauxA
LengauerC
WaldmanT
ZhouS
1998 Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282(5393) 1497 1501
18. WaldmanT
KinzlerKW
VogelsteinB
1995 p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 55(22) 5187 5190
19. HurleyPJ
WilskerD
BunzF
2007 Human cancer cells require ATR for cell cycle progression following exposure to ionizing radiation. Oncogene 26(18) 2535 2542
20. MailandN
FalckJ
LukasC
SyljuasenRG
WelckerM
2000 Rapid destruction of human Cdc25A in response to DNA damage. Science 288(5470) 1425 1429
21. SorensenCS
SyljuasenRG
FalckJ
SchroederT
RonnstrandL
2003 Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3(3) 247 258
22. ChanTA
HermekingH
LengauerC
KinzlerKW
VogelsteinB
1999 14-3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 401(6753) 616 620
23. PinesJ
1999 Four-dimensional control of the cell cycle. Nat Cell Biol 1(3) E73 9
24. AleemE
KiyokawaH
KaldisP
2005 Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat Cell Biol 7(8) 831 836
25. L'ItalienL
TanudjiM
RussellL
SchebyeXM
2006 Unmasking the redundancy between Cdk1 and Cdk2 at G2 phase in human cancer cell lines. Cell Cycle 5(9) 984 993
26. FungTK
MaHT
PoonRY
2007 Specialized roles of the two mitotic cyclins in somatic cells: Cyclin A as an activator of M phase-promoting factor. Mol Biol Cell 18(5) 1861 1873
27. GongD
PomereningJR
MyersJW
GustavssonC
JonesJT
2007 Cyclin A2 regulates nuclear-envelope breakdown and the nuclear accumulation of cyclin B1. Curr Biol 17(1) 85 91
28. MitraJ
EndersGH
2004 Cyclin A/Cdk2 complexes regulate activation of Cdk1 and Cdc25 phosphatases in human cells. Oncogene 23(19) 3361 3367
29. De BoerL
OakesV
BeamishH
GilesN
StevensF
2008 Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 27(31) 4261 4268
30. FurunoN
den ElzenN
PinesJ
1999 Human cyclin A is required for mitosis until mid prophase. J Cell Biol 147(2) 295 306
31. MooreJD
YangJ
TruantR
KornbluthS
1999 Nuclear import of Cdk/cyclin complexes: Identification of distinct mechanisms for import of Cdk2/cyclin E and Cdc2/cyclin B1. J Cell Biol 144(2) 213 224
32. YangJ
KornbluthS
1999 All aboard the cyclin train: Subcellular trafficking of cyclins and their CDK partners. Trends Cell Biol 9(6) 207 210
33. DonzelliM
DraettaGF
2003 Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep 4(7) 671 677
34. MatsuokaS
RotmanG
OgawaA
ShilohY
TamaiK
2000 Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A 97(19) 10389 10394
35. LiuQ
GuntukuS
CuiXS
MatsuokaS
CortezD
2000 Chk1 is an essential kinase that is regulated by atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14(12) 1448 1459
36. ZhaoH
Piwnica-WormsH
2001 ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21(13) 4129 4139
37. JazayeriA
FalckJ
LukasC
BartekJ
SmithGC
2006 ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 8(1) 37 45
38. CortezD
GuntukuS
QinJ
ElledgeSJ
2001 ATR and ATRIP: Partners in checkpoint signaling. Science 294(5547) 1713 1716
39. BorladoLR
MendezJ
2008 CDC6: From DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis 29(2) 237 243
40. Clay-FarraceL
PelizonC
SantamariaD
PinesJ
LaskeyRA
2003 Human replication protein Cdc6 prevents mitosis through a checkpoint mechanism that implicates Chk1. EMBO J 22(3) 704 712
41. HermandD
NurseP
2007 Cdc18 enforces long-term maintenance of the S phase checkpoint by anchoring the Rad3-Rad26 complex to chromatin. Mol Cell 26(4) 553 563
42. LauE
ZhuC
AbrahamRT
JiangW
2006 The functional role of Cdc6 in S-G2/M in mammalian cells. EMBO Rep 7(4) 425 430
43. LiuL
ChoiJH
YimH
ChoiJS
ParkBD
2009 ATR (AT mutated Rad3 related) activity stabilizes Cdc6 and delays G2/M-phase entry during hydroxyurea-induced S-phase arrest of HeLa cells. Int J Biochem Cell Biol 41(6) 1410 1420
44. OehlmannM
ScoreAJ
BlowJJ
2004 The role of Cdc6 in ensuring complete genome licensing and S phase checkpoint activation. J Cell Biol 165(2) 181 190
45. MailandN
DiffleyJF
2005 CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 122(6) 915 926
46. MelixetianM
BallabeniA
MasieroL
GaspariniP
ZamponiR
2004 Loss of geminin induces rereplication in the presence of functional p53. J Cell Biol 165(4) 473 482
47. KanQ
JinnoS
KobayashiK
YamamotoH
OkayamaH
2008 Cdc6 determines utilization of p21(WAF1/CIP1)-dependent damage checkpoint in S phase cells. J Biol Chem 283(26) 17864 17872
48. KanQ
JinnoS
YamamotoH
KobayashiK
OkayamaH
2008 ATP-dependent activation of p21WAF1/CIP1-associated Cdk2 by Cdc6. Proc Natl Acad Sci U S A 105(12) 4757 4762
49. LauE
ChiangGG
AbrahamRT
JiangW
2009 Divergent S phase checkpoint activation arising from prereplicative complex deficiency controls cell survival. Mol Biol Cell 20(17) 3953 3964
50. NevisKR
Cordeiro-StoneM
CookJG
2009 Origin licensing and p53 status regulate Cdk2 activity during G(1). Cell Cycle 8(12) 1952 1963
51. YoshidaK
SugimotoN
IwahoriS
YugawaT
Narisawa-SaitoM
2010 CDC6 interaction with ATR regulates activation of a replication checkpoint in higher eukaryotic cells. J Cell Sci 123(Pt 2) 225 235
52. FengD
TuZ
WuW
LiangC
2003 Inhibiting the expression of DNA replication-initiation proteins induces apoptosis in human cancer cells. Cancer Res 63(21) 7356 7364
53. CimprichKA
CortezD
2008 ATR: An essential regulator of genome integrity. Nat Rev Mol Cell Biol 9(8) 616 627
54. LovejoyCA
XuX
BansbachCE
GlickGG
ZhaoR
2009 Functional genomic screens identify CINP as a genome maintenance protein. Proc Natl Acad Sci U S A 106(46) 19304 19309
55. HalazonetisTD
GorgoulisVG
BartekJ
2008 An oncogene-induced DNA damage model for cancer development. Science 319(5868) 1352 1355
56. HarperJW
ElledgeSJ
2007 The DNA damage response: Ten years after. Mol Cell 28(5) 739 745
57. KastanMB
BartekJ
2004 Cell-cycle checkpoints and cancer. Nature 432(7015) 316 323
58. TopalogluO
HurleyPJ
YildirimO
CivinCI
BunzF
2005 Improved methods for the generation of human gene knockout and knockin cell lines. Nucleic Acids Res 33(18) e158
59. RagoC
VogelsteinB
BunzF
2007 Genetic knockouts and knockins in human somatic cells. Nat Protoc 2(11) 2734 2746
60. SmitsVA
ReaperPM
JacksonSP
2006 Rapid PIKK-dependent release of Chk1 from chromatin promotes the DNA-damage checkpoint response. Curr Biol 16(2) 150 159
61. van VugtMA
BrasA
MedemaRH
2004 Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Mol Cell 15(5) 799 811
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 2
- Souvislost haplotypu M2 genu pro annexin A5 s opakovanými reprodukčními ztrátami
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Příjem alkoholu a menstruační cyklus
Nejčtenější v tomto čísle
- Genome-Wide Association Study in Asian Populations Identifies Variants in and Associated with Systemic Lupus Erythematosus
- Nuclear Pore Proteins Nup153 and Megator Define Transcriptionally Active Regions in the Genome
- The Genetic Interpretation of Area under the ROC Curve in Genomic Profiling
- Nucleoporins and Transcription: New Connections, New Questions