
Inhibition of mTORC1 Enhances the Translation of Chikungunya Proteins the Activation of the MnK/eIF4E Pathway
The ongoing chikungunya epidemic outbreak in the Caribbean, Central and South America highlights how poor is our understanding of CHIKV pathogenesis and the urgent need for new strategies that may limit CHIKV spread. Immunological studies have suggested that dissemination of infection is largely determined by early events of viral-host cell interactions. In our prior study, we investigated the role of type I interferon responses and the autophagy pathway as mediators of viral control. Here, we evaluated the role of mTOR, making the surprising discovery that inhibition of mTORC1 enhances viral protein translation independently of type I IFN and autophagy. While the inhibition of mTORC1 has no impact on viral binding or entry, we observed an increased translation of both structural and nonstructural viral proteins. Interestingly, the positive impact of mTORC1 inhibition is restricted to viral proteins, as compared to host cap-dependent protein translation that remains suppressed. Further analysis demonstrates that this bypass pathway is mediated the activation of PI3K and MnKs, which in turn hyper-phosphorylate eIF4E, a critical initiation protein for translation. Notably, CHIKV replication enables this pathway as a means to efficiently replicate. Thus, our study provides an unexpected role for mTORC1 in the control of CHIKV infection and highlights a new strategy by which the expression of CHIKV proteins can bypass and/or use the inhibition of mTORC1.
Published in the journal:
. PLoS Pathog 11(8): e32767. doi:10.1371/journal.ppat.1005091
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005091
Summary
The ongoing chikungunya epidemic outbreak in the Caribbean, Central and South America highlights how poor is our understanding of CHIKV pathogenesis and the urgent need for new strategies that may limit CHIKV spread. Immunological studies have suggested that dissemination of infection is largely determined by early events of viral-host cell interactions. In our prior study, we investigated the role of type I interferon responses and the autophagy pathway as mediators of viral control. Here, we evaluated the role of mTOR, making the surprising discovery that inhibition of mTORC1 enhances viral protein translation independently of type I IFN and autophagy. While the inhibition of mTORC1 has no impact on viral binding or entry, we observed an increased translation of both structural and nonstructural viral proteins. Interestingly, the positive impact of mTORC1 inhibition is restricted to viral proteins, as compared to host cap-dependent protein translation that remains suppressed. Further analysis demonstrates that this bypass pathway is mediated the activation of PI3K and MnKs, which in turn hyper-phosphorylate eIF4E, a critical initiation protein for translation. Notably, CHIKV replication enables this pathway as a means to efficiently replicate. Thus, our study provides an unexpected role for mTORC1 in the control of CHIKV infection and highlights a new strategy by which the expression of CHIKV proteins can bypass and/or use the inhibition of mTORC1.
Introduction
Since 2005 there has been a recurrence of Chikungunya disease, with the initial outbreak occurring in the French territory La Réunion [1]. The epidemic has spread worldwide, with outbreaks in five continents [2,3]. Notably, in just nine months during, Chikungunya virus (CHIKV) spread to 22 countries in the Caribbean, Central and South America, resulting in hundreds of thousands of cases [4]. The treatment of CHIKV infections relies on symptomatic relief, as no effective anti-viral agents are available [3]. We therefore set out to investigate cellular pathways that regulate CHIKV replication and spread.
Like other alphaviruses, CHIKV contains a single positive stranded RNA genome of approximately 11.5 kB [5]. The genomic RNA is capped and polyadenylated, and encodes two open reading frames (ORFs). The 5' ORF encodes four nonstructural proteins that participate in genome replication [6]. It is expressed via cap-dependent translation as an nsP1–3 or nsP1–4 polyprotein that is cleaved by the nsP2-encoded protease. The structural proteins are encode by a single ORF within the subgenomic region and is also translated via a cap-dependent mechanism [7]. As observed for other alpahviruses (e.g., Sindbis), CHIKV infection induces several cell stress responses, which might be associated with pathogenesis [8]. In our previous work, we showed that cells infected by CHIKV exhibit phenotypic characteristics of oxidative stress, endoplasmic reticulum stress, interferon induction, autophagy and apoptosis [9]. Notably, these adaptations are due, in part, to modification of the regulator kinase mTOR.
The mammalian target of Rapamycin (mTOR) coordinates cellular catabolic and anabolic processes to promote growth, proliferation and survival signals [10]. mTOR elicits its pleiotropic functions in the context of two functionally distinct signaling complexes, termed mTOR complex 1 (mTORC1) and complex 2 (mTORC2). mTORC1, which contains mTOR, mLST8/GβL, Raptor and PRAS40, plays a key role in cap-dependent translation initiation by directly phosphorylating p70 S6 kinase 1 (S6K1) and eIF4E-binding protein 1 (4E-BP1), and is sensitive to Rapamycin [10]. S6K1 phosphorylate several proteins that are associated with mRNA translation or its control, including ribosomal protein S6 and eukaryotic initiation factor 4B (eIF4B) [10]. The 4E-BP1 are small phosphoproteins which bind to eIF4E at a site that overlaps its interaction site for eIF4G, preventing the formation of eIF4F complex essential for the initiation of capped mRNA [10]. mTORC2 shares mTOR and mLST8/GβL with mTORC1, but possesses three unique components, namely, rictor, mSin1 and PRR5/Protor [11]. Despite the presence of mTOR, mTORC2 is considerably less susceptible to Rapamycin inhibition [10].
As a master sensor of cellular homeostatic perturbations, several studies have investigated the relationship between mTOR activity and viral infection. Numerous viruses modify the activity of mTOR (or mTOR pathways) [12–14]. Regulation of mTOR induces virus-specific effects that are often with opposing action. For example, blocking of mTOR by Rapamycin or TORISEL (referred to be the class of drugs known as Rapalog) inhibit the replication of HCMV [12]; yet Rapalog treatment facilitates HEV replication [13]. During HCV infection of liver cells, activation of PI3K-PKB-mTOR mediates both viral supportive functions (e.g., prevention of apoptosis in HCV-infected cells), and the production of antiviral interferon [14,15].
Regarding CHIKV infection, we previously observed that transient inhibition of the mTORC1 pathway during the first hour of infection [9]. While we demonstrated that this transient inhibition of mTOR correlates with CHIKV-induced autophagy, the direct role of mTOR activity on CHIKV replication remained unknown. Herein, we report that inhibition of mTORC1 enhances CHIKV replication in a cell-intrinsic manner and promotes in vivo spread and worsening of disease. Moreover, we show that mTORC1 impacts CHIKV infection independently of type I IFN production and autophagy. Instead, its actions directly effect translation of viral proteins via the activation of the MnK/eIF4E pathway. These results reveals a role for mTORC1 as a host defense mechanism that limits CHIKV replication and highlights a new strategy by which the expression of CHIKV proteins can bypass the inhibition of mTORC1.
Results
mTORC1 regulates the magnitude of CHIKV infection
To determine the relationship between CHIKV infection and mTOR activity, mouse embryonic fibroblast (MEFs) cells were transfected with siRNA to suppress mTOR expression (Fig 1A), followed by infection with CHIKV (CHIKV-21, the La Réunion 2005 strain). Surprisingly, we observed a 3 fold increase in the number of infected cells as compared to control siRNA treatment (Fig 1B and 1C). Similar findings were found across different doses of input virus, following both the percentage of E2+ cells and extracellular viral load (Fig 1C and 1D). To confirm the impact of mTOR activity on CHIKV infection, we tested PP242, an ATP-competitive mTOR inhibitor that specifically targets the active site of mTOR kinase, and again observed enhanced CHIKV infection (S1A and S1B Fig). These results suggest that mTOR activity restricts CHIKV infection, and highlighted an important paradox. While viral mRNA employs cap-dependent translation to replicate [7], CHIKV infection is actually increased when mTOR is inhibited.
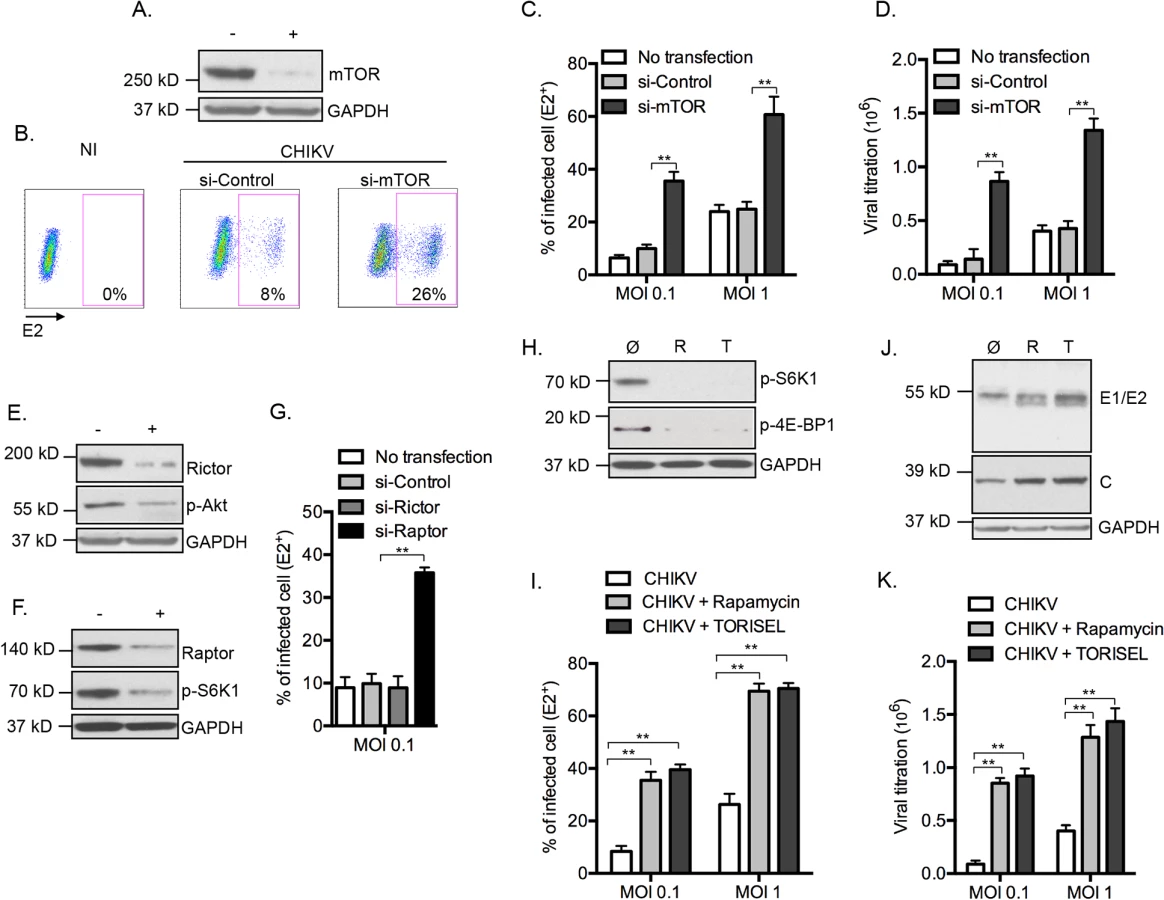
To ascertain the mTOR molecular complex responsible for these findings, we selectively inhibited mTORC1 and mTORC2 by silencing raptor or rictor, respectively (Fig 1E and 1F). Inhibition of gene expression of raptor, but not rictor, recapitulated the enhanced CHIKV infection (Fig 1G). Confirming these results, we observed enhanced E2+ cells, increased expression of CHIKV proteins and higher extracellular viral load when cells were treated with the mTORC1 inhibitors Rapamycin or TORISEL (Fig 1H–1K). Importantly, a similar outcome was observed in cells with reduced expression of rictor (S2 Fig), demonstrating that Rapalog affected CHIKV infection independently of mTORC2. Together, these data indicate that inhibition of mTORC1, but not mTORC2, enhances CHIKV infection in vitro.
Using a complementary approach, we next evaluated the impact of mTORC1 on CHIKV infection by enhancing mTORC1 activity. This was achieved by inhibiting the expression of tuberous sclerosis 2 (TSC2) gene, a physiologic inhibitor of mTORC1 (S3A Fig). When cells were treated with tsc2 siRNA, the percentage of E2+ cells was decreased, as compared to cells treated with si-control (S3B Fig). This was validated using real time microscopy, which indicated that reduced gene expression of tsc2 significantly restricted CHIKV propagation (S3C Fig). As an additional control for potential off-target effects, we demonstrated that Rapalog exposure overcame the inhibitory effect observed in tsc2 si-RNA treated cells (S3D Fig). These results provide evidence for mTORC1 as an antiviral mechanism in the context of CHIKV infection. Following these results, we were interested to examine the possible interplay between mTORC1 and two effector pathways that were previously shown to impact CHIKV replication: type I IFN production; and autophagy initiation.
Effect of mTORC1 on CHIKV infection is independent of type I interferon production
CHIKV induces rapid production of type I interferon (IFN) [16]. As several groups have reported that mTOR regulate interferon expression and mRNA translation of IFN-stimulated genes (ISGs) [17,18], we tested if mTORC1 inhibition increased CHIKV infection in a type I IFN dependent manner. We investigated the impact of Rapalog treatment in MEF deficient for both interferon regulatory factor (IRF) 3 and IRF7, two proteins essential for the production of type I IFN in CHIKV infected MEFs [16]. Remarkably, we observed that even in absence of irf3 and irf7 (irf3-/- / irf7-/-), Rapamycin or TORISEL treatment resulted in increased CHIKV infection (S4A Fig). Similar results were obtained using interferon-α/β receptor (IFNAR) deficient MEF (S4A Fig).
We also analyzed the impact of mTORC1 inhibition on NF-κB-mediated inflammation. Indeed, Rapalog treatment did not perturb the expression levels of IκBα or the activation state of NF-κB (p-p65) (S4B Fig). Moreover, NF-κB-mediated cytokines were secreted at a similar level in untreated or TORISEL-treated cells, supporting that mTORC1 inhibition did not influence NF-κB pathways (S4C Fig). To assess other potential host response pathways, we queried whether the TORISEL-mediated enhancement of CHIKV infection is dependent on host cell transcription. Infected cells were exposed to TORISEL in the presence or absence of actinomycin D (ActD). Remarkably, the fold induction of CHIKV-GFP expression stimulated by TORISEL was unaffected by inhibition of transcription (S4D and S4E Fig). Based on these data, we excluded type I IFN or other host induced immune responses as the mechanism by which mTORC1 regulates CHIKV infection.
Effect of mTORC1 on CHIKV infection is independent of autophagy
mTORC1 is a well-known inhibitor of macroautophagy (referred to as autophagy), a bulk degradation pathway that controls clearance and recycling of intercellular constituents for the maintenance of cellular survival [19]. Previously, we and others showed that CHIKV-induced autophagy serves as a mechanism of host defense by favoring cell survival and thereby restricting viral spread [9,20]. To test the hypothesis that mTORC1 enhances CHIKV infection in an autophagy dependent manner, we assessed viral infection in MEFs deficient for key autophagy genes Atg5 (autophagy-related gene 5) or Atg7. Interestingly, autophagy deficient cells still exhibited increased infection following Rapamycin or TORISEL treatment (S5A–S5D Fig). Expression of Atg5 and Atg7 genes were also silenced in human foreskin fibroblastic cell line (HFF), with results indicating that the mTOR phenotype is indeed independent of autophagy and that the effect is not species specific (S5E–S5G Fig). Of note, while segregated from the mTOR effect, we did confirm that inhibition of autophagy genes significantly increased CHIKV infection, as previously reported [9,21].
Rapalog treatment results in enhanced CHIKV disease
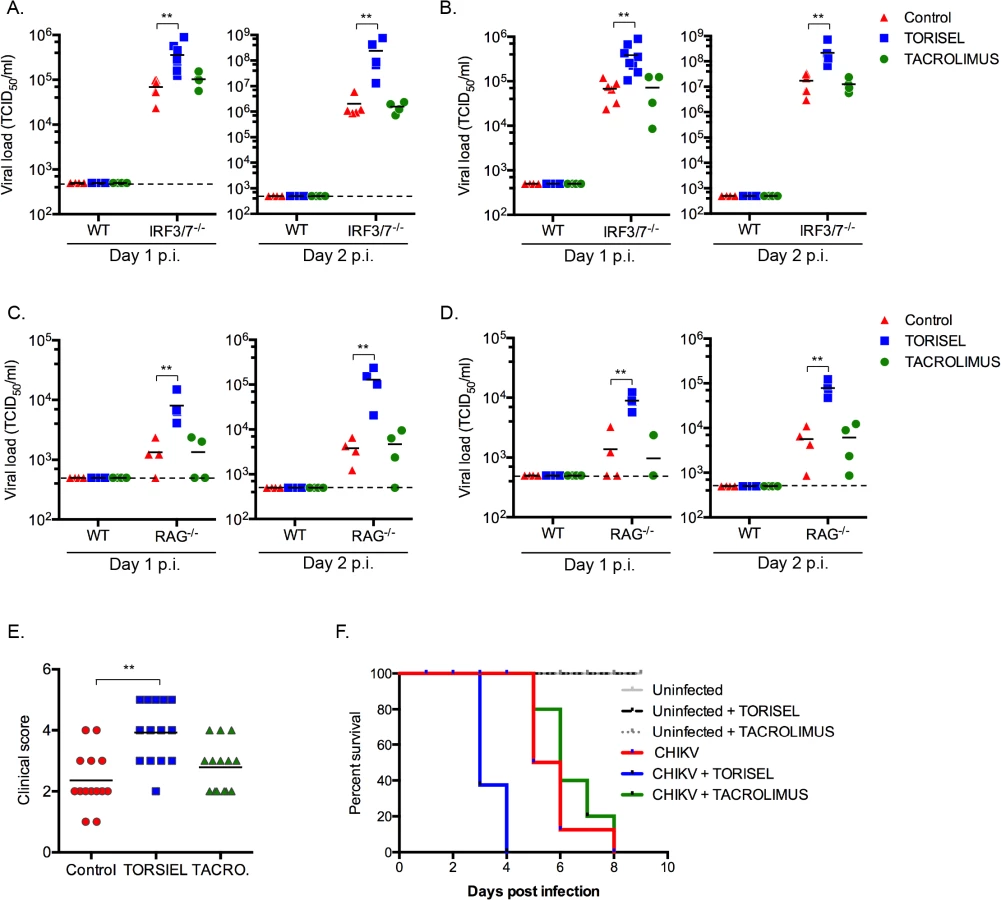
To study the impact of rapalog treatment on in vivo CHIKV pathogenesis we used mice lacking the ability to produce type I IFNs, as wild type (WT) adult animals are resistant to severe forms of infection [22]. With the knowledge that mTOR acts independently of type I IFN expression, it was possible to employ irf3-/- x irf7-/- double deficient mice as a means to evaluate the impact of mTOR inhibition. This mouse strain is highly sensitive to CHIKV infection, with adult mice succumbing by day 6 post-infection [21]. After 8 days of intra-peritoneal injection of TORISEL (10mg/kg, injected every 2 days), irf3-/- / irf7-/-mice were infected by CHIKV and tissues were analyzed at day 1 and 2 post-infection. We first validated that TORISEL inhibited mTOR activity in the skin and muscle of treated mice (S6 Fig). Consistent with our in vitro data, mice pre-treated with TORISEL had higher viral titers in both the skin (injection site) and muscle as compared to control mice, indicating that mTOR inhibition affects in vivo viral infection (Fig 2A and 2B). As one experimental caveat concerns the immunosuppressive effects of TORISEL, we nonetheless examined T and B cell inhibition as a confounding factor in our experiments. Notably, treatment with tacrolimus, a related FKBP-interacting immunosuppressive drug, did not affect viral titers in infected tissue (Fig 2A and 2B). These observations were further confirmed using mice deficient for rag2 (rag2-/-), and therefore lacking mature lymphocytes (Fig 2C and 2D). Finally, we observed a worsening of disease in TORSIEL treated animals, and a more rapid time to death of infected mice (Fig 2E and 2F). These experiments highlight the marked outcome of mTORC1 inhibition and enhanced viral replication, which lead to exacerbation of chikungnuya disease.
mTORC1 specifically influences the CHIKV replication phase
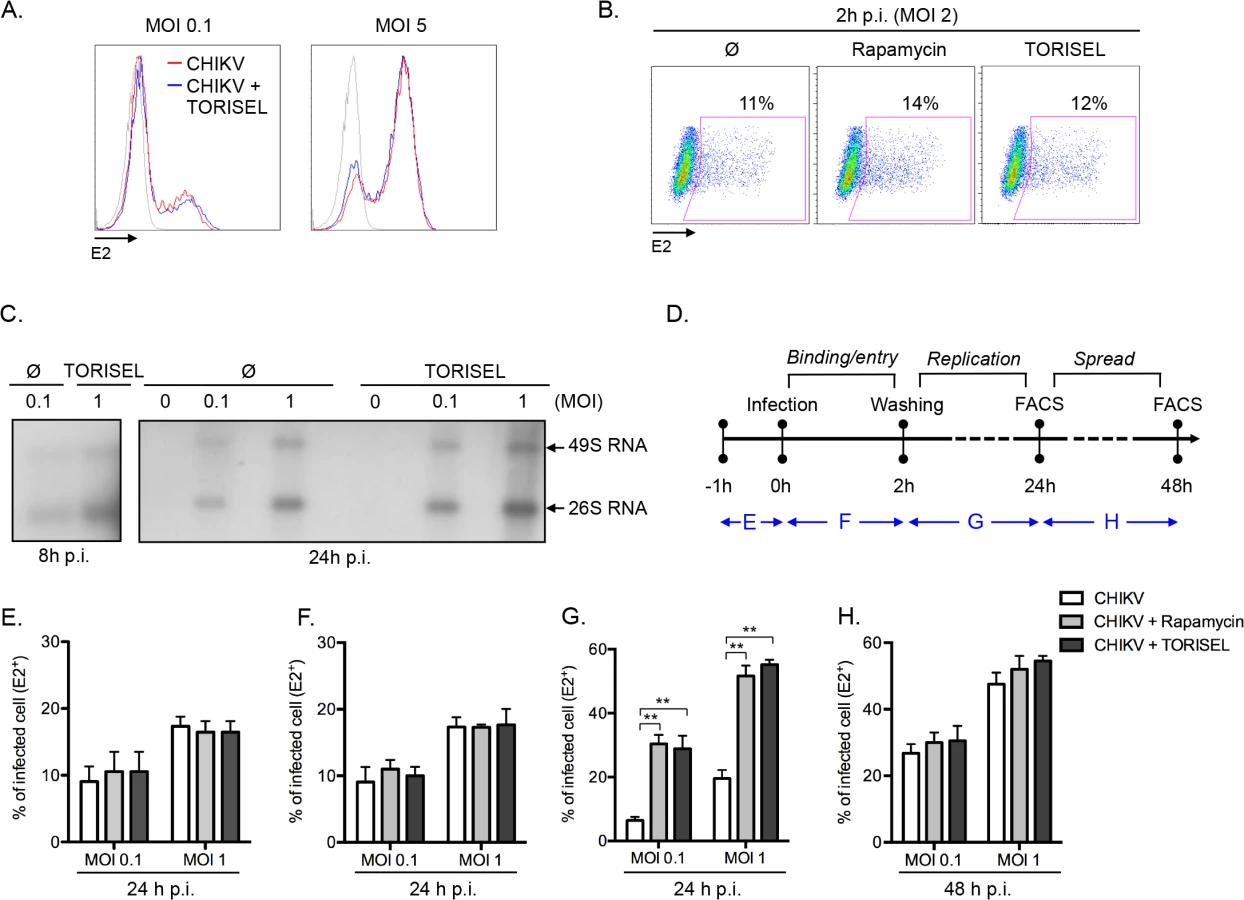
Having ruled out the two expected mechanisms (type I IFN and autophagy) by which mTORC1 could regulate viral infection, we addressed the mechanism of action using an unbiased approach, interrogating the effect of mTOR inhibition on the binding, entry, replication and/or spread of CHIKV. A direct analysis of viral binding (at 4°C) using a FACS-based assay showed that Rapalog treatment did not affect CHIKV binding (Fig 3A). Similarly, after a short period of infection (2h p.i. at 37°C), no difference was observed in intracellular staining of E2 (i.e., quantification of E2 present within the input virus) when comparing Rapalog-treated and untreated cells (Fig 3B). However, quantification of CHIKV genome during the first 24h of infection, showed a higher amount of both positive strand 49S genomic and subgenomic 26S viral mRNA in TORISEL-treated cells as compare to untreated cells (Fig 3C). Notably, a kinetic assessment of the timing for which Rapalog treatment influences CHIKV infection supports the conclusion that mTORC1 has its maximum impact on the CHIKV replication step (Fig 3D–3H). Indeed, binding and entry occur during the first hours of infection, and transient Rapalog exposure 1h pre-infection (Fig 3E), or two hours post-infection (Fig 3F), did not impact the eventual rate of infection. Only mTORC1 inhibition of during the first 24h of infection resulted in increased proportion of E2 positive cells (Fig 3G), yet treatment after the initial 24h of infection showed no additional impact (Fig 3H). These results suggest that mTORC1 activity specifically target the viral replication phase.
Inhibition of mTORC1 improves the translation of both nonstructural and structural proteins
Following the evidence for Rapalog exposure acting to increase viral replication, we investigated whether enhanced translation of nonstructural proteins (nsP) accounts for the higher viral mRNA replication. To monitor nonstructural protein translation, we used a reporter CHIKV encoding luciferase under the control of the genomic promoter (CHIKV-Luc, construct illustrated in Fig 4A). Strikingly, TORISEL exposure resulted in a 3–4-fold increase in luciferase activity for all viral doses tested (Fig 4A). Notably, these results were obtained 4h post-infection, a time point prior to the completion of CHIKV replication; thereby ensuring Luciferase activity is an accurate measure of nonstructural protein translation. These data support Rapalog treatment acting in a cell-intrinsic manner to enhance the translation of viral nonstructural proteins.
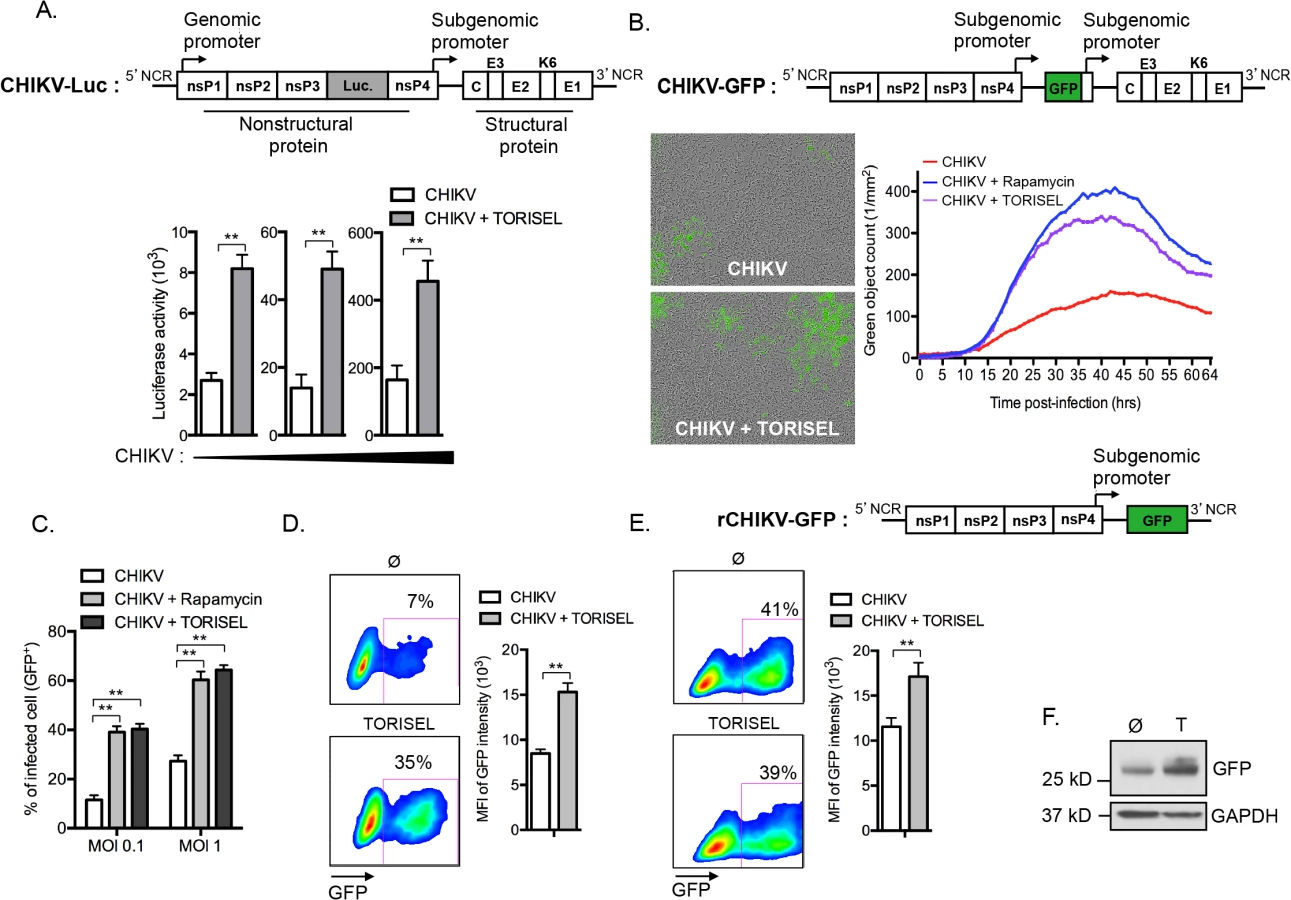
As the genomic ORF (encoding for nonstructural proteins) and subgenomic ORF (encoding for structural proteins) of CHIKV utilize different promoters, we next assessed if the inhibition of mTORC1 could also improve the translation of structural proteins. To accomplish this, we utilized a recombinant CHIKV expressing GFP under the control of the subgenomic promoter (CHIKV-GFP 5’ construct illustrated in Fig 4B), and performed hourly monitoring of infection using real time microscopy. Results confirmed that Rapalog exposure increases infection (Fig 4B and S1 and S2 Movies). Similar results were obtained by examining the percentage of GFP positive cells using cytometric analysis, showing that Rapalog treatment similarly affects the recombinant CHIKV-GFP 5’ and wild type CHIKV (Fig 4C as compared to Fig 1). Importantly, evaluation of GFP expression in infected MEF (GFP+ cells) indicated that TORISEL-treatment increased both the percentage of GFP expressing cells (7 to 35%, p-value = 0.025), and the per cell expression of GFP (8500 to 15200 MFI, p-value = 0.014, Fig 4D). We confirmed this result by using a truncated form of CHIKV, lacking the subgenomic region (rCHIKV-GFP construct illustrated in Fig 4E). This defective GFP reporter virus permitted us to monitor the translation of structural proteins at later time points. At 24h post-transfection, cells were treated with TORISEL and GFP expression was analyzed after an additional 24h (Fig 4E). While the efficiency of transfection was similar (41% vs. 39%), TORISEL-treated cells exhibited a higher expression of GFP protein as compared to untreated cells (11750 to 16450 MFI, p-value = 0,026, Fig 4E and 4F). Therefore, Rapalog treatment acts to enhance translation of both nonstructural and structural CHIKV proteins, and mediates its activity in a cell autonomous manner.
Alphavirus protein translation is enhanced despite a global reduction in cellular protein translation
Regarding the central role for mTORC1 on the activation of cap-dependent translation, our results showed a paradoxical effect of Rapalog on CHIKV proteins translation. To determine if this effect is selective to viral proteins, we investigated the global state of host protein translation (Fig 5A). Using the SUnSET method [23], we showed that TORISEL exposure decreased the global state of host protein translation, in both uninfected and infected cells. Confirming our findings, we show that in the same experiment, TORISEL treated cells expressed higher amount of CHIKV E1 and E2 (Fig 5A). These results suggest that CHIKV translation, despite it being cap-dependent, has evolved a mechanism to bypass mTORC1 inhibition.
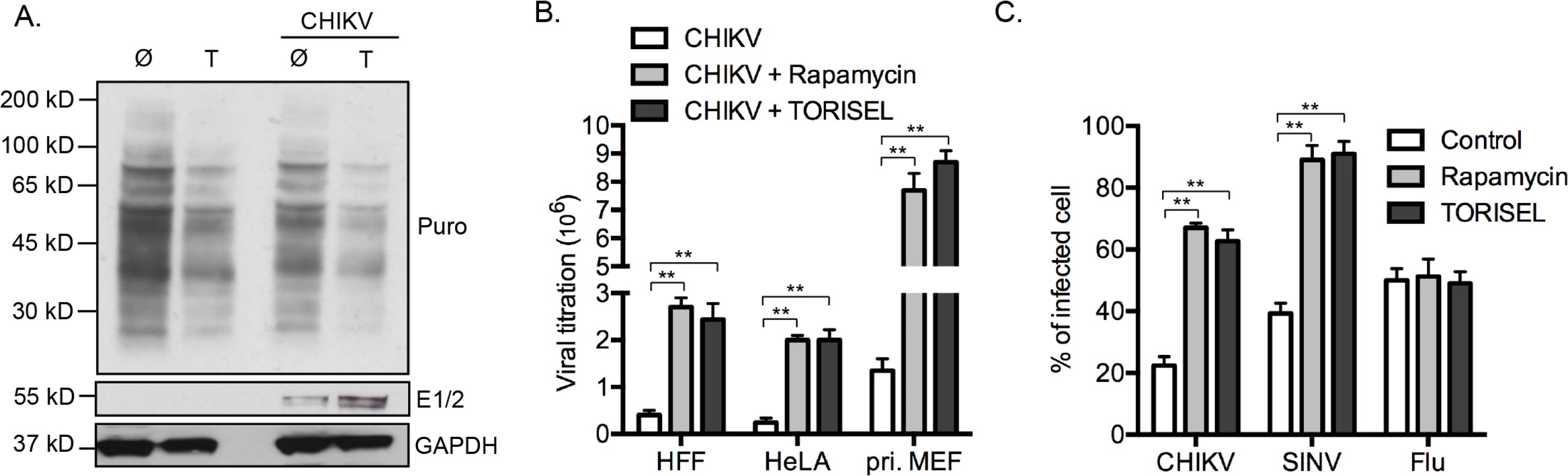
Our results were extended to both human cell lines as well as primary human and mouse fibroblasts. Specially, human foreskin fibroblasts (HFF), human epithelial cells line (HeLa) and primary mouse fibroblast (pri. MEF) were infected by CHIKV, which all showed a similar increase in viral load when mTORC1 was inhibited (Fig 5B). Interestingly, the enhancement of virus titer was more pronounced in primary MEF as compared to cell lines (9–10-fold increase as compared to 3–4-fold increase), indicating a role for mTORC1 in physiologic conditions.
We next investigated if a similar effect of Rapalog treatment could be observed with other viral infection. We chose to study two different viruses: sindbis (SINV), a second member of alphavirus family with a replication cycle similar to CHIKV; and influenza A (Flu, strain A/Puerto Rico/8/1934 H1N1), a member of orthomyxoviridae family. Interestingly, while Rapalog exposure enhanced CHIKV and SINV infection, no effect was observed for Flu (Fig 5C). These results suggest that different viruses have established unique strategies for modulating mTORC1 activity and/or overcoming translational stop mediated by mTOR inhibition.
CHIKV protein translation requires eIF4E activity
Several classes of proteins use a cap-independent translation mechanism which includes the expression of an internal ribosome entry site (IRES) or IRES-like structures [24]. Indeed, IRES are often used by viruses as a means to ensure that viral protein translation is active during cellular stress or other conditions leading to mTORC1 inhibition. For both the structural and nonstructural polyprotein ORF of CHIKV, no IRES or IRES-like structures have been identified identified, suggesting that CHIKV proteins are translated via a cap-dependent mechanism [5]. To test this prediction, we investigated infection efficiency in cells silenced for eIF4E, a protein essential for the initiation of capped mRNA [25]. MEF cells were pretreated with siRNA that targeted eif4e mRNA and infection efficiency was analyzed by flow cytometry and real time imaging (S7A and S7B Fig). Reduced eIF4E expression decreased the amount of CHIKV infected cells. Similarly, inhibition of the interaction between eIF4E and eIF4G, using the inhibitor 4EGI-1, markedly limited the translation of both structural and nonstructural CHIKV protein (S7C and S7D Fig). These results demonstrated a key role for eIF4E protein in CHIKV infection and supported the prior assumption that CHIKV proteins require a cap-dependent translation mechanisms to be processed.
CHIKV protein expression bypasses mTORC1 inhibition via MnK / eIF4E mediated translation
A Rapalog resistant mechanism is present in some tumor cells to maintain translation of capped mRNA when mTORC1 is inhibited [26,27]. This bypass mechanism requires phosphorylation of eIF4E at serine 209, increasing its binding affinity for the capped mRNA, and thereby favoring formation of translation initiation complexes [28]. To investigate the role of mTORC1 activity on eIF4E phosphorylation in our model, we first analyzed the amount of p-eIF4E in TORISEL-treated MEF (Fig 6A). As shown, cells treated with TORISEL expressed higher amount of p-eIF4E as compare to untreated cells, demonstrating that Rapalog treatment leads to increased eIF4E activation (Fig 6A). Importantly, similar results were observed at 24h post-infection, demonstrating that Rapalog exposure enhances the phosphorylation of eIF4E in uninfected and CHIKV infected cells (Fig 6A). The activation of eIF4E was directly linked to Rapalog-induced CHIKV infection by showing that si-eif4e transfected cells were resistant to Rapalog treatment (Fig 6B). These results demonstrated eIF4E activity is critical for increased CHIKV replication when mTORC1 is inhibited. Of note, despite TORISEL treatment leading to an increase in p-eIF4E in uninfected cells, the global amount of host protein translation was diminished (Fig 4A), suggesting that eIF4E activity is being preferentially co-opted by CHIKV.
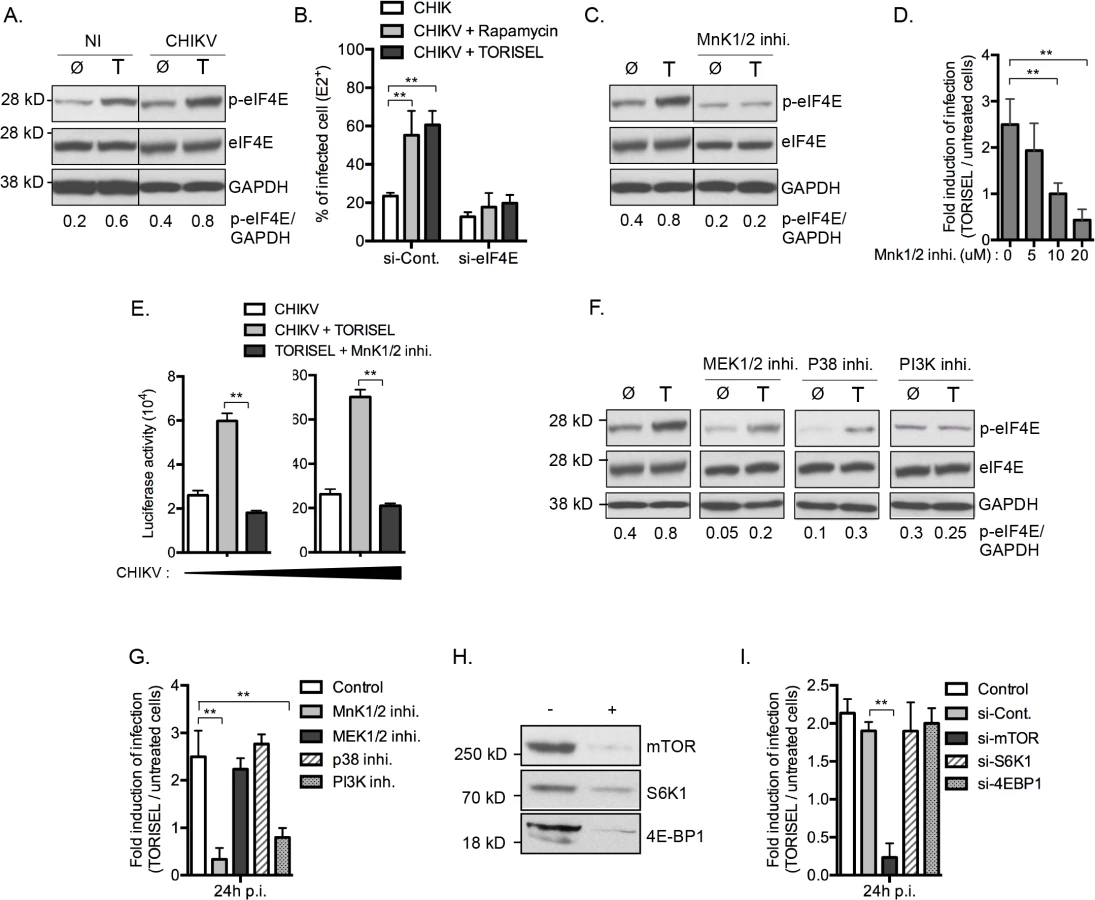
MnK1/2 are the major kinases mediating phosphorylation of eIF4E [29]. We therefore asked whether TORISEL-induced phosphorylation of eIF4E was dependent on MnK proteins. This was performed using as an inhibitor for both MnK1 and MnK2 (CGP57380), which prevented the increase of p-eIF4E observed after TORSIEL treatment (Fig 6C). These results are consistent with prior reports of Rapalog treatment enhancing eIF4E phosphorylation via the activation of MnKs [28,30–32]. Importantly, using the recombinants CHIKV-GFP 5’ or CHIKV-luciferase, we demonstrated that infection and/or translation of CHIKV proteins were not influenced by TORISEL exposure in cells pretreated with MnKs inhibitor (CGP57380 or MnK1/2 inhibitor II) (Fig 6D and 6E). These findings indicate that mTORC1 inhibition favors CHIKV protein translation by an increased MnK-dependent phosphorylation of eIF4E.
We next studied activators of MnK1/2 in order to define the molecular pathway by which eIF4E is engaged. Strikingly, neither of two known kinases responsible for MnK activation, mitogen-activated protein kinase kinase (MAPKK also known as MEK) and p38 MAPK, were required for the enhanced phosphorylation of eIF4E or for the increase in CHIKV proteins translation (Fig 6F and 6G). Cross talk between the phosphatidylinositol-3 kinase (PI3K) and MnKs signaling has been previously reported in human cancer cells [32]. In the context of viral infection, we show that the PI3K inhibitor LY294002 blocked the Rapalog-induced activation of eIF4E and the enhancement of CHIKV (Fig 6F and 6G), suggesting that mTOR inhibition increases eIF4E phosphorylation and subsequently the CHIKV infection through a PI3K-dependent and MnK-mediated mechanism.
To investigate potential co-regulation of mTORC1 among the different effector pathways, we analyzed the role of S6K1 and 4E-BP1. Notably, silencing of these respective genes did not impact Rapalog-induced CHIKV infection (Fig 6H and 6I). Together, these data indicate that mTORC1 has a direct impact on PI3K and the MnK/p-eIF4E pathway.
CHIKV-mediated inhibition of mTORC1 favors infection through a MnKs/p-eIF4E dependent pathway
To demonstrate the physiologic relevance of our discovery, we investigated the role of CHIKV-mediated inhibition of mTORC1 on infection. Interestingly, inhibition of mTORC1 correlated with an increase of eIF4E activity, with peak phosphorylation occurring at 6 h post-infection (Fig 7A). These data advances our previous report of a rapid and transient inhibition of mTORC1 during CHIKV infection, regulated by CHIKV-mediated ROS production and AMPK activation [9]. We also demonstrate that preventing CHIKV-mediated inhibition of mTOR, using ROS inhibitor (N-acetyl-L-cysteine), abrogated the enhanced p-eIF4E (Fig 7B). Moreover, inhibition of PI3K or MnKs, using respective inhibitors, abolished the CHIKV-mediated phosphorylation of eIF4E (Fig 7C). Together, these results show that CHIKV infection increases the phosphorylation of eIF4E through a PI3K and Mnk1/2 dependent.
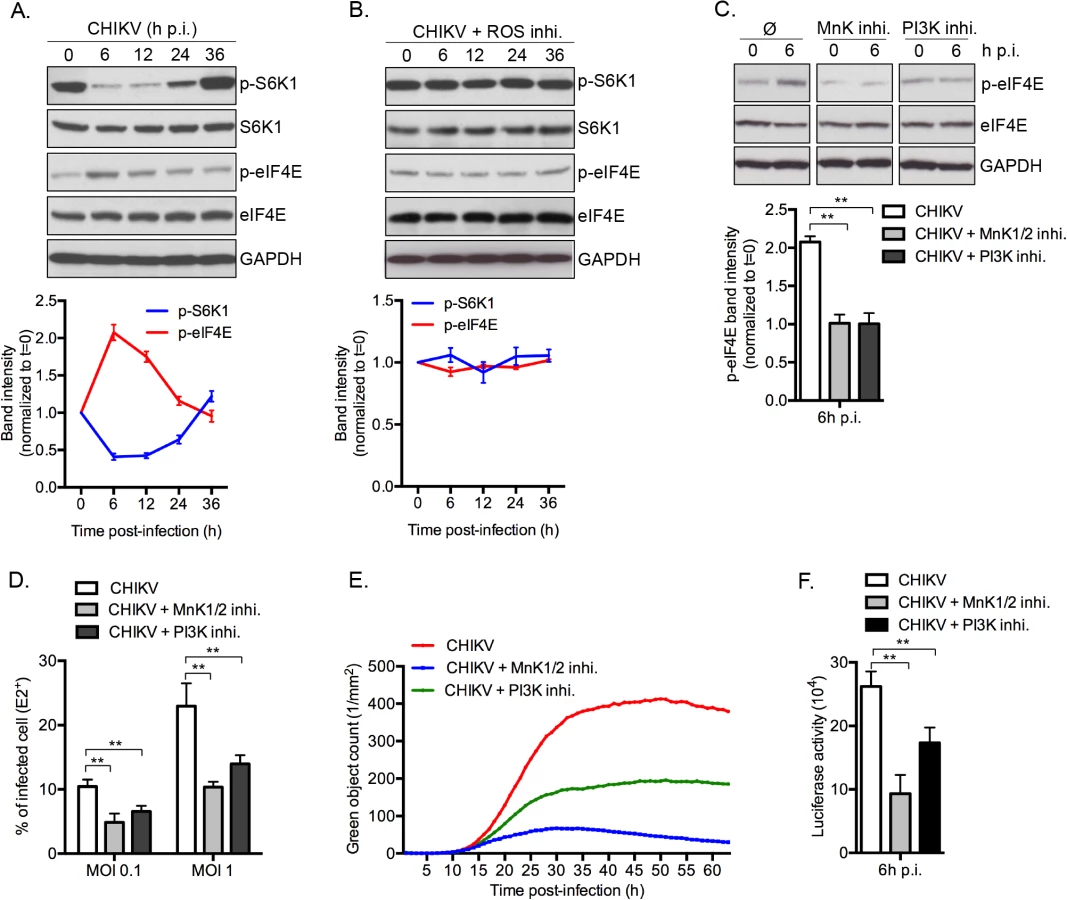
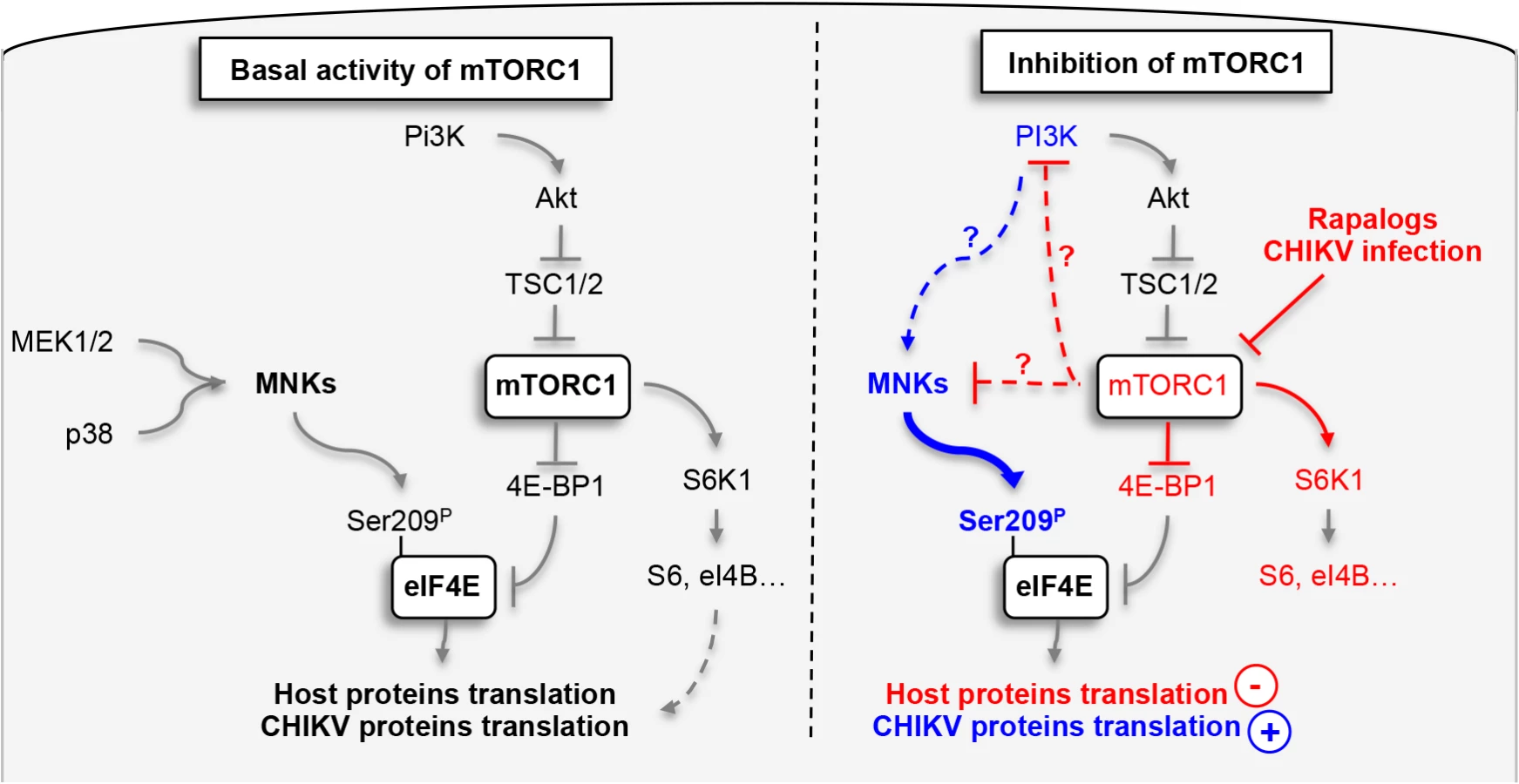
Finally, to define the impact of phosphorylation of eIF4E on natural CHIKV infection, we tested the direct impact of MnK or PI3Ks inhibitors on viral protein expression and viral spread (Fig 7D–7F). Interestingly, inhibition of either Mnk or PI3K significantly decreased the percentage of E2 positive cells (Fig 7D), the translation of structural (Fig 7E) and nonstructural proteins (Fig 7F). These data support our conclusion that CHIKV-induced activation of eIF4E favors infection during natural infection, and also explains the enhancement of viral infection when mTORC1 is pharmacologically inhibited by Rapalog treatment (Fig 8).
Discussion
Herein, we evaluated the role of the metabolic regulator mTOR during CHIKV infection. To our surprise, inhibition of mTORC1 enhanced viral protein translation via a mechanism that is independent of autophagy and type I IFN signaling. Being an important initiator of cap-dependent protein translation, the inhibitory effect of Rapalog treatment on viral has been well documented in instances where the virus possesses capped mRNA [14,33]. The results from our current study, however, identify an unexpected effect of mTORC1. During natural CHIKV infection, mTORC1 limits viral replication despite CHIKV requiring a host mRNA cap. This was further evident when cells were exposed to Rapalog, which resulted in a massive induction of CHIKV protein expression. Indeed, our study provides the first evidence for direct enhancement of viral protein translation during mTORC1 inhibition, thus revealing a new strategy developed by CHIKV to maintain viral protein expression in the context of cell stress.
A new viral bypass for the host translational machinery
As a metabolic sensor, mTOR activity is perturbed during viral infection [12]. To ensure viral protein translation, viruses have evolved strategies to bypass cellular programs that limit the ribosomal machinery. Regulation by mTORC1 has been a major focus and prior work has illustrated the different mechanisms evolved in the context of the host / microbe détente. For example, HSV-1 enhances mTORC1 activity; whereas Poliovirus, HIV-1, Sindbis and CHIKV inhibit this complex [12]. For viruses inducing a stop in protein translation, they have means to ensure translation of their own mRNAs. Most strategies identified thus far involve the use of non-canonical translation mechanisms, including IRES, ribosome shunting or VPg [24]. These processes permit efficient viral replication in absence of the cap-dependent translation initiation protein eIF4E.
To demonstrate that CHIKV proteins use a classical cap-dependent process, we analyzed the dependence on the active form of eIF4E. Indeed, absence of eIF4E expression lost of eIF4E phosphorylation or abrogation of eIF4E/eIF4G complex formation led to a drastic inhibition in CHIKV infection (Figs 7 and S7). Thus CHIKV has evolved a newly discovered mechanism of bypassing mTORC1 inhibition. Notably, we show that a similar strategy seems to be employed by Sindbis (Fig 5C)–consistent with its replication requiring an active form of eIF4E [18]–but not by influenza A (Fig 5C), whose infection proceeds normally even when eIF4E is functionally impaired [34].
To achieve translation initiation, CHIKV engages an MnK-dependent hyper-phosphorylation of eIF4E. This mechanism has been reported in tumors, providing a means to support cap-dependent protein translation under conditions of cell stress [30,31]. In our model, however, hyper-phosphorylation of eIF4E does not restore translation of host proteins, which is shutdown after Rapalog exposure (Fig 5A). This defines a selective strategy by CHIKV to enhance its viral protein translation via p-eIF4E, overcoming mTORC1 inhibition of conventional cap-dependent pathways.
Molecular connections between mTOR and MNKs pathways
The phosphorylation of eIF4E and the formation of the eIF4E/eIF4G complex are tightly regulated by signal transduction pathways that converge on MnK1/2 and mTOR [35]. While both pathways are known to positively impact translation initiation, interconnections between the two signaling mechanisms have remained unclear. Several investigations suggested that the MnKs are activated by Rapalog treatment, resulting in the maintenance of cap-dependent translation during inhibition of mTORC1 [30,31], however the molecular scaffolds are not known.
In the present study, we demonstrated that activation of MnK1/2, induced by Rapalog or CHIKV-mediated mTOR inhibition, results in upregulation of viral protein translation via the hyper-phosphorylation of eIF4E. We show that translation of CHIKV proteins are not only resistant to mTORC1 inhibition, but can be enhanced by this process via the activation of PI3K and the subsequent engagement of the MnK/eIF4E pathway. The effect of mTOR on CHIKV infection was independent of both S6K1 and 4E-BP1 signaling. These observations align with previous work published by Wang and colleagues, who reported that Rapamycin-induced activation of MnK is independent of S6K1 activity [32]. 4E-BP1 hypo-phosphorylation increases its binding to eIF4E and prevents the formation of eIF4F complex [10]. While we clearly show that Rapalog treatment induces hypophosphorylation of 4E-BP1, eIF4E-dependent translation is increased (Fig 1). Interestingly, in our model, PI3K inhibition was able to restrict CHIKV infection and limit the effect of Rapalog on virus replication. Since the classical downstream signals of mTORC1 (S6Ks and 4E-BPs) and upstream signal of MnKs (MEK and p38) are not involved in the Rapalog effect, molecular connections between mTOR, PI3K and the MnK pathway must be examined further.
Role of mTORC1 activity during an acute CHIKV infection
In our previous report, we described a transient inhibition of mTORC1 pathway during the first hour of infection that led to an induction of autophagy that in turn limits CHIK-induced apoptosis [9]. In infected cells, mTORC1 is inhibited through the intrinsic production of ROS leading to the activation of both AMPK and the TSC1/TSC2 complex [36]. Herein, we confirmed the transient inhibition of mTORC1 during CHIKV infection and discovered a new function of mTOR as a direct regulator for viral protein translation. Indeed, preventing mTOR-dependent hyper-phosphorylation of eIF4E using MnK or PI3K inhibitors significantly decrease infection efficiency (Fig 7). Integrating our new findings, we suggest that CHIKV-mediated inhibition of mTORC1 benefits the virus by increasing the efficiency and timing of translation, via the activation of eIF4E and autophagy respectively.
Strikingly, while ROS inhibition prevents the transient inhibition mTORC1 (Fig 7B), we did not observe a significant difference for CHIKV infection in ROS inhibitor-treated versus untreated cells (S8 Fig). These results could be explained by the fact that ROS itself has an mTOR-independent antiviral effect on CHIKV infection, as illustrated by ROS inhibition leading to a marked increase of infection in siRNA mtor or Rapalog treated cells (S8 Fig). Having this new knowledge, we postulate that inhibition of mTORC1 balances the antiviral effect of CHIKV-mediated ROS production.
mTOR participates in restricting viral replication
mTOR has been considered in other infectious models as important for host response. Pulendran and colleagues showed that the TLR9 ligand CpG-A triggered phosphorylation of mTOR and its downstream targets 4E-BPs and S6Ks [37]. Accordingly, TLR9-mediated production of type I IFN, tumor necrosis factor (TNF) and interleukin 6 (IL-6) was suppressed in human and mouse plasmacytoid dendritic cells (pDC) treated with Rapamycin [37]. Another strategy by which mTOR has been associated to immune antiviral response relates to its cross-talk with the autophagy pathway. Indeed, several viruses have developed strategies to use autophagososmes as a membrane support for viral replication and/or release, including HCV, poliovirus and HIV-1 [38,39]. In this context, inhibition of autophagy mediated by mTORC1 could limit viral infection.
Our results highlight a new antiviral mechanism for mTOR that is independent of IFN production and autophagy. We showed that inhibition of mTORC1 increased CHIKV infection in both in vitro and in vivo models. This process was based on direct regulation of viral protein translation and is independent of new transcriptional activity, as the effect of Rapalog treatment was unaffected by ActD exposure. Importantly, we also confirmed the impact of mTOR on CHIKV infection by increasing the activity of endogenous mTORC1. These results suggest that strategies aimed at enhancing activation of mTOR (e.g., specific diets or insulin injection) may be a means of controlling CHIKV infection.
In sum, we have provided evidence for a novel and unexpected mechanism by which CHIKV adapts to mTORC1 inhibition. In the context of acute CHIKV infection, eIF4E is important for the translation of viral proteins, and CHIKV-mediated mTORC1 inhibition increases the phosphorylation of eIF4E, thus favoring viral replication. These data also suggests that targeted engagement of upstream activator of mTORC1 (e.g., Insulin receptor or Akt) or blocking inhibitors of the mTORC1 pathway (e.g., TSC2) may constitute useful strategies for limiting the pathogenesis of acute Chikungunya disease.
Materials and Methods
Cells and mice
Wild-type mouse embryonic fibroblasts (MEF) were obtiained from the Korsmeyer laboratory (Farber, Boston, MA, USA). Atg5-/- and atg7-/- MEF were generous gifts of the Kroemer Laboratory (INSERM U848, Institut Gustave Roussy, Villejuif, France). Primary human foreskin fibroblasts (HFF) were obtained from America Type Culture Collection. All cell lines were mycoplasma free and maintained at 37°C in humidified atmosphere containing 5% CO2 in medium supplemented with 10% heat inactivated fetal calf serum, 100 μg/ml penicillin (Invitrogen); 100 U/ml streptomycin (Invitrogen), and MEM nonessential amino acid (Invitrogen). irf3-/- / irf7-/- mice were generated by Michael Diamond, with the original strains being provided by Tadatsugu Taniguchi.
CHIKV infection and drugs treatments in cell lines
The preparation of CHIKV from clinical samples has been previously described (Schuffenecker et al., 2006). CHIKV-21 strain was propagated in C6/36 cells and supernatants were harvested and frozen at -80°C before titration and further use. Recombinant CHIKV expressing GFP under the subgenomic promoter (CHIKV-GFP 5’) was generated using a full-length infection cDNA clone provided by S. Higgs (Vanlandingham et al., 2005). Recombinant CHIKV expressing Luciferase under genomic promoter (CHIKV-Luc.) was a gift from Philippe Despres and was generated as previously described [40]. The CHIKV replicon expressing EGFP (rCHIKV-GFP) was a gift from Gorben Pijlman and was generated as previously describe [41]. MEF or HFF cells (plate at ∼ 50% confluence in 24- or 96-wells plates) were exposed to the indicated viruses for 2h at 37°C, extensively washed with PBS and cultivated for various periods of time in presence of drugs before further analysis. The MOI was defined as the amount of CHIKV infectious units (calculated on BHK cells as PFU) per target cell. In indicated experiments, Rapamycin (100 nM—Calbiochem), TORISEL (0.1 mg/ml—Biovision), PP242 (0.1 μM—Euromedex), 4EGI-1 (25 μM–VWR International), CPG57380 (20 μM–R & D Systems Europe), MNK inhibitor-II (5 μM–Merck Chemicals LTD), PD 0325901 (1 μg/ml–Sigma Aldrich), SB203580 (1 μM–Sigma Aldrich) or LY294002 (25 μM—Ozyme) were used.
siRNA treatment
Smartpool siRNA targeting atg5, atg7, mTOR, raptor, rictor, eIF4E, S6K1, 4EBP-1 and control siRNA were from Dharmacon (Perbio, Berbères, France). MEF or HFF cells (0.1 × 106) were cultured in 6-well plates for 1 day in OptiMEM (Invitrogen) containing 10% FCS and transfected with 30 nM of indicated siRNA using lipofectamine RNAiMAX (Invitrogen). For all experiments, CHIKV infection was performed after 3d siRNA incubation. In all experiments, protein expression of targeted gene was confirmed to be knocked down to <90% of WT expression. Where inducated functional inhibition was evaluated.
Western blot analysis
Lysates were prepared in 1x Dulbecco’s Phosphate Buffer Saline (DPBS—Invitrogen) containing 1% Nonidet P 40 substitue (NP40 –Signa-Aldrish, MO, USA) and protease inhibitor cocktail (Roche Diagnostics, IN, USA). Total protein was determined by Lowry’s method and 25 mg was loaded on a 4–12% gradient SDS–polyacrylamide gel electrophoresis (Invitrogen). Proteins were transferred to 2 μM nitrocellulose membrane using the Trans-blot turbot kit (Bio-Rad) and blotted over-night with anti-mTOR (rabbit polyclonal, abcam), anti-Rictor (rabbit polyclonal, Cell Signaling), anti-Raptor (rabbit polyclonal, Cell Signaling), anti-Atg5 (mouse monoclonal, Cell Signaling), anti-Atg7 (mouse monoclonal, Cell Signaling), anti-pS6K1 (rabbit polyclonal, Abcam), anti-S6K1 (rabbit polyclonal, Abcam), anti-peIF4E (Ser209) (rabbit polyclonal, Cell Signaling), anti-eIF4E (rabbit polyclonal, Cell Signaling), anti-E1/E2 (rabbit polyclonal, gift from Olivier Schwartz laboratory), anti-C (monoclonal antibody, gift from Olivier Schwartz laboratory), anti-GFP (rabbit polyclonal, Cell Signaling) or anti-GAPDH (rabbit polyclonal, Cell Signaling). Secondary HRP-coupled Abs was detected using ECL Plus (Amersham Pharmacia Biotech).
Northern blot analysis
MEF were infected with CHIKV-21 at indicated MOI in presence of TORISEL. After 8h and 24h of infection, cells were washed twice in PBS and RNA was TRIzol (Invitrogen) extracted, quantified and diluted to the same concentration. Samples were prepared in NorthernMax formaldehyde loading dye (Ambion) with 1μl of ethidium bromide, heated to 65°C for 10 minutes, then separated on a 1.2% agarose (Lonza) gel containing 1x morpholinepropanesulfonic acid (MOPS), running buffer (Ambion) and 6.7% formaldehyde. RNA was transferred onto nitrocellulose membrane, cross-linked by ultraviolet irradiation (UVP), and prehybridized at 68°C for 1h in ULTRAhyb ultrasensitive hybridization buffer (Ambion). A plasmid used for the expression of CHIKV RNA probes corresponding to the 3′ portion of the E2 glycoprotein was generated by first amplifying the region of the CHIKV genome from 8703 (5′-GAAGCGACAGACGGGACG-3′) to 9266 (5′-GTTACATTTGCCAGCGGAA-3′) by PCR and subsequently TOPO-TA cloning the PCR product into the pCRTOPO-II vector. RNA probes complementary to positive strand RNA were labeled with 32P using the MAXIscript SP6 In Vitro Transcription Kit (Ambion), unincorporated nucleotides were removed using illustra MicroSpin S200 HR columns (GE healthcare), and probe was hybridized to the membrane overnight at 68°C. Membranes were washed several times at 68°C with 0.1× SSC with 0.1% SDS, then imaged using Amersham Hyperfilm MP autoradiography film (GE Healthcare).
CHIKV staining and flow cytometry
MEF or HFF cells were infected with CHIKV-21 or CHIKV-GFP 5’ at the indicated MOI for 24h and fixed with 4% PFA for 20 min. After fixation, cells infected with CHIKV-21 were permeabilized with BD Cytofix/Cytoperm (BD kit, BD Bioscniences) before labeling with anti-E2 (gift form Lecuit lab, Microorganismes et barrières de l’hôte, Institue Pasteur, France). The percentage of E2+ or GFP+ cells was measured by flow cytometery using FACSCanto (BD Biosciences, MD, USA) and FlowJo software (Tree Star, Inc.).
Live-cells imaging
MEFs were infected with CHIKV-GFP 5’ in 24-well or 96-well pate and imaged using IncuCyte HD system (Essen BioScience). Frames were captured at 1-hour intervals from 4 separate 950 × 760–μm2 regions per well using a 20× objective. Cultures were maintained at 37°C in a Hera cell 240 chamber (Thermo Electron Cormpration) throughout, with all experiments run in triplicate. GFP+ cells were counted using IncuCyte ZOOM software (Essen BioScience) and results are represented as green object count per mm2. Values from all 4 regions of each well were pooled and averaged across the 3 replicates. Movies were extracted directly from IncuCyte ZOOM software.
CHIKV titers in cell lines
MEF or HFF cells were infected with CHIKV-21 and supernatants were recovered at 24h after infection. Viral samples were titrated as TCID50 endpoint on Vero cells using a standard procedure. Serial 10-fold dilutions (100 μgl) of supernatants were added in six replicates in 96-well plates seeded with 104 Vero cells. The cytopathic effect was scored 5 days after infection and the titers was calculated by determining the last dilution giving 50% of wells with cells displaying a cytopathic effect. Results were expressed as TCID50/ml.
In vivo Rapalog treatment, infection and viral titration
irf3-/- / irf7-/- mice were treated with intra-peritoneal injection of 100 μL solution containing TORISEL (10mg/kg) (n = 32) or PBS (n = 31) every two days for 8 days. At day 8, mice were infected with 1x106 PFU CHIKV-21 subcutaneously (s.c.) in the bottom chest. For viral titration, skin and muscle were collected after days 1 (n = 6 for PBS-treated mice; n = 7 for TORISEL-treated mice), 2 (n = 5 for PBS-treated mice; n = 6 for TORISEL-treated mice), and 3 (n = 6 for PBS-treated mice; n = 5 for TORISEL-treated mice) of infection, homogenized, and viral samples were titrated as TCID50 endpoint on Vero cells using a standard procedure. Clinical score (n = 14) was determined at day 2 of infection, based on EAE from K. Racke (0 = nothing; 1 = limp tail; 2 = mouse don’t grasp the cage with toes but with the ankle; 3 = mouse is unable to return and land on its feet when flipped over; 4 = hindlimb drag behind are not used by the mouse for movement; 5 premoribund stat). Lethality of mice (n = 14) was followed for 10 days post-infection.
Nonradioactive measurements of protein synthesis with SUnSET
SunSet experiments were performed as previously described [23]. To summarize, cells were infected with CHIKV in the presence of TORISEL for 24 hrs. Then cells were cultivated with serum free media containing puromycin (10 υg/ml) for 30 min, washed with PBS and kept in culture for an additional 1h with normal media. Western blot was performed as previously described using an antibody against puromycin (clone 12D10, 1/5000, Millipore).
Ethics statement
Mouse studies were performed in strict accordance with the Institutional Guiding Principles for Biomedical Research Involving Animals and all experiments were performed in an A3 containment facility. The protocols were approved by the Institutional Committees on Animal Welfare of the Pasteur Institute (OLAW assurance #A5476-01). All efforts were made to minimize suffering.
Supporting Information
Zdroje
1. The 2005–2007 Chikungunya epidemic in Réunion: ambiguous etiologies, memories, and meaning-making. (2013) The 2005–2007 Chikungunya epidemic in Réunion: ambiguous etiologies, memories, and meaning-making. 32: 174–189. Available: http://www.tandfonline.com/doi/abs/10.1080/01459740.2012.679981. doi: 10.1080/01459740.2012.679981 23406067
2. Chikungunya outbreak in bueng kan province, Thailand, 2013. (2014) Chikungunya outbreak in bueng kan province, Thailand, 2013. 20: 1404–1406. Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=25061989&retmode=ref&cmd=prlinks. doi: 10.3201/eid2008.140481 25061989
3. Re-emergence of chikungunya virus. (2014) Re-emergence of chikungunya virus.: JVI.01432–14. Available: http://jvi.asm.org/cgi/doi/10.1128/JVI.01432-14.
4. Notes from the field: chikungunya virus spreads in the Americas—Caribbean and South America, 2013–2014. (2014) Notes from the field: chikungunya virus spreads in the Americas—Caribbean and South America, 2013–2014. 63: 500–501. Available: http://eutils.ncbi.nlm.nih.gov/entrez/eutils/elink.fcgi?dbfrom=pubmed&id=24898168&retmode=ref&cmd=prlinks.
5. Proteomic analysis of chikungunya virus infected microgial cells. (2012) Proteomic analysis of chikungunya virus infected microgial cells. 7: e34800. Available: http://dx.plos.org/10.1371/journal.pone.0034800. doi: 10.1371/journal.pone.0034800 22514668
6. An in vitro assay to study chikungunya virus RNA synthesis and the mode of action of inhibitors. (2014) An in vitro assay to study chikungunya virus RNA synthesis and the mode of action of inhibitors.: vir.0.069690–vir.0.069690. Available: http://vir.sgmjournals.org/content/early/2014/08/17/vir.0.069690-0.abstract.
7. Chikungunya virus and prospects for a vaccine. (2012) Chikungunya virus and prospects for a vaccine. 11: 1087–1101. Available: http://informahealthcare.com/doi/abs/10.1586/erv.12.84. doi: 10.1586/erv.12.84 23151166
8. Krejbich-Trotot P, Denizot M, Hoarau JJ, Jaffar-Bandjee MC, Das T, et al. (2011) Chikungunya virus mobilizes the apoptotic machinery to invade host cell defenses. The FASEB Journal 25: 314–325. doi: 10.1096/fj.10-164178 20881210
9. Joubert PE, Werneke SW, la Calle de C, Guivel-Benhassine F, Giodini A, et al. (2012) Chikungunya virus-induced autophagy delays caspase-dependent cell death. Journal of Experimental Medicine 209: 1029–1047. doi: 10.1084/jem.20110996 22508836
10. Influence of mTOR in energy and metabolic homeostasis. (2014) Influence of mTOR in energy and metabolic homeostasis. Available: http://linkinghub.elsevier.com/retrieve/pii/S0303720714002196.
11. mTORC2 in the center of cancer metabolic reprogramming. (2014) mTORC2 in the center of cancer metabolic reprogramming. 25: 364–373. Available: http://linkinghub.elsevier.com/retrieve/pii/S1043276014000721. doi: 10.1016/j.tem.2014.04.002 24856037
12. Martin S, Saha B, Riley JL (2012) The Battle over mTOR: An Emerging Theatre in Host–Pathogen Immunity. PLoS Pathog 8: e1002894–e1002895. doi: 10.1371/journal.ppat.1002894 23028309
13. Zhou X, Wang Y, Metselaar HJ, Janssen HLA, Peppelenbosch MP, et al. (2014) Rapamycin and everolimus facilitate hepatitis E virus replication: Revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. JOURNAL OF HEPATOLOGY: 1–9. doi: 10.1016/j.jhep.2014.05.026
14. Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. (2012) Hepatitis C virus activates the mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. 86: 6315–6322. Available: http://jvi.asm.org/cgi/doi/10.1128/JVI.00050-12. doi: 10.1128/JVI.00050-12 22457523
15. Li Q, ne VERPE, Krishnamurthy S, Cha H, Liang TJ (2013) Hepatitis C virus infection activates an innate pathway involving IKK-α in lipogenesis and viral assembly. Nature Medicine 19: 722–729. doi: 10.1038/nm.3190 23708292
16. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. (2010) Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. 207: 429–442. Available: http://www.jem.org/cgi/doi/10.1084/jem.20090851. doi: 10.1084/jem.20090851 20123960
17. RAPping production of type i interferon in pDCs through mTOR (2008) RAPping production of type i interferon in pDCs through mTOR: 1–3.
18. Herdy B, Jaramillo M, Svitkin YV, Rosenfeld AB, Kobayashi M, et al. (2012) Translational control of the activation of transcription factor NF-κB and production of type I interferon by phosphorylation of the translation factor eIF4E. Nature Immunology 13: 543–550. doi: 10.1038/ni.2291 22544393
19. Kinase mTOR: regulation and role in maintenance of cellular homeostasis, tumor development, and aging. (2014) Kinase mTOR: regulation and role in maintenance of cellular homeostasis, tumor development, and aging. 79: 88–101. Available: http://link.springer.com/10.1134/S0006297914020023. doi: 10.1134/S0006297914020023 24794724
20. Judith D, Mostowy S, Bourai M, Gangneux N, Lelek MEL, et al. (2013) Species-specific impact of the autophagy machinery on Chikungunya virus infection. Nature Publishing Group: 1–11. doi: 10.1038/embor.2013.51
21. Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. (2012) Interferon response factors 3 and 7 protect against Chikungunya virus hemorrhagic fever and shock. 86: 9888–9898. Available: http://jvi.asm.org/cgi/doi/10.1128/JVI.00956-12. doi: 10.1128/JVI.00956-12 22761364
22. A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. (2008) A mouse model for Chikungunya: young age and inefficient type-I interferon signaling are risk factors for severe disease. 4: e29. Available: http://dx.plos.org/10.1371/journal.ppat.0040029. doi: 10.1371/journal.ppat.0040029 18282093
23. Schmidt EK, Clavarino G, Ceppi M, Pierre P (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat Meth 6: 275–277. doi: 10.1038/nmeth.1314
24. Structural and functional diversity of viral IRESes. (2009) Structural and functional diversity of viral IRESes. 1789: 542–557. Available: http://linkinghub.elsevier.com/retrieve/pii/S1874939909000820. doi: 10.1016/j.bbagrm.2009.07.005 19632368
25. eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. (2008) eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. 86: 178–183. Available: http://www.nrcresearchpress.com/doi/abs/10.1139/O08-034. doi: 10.1139/O08-034 18443631
26. Adaptation to chronic mTOR inhibition in cancer and in aging. (2013) Adaptation to chronic mTOR inhibition in cancer and in aging. 41: 956–961. Available: http://www.biochemsoctrans.org/bst/041/bst0410956.htm. doi: 10.1042/BST20130080 23863163
27. Cope CL, Gilley R, Balmanno K, Sale MJ, Howarth KD, et al. (2014) Adaptation to mTOR kinase inhibitors by amplification of eIF4E to maintain cap-dependent translation. Journal of Cell Science 127: 788–800. doi: 10.1242/jcs.137588 24363449
28. Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. (2008) Phosphorylation of eIF4E by MNKs supports protein synthesis, cell cycle progression and proliferation in prostate cancer cells. 29: 2279–2288. Available: http://www.carcin.oxfordjournals.org/cgi/doi/10.1093/carcin/bgn221. doi: 10.1093/carcin/bgn221 18809972
29. The oncogene eIF4E: using biochemical insights to target cancer. (2013) The oncogene eIF4E: using biochemical insights to target cancer. 33: 227–238. Available: http://online.liebertpub.com/doi/abs/10.1089/jir.2012.0142. doi: 10.1089/jir.2012.0142 23472659
30. Stead RL, Proud CG (2013) Rapamycin enhances eIF4E phosphorylation by activating MAP kinase-interacting kinase 2a (Mnk2a). FEBS Letters 587: 2623–2628. doi: 10.1016/j.febslet.2013.06.045 23831578
31. Grzmil M, Huber RM, Hess D, Frank S, Hynx D, et al. (2014) MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J Clin Invest 124: 742–754. doi: 10.1172/JCI70198 24401275
32. Wang X, Yue P, Chan CB, Ye K, Ueda T, et al. (2007) Inhibition of Mammalian Target of Rapamycin Induces Phosphatidylinositol 3-Kinase-Dependent and Mnk-Mediated Eukaryotic Translation Initiation Factor 4E Phosphorylation. Molecular and Cellular Biology 27: 7405–7413. doi: 10.1128/MCB.00760-07 17724079
33. McNulty S, Flint M, Nichol ST, Spiropoulou CF (2012) Host mTORC1 Signaling Regulates Andes Virus Replication. Journal of Virology 87: 912–922. doi: 10.1128/JVI.02415-12 23135723
34. Influenza virus mRNA translation revisited: is the eIF4E cap-binding factor required for viral mRNA translation? (2007) Influenza virus mRNA translation revisited: is the eIF4E cap-binding factor required for viral mRNA translation? 81: 12427–12438. Available: http://jvi.asm.org/cgi/doi/10.1128/JVI.01105-07. 17855553
35. Many roads from mTOR to eIF4F. (2013) Many roads from mTOR to eIF4F. 41: 913–916. Available: http://www.biochemsoctrans.org/bst/041/bst0410913.htm. doi: 10.1042/BST20130082 23863155
36. JOUBERT P-E, Werneke S, la Calle de C, Guivel-Benhassine F, Giodini A, et al. (2014) Chikungunya-induced cell death is limited by ER and oxidative stress-induced autophagy. Autophagy 8: 1261–1263. doi: 10.4161/auto.20751
37. Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, et al. (2008) Toll-like receptor–mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nature Immunology 9: 1157–1164. doi: 10.1038/ni.1645 18758466
38. Deretic V, Levine B (2009) Autophagy, Immunity, and Microbial Adaptations. Cell Host & Microbe 5: 527–549. doi: 10.1016/j.chom.2009.05.016
39. Blanchet FP, Moris A, Nikolic DS, Lehmann M, Cardinaud S, et al. (2010) Human Immunodeficiency Virus-1 Inhibition of Immunoamphisomes in Dendritic Cells Impairs Early Innate and Adaptive Immune Responses. Immunity 32: 654–669. doi: 10.1016/j.immuni.2010.04.011 20451412
40. Gad HH, Paulous S, Belarbi E, Diancourt L, Drosten C, et al. (2012) The E2-E166K substitution restores Chikungunya virus growth in OAS3 expressing cells by acting on viral entry. Virology 434: 27–37. doi: 10.1016/j.virol.2012.07.019 22889614
41. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. (2010) Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. 84: 10877–10887. Available: http://jvi.asm.org/cgi/doi/10.1128/JVI.00949-10. doi: 10.1128/JVI.00949-10 20686047
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 8
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- Human Non-neutralizing HIV-1 Envelope Monoclonal Antibodies Limit the Number of Founder Viruses during SHIV Mucosal Infection in Rhesus Macaques
- Type VI Secretion System Toxins Horizontally Shared between Marine Bacteria
- Are Human Intestinal Eukaryotes Beneficial or Commensals?
- Hepcidin and Host Defense against Infectious Diseases