
CR3 and Dectin-1 Collaborate in Macrophage Cytokine Response through Association on Lipid Rafts and Activation of Syk-JNK-AP-1 Pathway
The incidence of life-threatening fungal infections is increasing during the last decades. A better understanding of the interactions between fungal pathogen and its host cell is important to the development of new therapeutic strategies against fungal infections. Dimorphic fungus Histoplasma capsulatum becomes disseminated and threatens life in immunocompromised individuals. This fungal pathogen utilizes complement receptor 3 (CR3) and Dectin-1, two pattern recognition receptors on the surface of innate immune cells, to induce macrophage cytokine response. In this study, we demonstrated that CR3 and Dectin-1 act collaboratively to induce macrophage TNF and IL-6 response through a mechanism dependent on activation of the Syk-JNK-AP-1 signaling axis. CR3 and Dectin-1 are recruited and form clusters on lipid raft microdomains upon stimulation by H. capsulatum, leading to activation of their signaling convergence at Syk kinase and induction of subsequent cytokine response. In addition, we showed that CR3 and Dectin-1 cooperatively instruct the adaptive antifungal immunity to defense against H. capsulatum infection. Our findings define the molecular mechanisms underlying receptor crosstalk between CR3 and Dectin-1 and provide a valuable model for receptor collaboration in the context of host-fungus interactions.
Published in the journal:
. PLoS Pathog 11(7): e32767. doi:10.1371/journal.ppat.1004985
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004985
Summary
The incidence of life-threatening fungal infections is increasing during the last decades. A better understanding of the interactions between fungal pathogen and its host cell is important to the development of new therapeutic strategies against fungal infections. Dimorphic fungus Histoplasma capsulatum becomes disseminated and threatens life in immunocompromised individuals. This fungal pathogen utilizes complement receptor 3 (CR3) and Dectin-1, two pattern recognition receptors on the surface of innate immune cells, to induce macrophage cytokine response. In this study, we demonstrated that CR3 and Dectin-1 act collaboratively to induce macrophage TNF and IL-6 response through a mechanism dependent on activation of the Syk-JNK-AP-1 signaling axis. CR3 and Dectin-1 are recruited and form clusters on lipid raft microdomains upon stimulation by H. capsulatum, leading to activation of their signaling convergence at Syk kinase and induction of subsequent cytokine response. In addition, we showed that CR3 and Dectin-1 cooperatively instruct the adaptive antifungal immunity to defense against H. capsulatum infection. Our findings define the molecular mechanisms underlying receptor crosstalk between CR3 and Dectin-1 and provide a valuable model for receptor collaboration in the context of host-fungus interactions.
Introduction
Diseases caused by fungal pathogens have become an important cause of morbidity and mortality over the last decades due to the increasing number of immunocompromised patients [1]. To reveal the cellular and molecular mechanisms of the interaction between host and fungal pathogens will be helpful for the development of new therapeutic strategies. Innate immune cells recognize pathogen-associated molecular patterns (PAMPs) on fungi through pattern recognition receptors (PRRs) [2, 3]. The fungal cell wall is composed predominantly of glucans, chitin, mannose and other covalently-linked proteins with the composition varies between species and even between the different strains and morphological forms of the same species [4, 5]. Since a single pathogen is composed of multiple PAMPs, innate immune cells are likely to simultaneously or sequentially utilize a complex set of PRRs to interact with a specific pathogen. The coordination between PRRs leading to activation or inhibition of downstream signaling is referred to as “receptor crosstalk” [6].
PRR collaboration is known to be important for the host to control invading pathogens. It has been reported that Dectin-1 functions synergistically with TLR2 in amplifying innate cell cytokine response when stimulated with their respective ligands [7–10]. Collaboration between Dectin-1 and TLR2 is mediated by the activation of both Dectin-1/Syk and TLR/Myd88 pathways which results in increased NF-κB activity [7, 9]. Receptor crosstalk between C-type lectin receptors (CLRs) has also been reported. Dectin-1 collaboration with SIGNR1 or with Dectin-2 is largely dependent on the activation of Syk kinase to enhance immune responses against Candida albicans [11, 12]. Stimulation with C. albicans induces colocolization and physical association of Dectin-1 and SIGNR1 whose collaboration results in macrophage oxidative response [11]. Aside from collaborating with TLRs and CLRs, Dectin-1 is also known to collaborate with CR3 in macrophage cytokine response to H. capsulatum in a Syk kinase-dependent manner [13]. However, the coordinated mechanisms in CR3 and Dectin-1 collaboration leading to cytokine production are undefined.
CR3 (Mac-1, αMβ2, or CD11b/CD18) is the principal β2 integrin known to contribute to fungal recognition in innate immune cells [3]. CR3 contains two ligand binding sites, I domain and lectin-like domain, which bind to protein ligands (such as iC3b, ICAM-1, and fibronectin) and β-glucan, respectively [14]. CR3 is an enigmatic receptor which transduces diverse and distinct signaling upon engagement with different ligands [15, 16]. Activation of CR3 is mediated by conformation change and regulated by inside-out and outside-in signals [17]. The inside-out signaling to activate CR3 can be initiated from other receptors, such as TLRs and Dectin-1 [18, 19]. Engagement of CR3 also elicits outside-in signaling which activates innate immune effector functions, such as phagocytosis, cytotoxic killing, and cytokine production [17]. Despite many studies revealing the molecular mechanisms regulating CR3 phagocytosis [20], the signaling pathway(s) responsible for its cytokine response is yet to be addressed.
In contrast to CR3, the understanding of Dectin-1 signaling is growing during the last decade. Dectin-1 engagement induces the phosphorylation of its intracellular ITAM-like motif leading to the recruitment and activation of Syk [21]. Syk facilitates the phosphorylation of PLCγ2, allowing subsequent activation of MAPKs, AP-1 and NFAT and the assembly of Card9-Bcl10-Malt1 signalsome which mediates the canonical NF-κB activation [22–24]. A recent study also showed that Card9 bridges the interaction between Ras-GRF-1 and H-Ras, leading to downstream ERK activation upon Dectin-1 ligation [25]. In addition, Dectin-1 triggers Syk-independent Raf-1 activation through which antagonizes Syk-induced noncanonical NF-κB activation [26]. The requirement for Syk and the use of Card9 in Dectin-1 signaling differs in different macrophage and dendritic cell (DC) populations [27, 28]. Thus, signaling pathway downstream of Dectin-1 and signaling crosstalk between Dectin-1 and heterologous PRRs can very well be cell-type specific.
Here we used H. capsulatum and particulate ligands to study the molecular mechanism of collaboration between CR3 and Dectin-1 in macrophages. Our findings clearly showed that collaboration between CR3 and Dectin-1 in induction of TNF and IL-6 production was through synergistic activation of their downstream Syk-JNK-AP-1 signaling axis. In addition, while both CR3 and Dectin-1 were recruited and clustered on lipid raft microdomains upon encountering H. capsulatum, disruption of lipid raft hampered their collaboration in signaling activation and the subsequent cytokine response. Interestingly, CR3 and Dectin-1 cooperatively participated in host defense against disseminated histoplasmosis. Taken together, our results revealed the molecular mechanism underlying crosstalk between CR3 and Dectin-1 in enhancing cytokine response and demonstrated that they orchestrate adaptive antifungal immune response.
Results
CR3 and Dectin-1 collaborate to activate Syk in macrophage cytokine response
By use of blocking antibodies we previously showed that macrophage utilizes CR3 to phagocytose and both CR3 and Dectin-1 to mediate cytokine response to H. capsulatum [13]. Employing Itgam-/-, Clec7a-/-, and Itgam-/-Clec7a-/- macrophages here we investigated the mechanism of receptor crosstalk between CR3 and Dectin-1 (S1A Fig). While losing either or both CR3 and Dectin-1 did not affect the propagation of intracellular H. capsulatum (S2A Fig), TNF and IL-6 responses to either heat-killed or viable H. capsulatum were reduced in Itgam-/- and Clec7a-/- macrophages and the reduction was further enhanced in macrophages deficient in both receptors (Figs 1A and S3A, and also refer to S2B Fig). Blocking Dectin-1 in Itgam-/- macrophages or blocking CR3 in Clec7a-/- macrophages also revealed that signals from these two receptors had additive effect in TNF and IL-6 production (S4A Fig). It is worth noting that heat treatment did not change H. capsulatum recognition by macrophages.
Both CR3 and Dectin-1 are known to signal through activation of Syk kinase [21, 29]. We examined whether Syk is involved in the collaborative cytokine response induced by CR3 and Dectin-1. Stimulation with heat-killed or viable H. capsulatum activated Syk kinase and Syk activation was dampened in Itgam-/- and Clec7a-/- macrophages as well as in WT macrophages blocked by anti-CR3 or anti-Dectin-1 antibody (Figs 1B, S3B, S5A and S4B). The level of phosphorylated Syk was further reduced in Itgam-/-Clec7a-/- macrophages and in WT macrophages blocked by both anti-CR3 and anti-Dectin-1 antibodies (Figs 1B, S3B and S4B). These results suggest that signals from CR3 and Dectin-1 cooperatively activate Syk kinase. Treatment with Syk inhibitors significantly reduced H. capsulatum-induced TNF and IL-6 production and the reduction was not due to the cytotoxic effect of inhibitors (Figs 1C and S6). While Syk deficiency did not affect the expression of CR3 and Dectin-1, the deficiency almost completely abolished the production of TNF (Figs 1D and S1B). These results clearly demonstrate that CR3 and Dectin-1 act in concert in macrophage cytokine response to viable as well as heat-killed H. capsulatum by intensifying Syk activation.

To verify the nature of collaboration between receptors, specific particulate ligands for CR3 (iC3b-coated bead) and Dectin-1 (depleted zymosan) were used to stimulate cells. iC3b-coated beads, but not uncoated beads, and depleted zymosan additively induced TNF and IL-6 production in WT macrophages while Itgam-/- macrophages did not respond to stimulation by iC3b-coated beads neither did Clec7a-/- macrophages to depleted zymosan (Figs 1E and S7A). Co-stimulation of macrophages with iC3b-coated beads and depleted zymosan enhanced the level of phosphorylated Syk compared to stimulation by either ligand alone (Figs 1F and S7B) and treatment with Syk inhibitors abolished TNF and IL-6 response induced by these two ligands (Fig 1G). These data collectively reveal that Syk is the point where signals from CR3 and Dectin-1 converge to mediate collaborative cytokine response.
CR3 and Dectin-1 clustering on lipid rafts facilitates their coordinated actions
It has been demonstrated that localization of Dectin-1 and CR3 to lipid raft microdomains is critical for signaling activation of each respective receptor [30, 31]. While both CR3 and Dectin-1 diffusely distributed in the cytosol and on cell membrane in unstimulated macrophages, they were recruited and colocalized on lipid raft microdomains at the interface between macrophage and H. capsulatum yeasts upon stimulation (Fig 2A). Isolation of lipid rafts by sucrose gradient confirmed that CR3 and Dectin-1 translocated to flotillin-1-enriched membrane microdomains (Fig 2B). Stimulating macrophages with H. capsulatum induced the phosphorylation of Syk which was associated with lipid raft microdomains (Fig 2C). It is noted that Syk was present in the lipid raft fractions on H. capsulatum-stimulated as well as on unstimulated cells, suggesting that rather than recruitment, H. capsulatum stimulation triggers phosphorylation of Syk that was already present on lipid rafts (S8A and S8B Fig). Disruption of lipid rafts by methyl-β-cyclodextrin (MβCD) significantly reduced TNF and IL-6 production and cholesterol replenishment rescued the ability to produce TNF but not IL-6 (Fig 2D). H. capsulatum-induced Syk phosphorylation was diminished in cells treated with MβCD and it was partially restored by cholesterol replenishment (Fig 2E). These results demonstrate that activated Syk is associated with lipid raft microdomains where CR3 and Dectin-1 cluster and the integrity of lipid rafts is important to macrophage cytokine response and signaling activation induced by H. capsulatum.

Micro-Western Array analysis reveals the signaling pathways activated by H. capsulatum
We used Micro-Western Arrays (MWAs) by employing 92 antibodies to probe for phosphorylated (84 antibodies) and non-phosphorylated (8 antibodies) signaling proteins known to be involved in the pathways downstream of PRRs for phagocytosis and cytokine production to screen for signaling molecules activated by H. capsulatum (S9 Fig and S1 Table). The heat map shows that H. capsulatum induced phosphorylation of Syk, Raf-1, PLCγ2, PKC and molecules in the PI3K/Akt, NF-κB, and MAPK pathways at as early as 15 min and c-Jun and c-Fos (two components of AP-1) at 60 min after stimulation (Figs 3A and S9). Activated signaling molecules were validated by conventional Western blot analysis. Consistent with the MWA data, H. capsulatum stimulation caused the increase of phosphorylated Syk, Akt, Raf-1, JNK, ERK, p38, IKKα/IKKβ, IκBα and NF-κBp65 at 15 min, while phosphorylation of c-Jun and c-Fos occurred at 60 min after stimulation (Fig 3B). However, the amount of phosphorylated PLCγ2 and PKC isoforms (PKCε, PKCη, PKCθ) were below the limit of detection. Taken together, our data show that H. capsulatum stimulation leads to activation of Syk, Raf-1, AP-1, as well as molecules involved in the Akt/PI3K, NF-κB and MAPK pathways.

JNK is involved in the crosstalk between CR3 and Dectin-1
We used pharmacological kinase inhibitors to identify the signaling molecule(s) that participates in macrophage cytokine response to H. capsulatum. Treatment with PI3K, JNK and ERK inhibitors greatly diminished TNF and IL-6 production, yet inhibiting Raf-1 enhanced TNF and IL-6 responses (Fig 4A). Interestingly, p38 inhibitor had disparate effects on TNF and IL-6 production (Fig 4A). LDH assay showed that pharmacological inhibitors at the concentrations we used did not have cytotoxic effect on the cells (S6 Fig). Interestingly, Syk deficiency abrogated phosphorylation of Raf-1 and JNK but not that of Akt, ERK or p38 (Fig 4B) although inhibiting their activation diminished TNF and IL-6 production (Fig 4A). Results in Fig 4C show that inhibition of JNK activation in cells stimulated with iC3b-coated beads and depleted zymosan significantly reduced TNF and IL-6 production yet inhibition of Raf-1 did not affect the production of either cytokine. These results indicate that JNK, a signaling molecule downstream of Syk, plays an important role in the coordinated CR3 and Dectin-1cytokine response.

We next investigated the link between CR3 and Dectin-1 engagement and JNK activation. Results showed that JNK phosphorylation was reduced to about 50% of WT control in macrophages lacking either receptor across all time points after stimulation with heat-killed or viable H. capsulatum and JNK phosphorylation was almost completely abolished in Itgam-/-Clec7a-/- macrophages (Figs 4D, S3C and S5B). Similarly, blocking either receptor reduced JNK phosphorylation and blocking both receptors reduced JNK phosphorylation even further (Fig 4E). Stimulation of WT macrophages with both iC3b-coated beads and depleted zymosan increased the level of JNK phosphorylation compared to stimulation with either ligand alone (Fig 4F) and the additive effect of these two ligands was diminished in macrophages deficient in either receptor (Fig 4G). Taken together, our results show that engagement of CR3 and Dectin-1 separately induces JNK phosphorylation and engagement of both have an additive effect on JNK activation which is downstream of Syk.
AP-1 mediates the collaborative cytokine response upon CR3 and Dectin-1 engagement
Results in Fig 3 show that stimulation of macrophages with H. capsulatum triggered the activation of both NF-κB and AP-1. To identify whether NF-κB or AP-1 mediates the collaboration between CR3 and Dectin-1 for cytokine production, we first clarified whether they were activated in Syk-/- macrophages. Fig 5A shows that H. capsulatum-induced c-Fos and c-Jun phosphorylation was greatly diminished in Syk-/- macrophages while IκBα phosphorylation and degradation was not affected, indicating that AP-1, but not NF-κB, is the transcription factor downstream of Syk. In addition, while IκBα and NF-κBp65 phosphorylation was not affected in single and double knockout macrophages, activation of c-Fos and c-Jun was dampened in Itgam-/- and Clec7a-/- macrophages and almost completely abolished in Itgam-/-Clec7a-/- macrophages (Figs 5B, 5C, S3D and S5C). Stimulation of WT macrophages with iC3b-coated beads and depleted zymosan together had an additive effect over stimulation with either one alone on c-Jun and c-Fos but not NF-κB activation (Fig 5D and 5E). The additive effect was abolished in single knockout macrophages (Fig 5F). When c-Jun and c-Fos were silenced by respective siRNA separately, TNF and IL-6 production was greatly reduced (Fig 5G and 5H). Together these data show that AP-1, but not NF-κB, is the transcription factor that mediates the collaborative cytokine response induced by CR3 and Dectin-1.
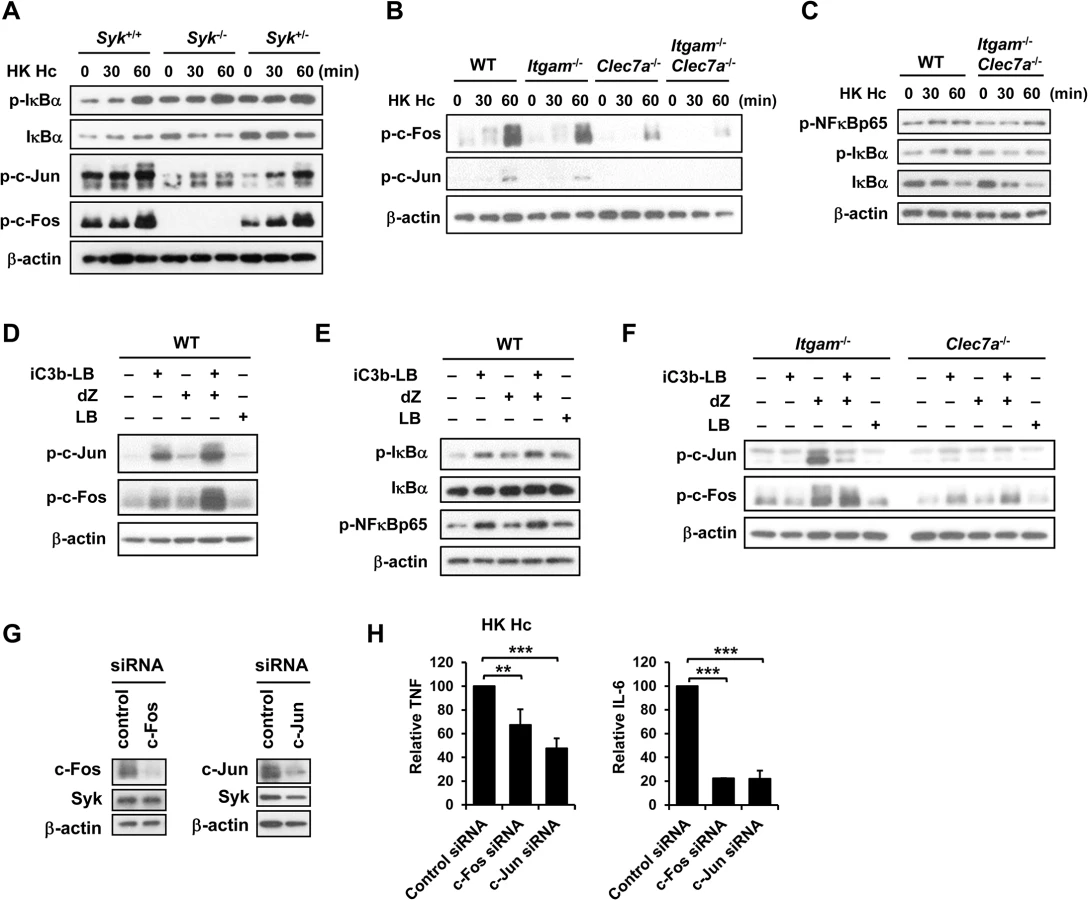
CR3 and Dectin-1 concert in host defense against H. capsulatum infection
We employed disseminated histoplasmosis model, which is characterized by splenomegaly with large numbers of macrophages infiltrating the spleen, to investigate the contribution of CR3 and Dectin-1 in host defense against H. capsulatum infection [32]. WT, Itgam-/-, Clec7a-/- and Itgam-/-Clec7a-/- mice were intravenously infected with a low sublethal dose of H. capsulatum (2.5 × 105). While the fungal loads in Itgam-/- and Clec7a-/- single knockout mice were comparable to (Itgam-/-) or slightly higher than (Clec7a-/-) WT mice, that in double knockout mice were significantly higher than either of the single knockout mice on 7 days after infection (Fig 6A). Accompanying higher fungal loads, TNF, IFN-γ and IL-17 levels in splenocyte cultures of double knockout mice were lower than in either of the single knockout mice and IL-6 levels were lower than in Clec7a-/- mice (Fig 6B). Interestingly, the percentages of IFN-γ-producing CD4+ and CD8+ cells were significantly reduced in Itgam-/- and Clec7a-/- mice compared to WT mice, and they were reduced even further in double knockout mice (Fig 6C). Consistent with in vivo IFN-γ response, Itgam-/-, Clec7a-/- and Itgam-/-Clec7a-/- dendritic cells stimulated with H. capsulatum produced significantly less IL-12p35 transcripts and IL-12p40 transcripts was reduced in Itgam-/-Clec7a-/- dendritic cells compared to WT cells (S10A and S10B Fig). These results together reveal that CR3 and Dectin-1 act in concert in instruction host adaptive immunity against H. capsulatum.
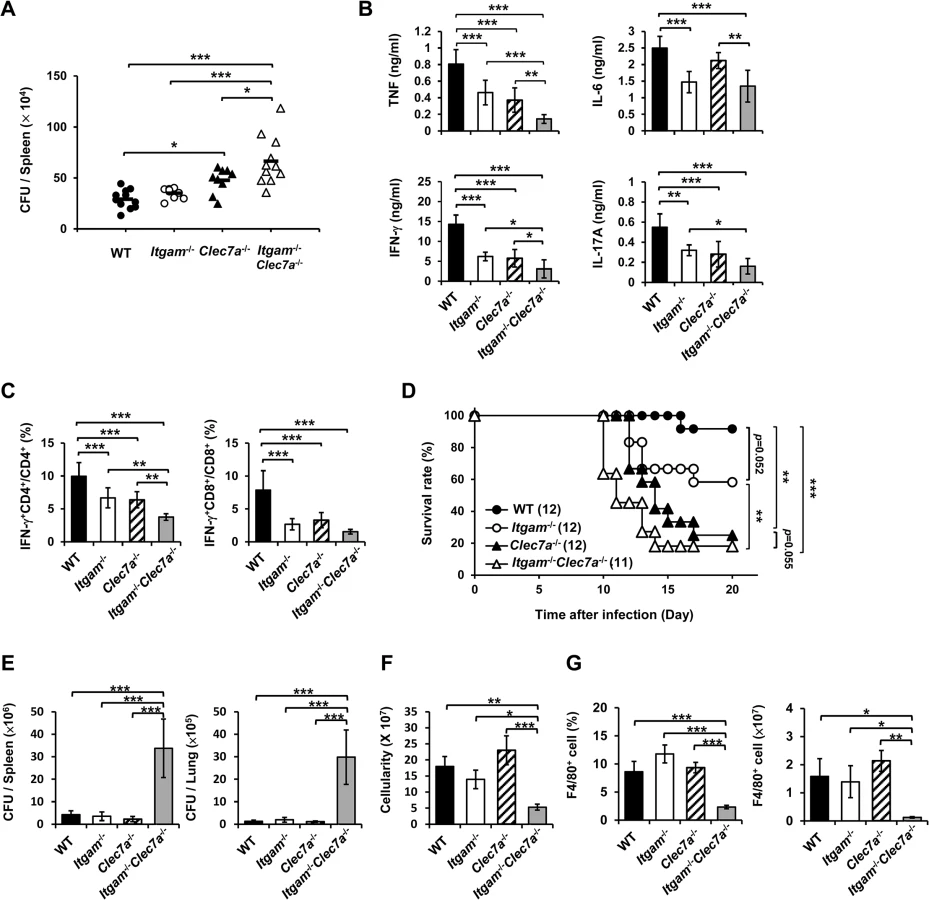
Infection with a higher dose of H. capsulatum (5 × 106), Itgam-/-Clec7a-/- mice had significantly greater fungal burden, higher mortality and succumbed at an earlier time point (10 to14 days) compared to Itgam-/-, Clec7a-/- or WT mice (Fig 6D and 6E). The number of total splenocytes in infected mice was significantly lower in Itgam-/-Clec7a-/- compared to either WT or single knockout mice (Fig 6F). Impressively, infection greatly reduced the proportion and number of F4/80+ cell population in Itgam-/-Clec7a-/- mice, which was significantly lower than in either of the single knockout mice and WT mice (Fig 6G and also refer to S11A and S11B Fig). These results suggest that macrophages in Itgam-/-Clec7a-/- mice fail to respond to H. capsulatum infection and/or rapidly undergo apoptosis. Macrophages being the major player in the interaction between the host and the fungal pathogen H. capsulatum, deficiency in innate receptors that recognize the pathogen for phagocytosis (CR3) and proinflammatory cytokine production (CR3 and Dectin-1) [13] greatly affects host defense against this pathogen. Furthermore, these two innate receptors CR3 and Dectin-1, collaboratively function not only in innate immune response but also orchestrate adaptive immune response against disseminated histoplasmosis.
Discussion
Recognition of invading pathogens by PRRs triggers innate immune responses and shapes the adaptive immunity. Fungal cell wall is complex in its composition which varies between species, strains, and morphological forms. Innate immune cells use a unique set of PRRs to recognize and respond to a given fungus. Therefore, there is a lot of interest to dissect the molecular mechanism of coordination between heterogeneous PRRs. In the present study, we showed that CR3 and Dectin-1 collaborate in regulating macrophage pro-inflammatory cytokine response. Engagement of CR3 and Dectin-1 induces their clustering on lipid raft microdomains which function as a platform for downstream signaling. Utilizing MWA, genetic approach and pharmacological inhibitors, we demonstrated that receptor crosstalk between CR3 and Dectin-1 enhances the activation of their downstream Syk-JNK-AP-1 signaling pathway. Furthermore, our results showed that CR3 and Dectin-1 both participate and function cooperatively in host defense against H. capsulatum by facilitating adaptive antifungal immunity.
The pathogenic fungus H. capsulatum is assorted to chemotype I and II based on the absence and presence of α-(1,3)-glucan in the yeast cell wall [4]. H. capsulatum strain 505 we used in this study having its β-(1,3)-glucan readily exposed does not express α-(1,3)-glucan on the yeast cell surface which makes it likely to be assorted to chemotype I (S12A Fig). Rappleye et al. showed that H. capsulatum yeast strain G186A AGS (+) expressing α-(1,3)-glucan on the outer cell wall layer (S12B Fig) is not recognized by Dectin-1 [33]. The isogenic strain ags1-null mutant lacking α-(1,3)-glucan is recognized by Dectin-1-expressing cells and induces TNF production in phagocytic cells [33]. We showed in a previous study and here that strain 505 induces macrophage TNF and IL-6 production through both CR3 and Dectin-1, a phenomenon possibly unique to H. capsulatum that classified as chemotype I [13]. Interestingly, unlike what is reported in C. albicans [34], heat treatment does not alter β-glucan expression on H. capsulatum strain 505 (S12A and S12C Fig). We also discovered that Syk-JNK-AP-1 signaling and TNF and IL-6 production triggered by heat-killed H. capsulatum are comparable to that induced by viable organism.
In the case of PRR crosstalk, previous studies showed that Syk is required for the synergy between TLR and Dectin-1 and acts as the convergence point of Dectin-1 and Dectin-2 signaling, both of which situations lead to NF-κB activation [7, 9, 12, 35]. We demonstrated here that signaling crosstalk between CR3 and Dectin-1 through stimulation with H. capsulatum initiates at and converges on Syk. However, unlike receptor crosstalk between TLR and Dectin-1 and that between Dectin-1 and Dectin-2, activation of Syk through CR3 and Dectin-1 does not connect to NF-κB pathway. Instead, Syk leads to activation of JNK and AP-1. Consistent with H. capsulatum stimulation, co-stimulation with iC3b-coated beads and depleted zymosan enhances activation of Syk, JNK and AP-1, but not NF-κB. Our findings clearly define Syk-JNK-AP-1 axis as the signaling pathway downstream of the collaborative interaction between CR3 and Dectin-1.
CR3 is unique among the members of β2 integrin family that it contains two ligand binding sites, I domain and lectin-like domain. Single or dual ligation of I domain and lectin-like domain in CR3 triggers disparate signaling pathways and cellular responses [15, 17]. We have previously shown that CR3 recognizes H. capsulatum through both I domain and lectin-like domain within CD11b, demonstrating that H. capsulatum mediates dual CR3 ligation [13]. Here we observed that stimulation of macrophages with iC3b-coated beads (single CR3 ligation of I domain) does not induce Syk phosphorylation, while co-stimulation with iC3b-coated beads and depleted zymosan (dual CR3 ligation and Dectin-1 ligation) triggers Syk phosphorylation and the level of phosphorylation is greater than stimulation by depleted zymosan alone (single CR3 ligation of lectin-like domain and Dectin-1ligation). Interestingly, stimulating Clec7a-/- macrophages with both iC3b-coated beads and depleted zymosan only minimally activates Syk. The lack of inside-out signaling provided by Dectin-1 may account for the failure for iC3b-coated beads and depleted zymosan to activate CR3 [19]. Together, our results show that dual ligation of CR3 optimizes the signaling synergy induced by simultaneous engagement of CR3 and Dectin-1. An early study showed that H. capsulatum yeasts activate the alternative complement pathway which may lead to iC3b deposition [36]. Although CR3 can directly recognize and respond to H. capsulatum, whether complement opsonization would enhance the collaboration between CR3 and Dectin-1 or change PRR usage in macrophage interaction with H. capsulatum needs to be investigated.
PRR clustering in membrane lipid microdomains is crucial for host cells to optimize detection of fungal pathogens by formation of phagocytic synapse and serving as a platform for signaling synergy [31, 37, 38]. In this study, we show that the spatial nearness of CR3 and Dectin-1 and that both of them being mobilized to and associated with lipid rafts are important to achieve their collaboration upon engagement with H. capsulatum. However, little is known about how PRRs are recruited to lipid rafts and how signaling crosstalk is induced by heterogeneous PRRs. It has been reported that intracellular osteopontin (iOPN) is essential for clustering of heterologous PRRs, including Dectin-1, TLR2 and mannose receptor, that recognize Pneumocystis [38]. In addition, iOPN is involved in Dectin-1 and TLR2 downstream signaling by acting as a scaffold protein which associates with their respective downstream molecule Syk and IRAK1 [38]. It remains to be answered whether the steric assembly and the signaling synergy of CR3 and Dectin-1 induced by H. capsulatum is also mediated by iOPN, or by other adaptor molecule(s).
Macrophage plays multiple roles in host defense against H. capsulatum. Besides being a host cell, it also functions as cytokine/chemokine-producing cell and antigen donor cell, and it serves as effector cell when stimulated with IFN-γ, IL-17 and GM-CSF [39–42]. However, little is known about the signaling pathways downstream of functional receptors after macrophage encountering H. capsulatum. By MWA, this study is the first to uncover signaling pathways activated by H. capsulatum in macrophages. Our results reveal that PI3K, NF-κB, MAPK and AP-1 signaling pathways are activated by H. capsulatum. It is interesting to note that although PI3K, ERK and p38 activities modulate TNF and IL-6 production, they do not function downstream of Syk. In addition, inhibition of NF-κB does not affect H. capsulatum-induced TNF and IL-6 responses (S13 Fig). Thus, we postulate that other pathways may act in parallel with Syk-JNK-AP-1 pathway to regulate H. capsulatum-induced TNF and IL-6 response in macrophages.
We show that H. capsulatum induces Raf-1 activation in a Syk-dependent manner. Inhibition of Raf-1 increases H. capsulatum-induced TNF and IL-6 production. Our results define Raf-1 as a negative regulator in macrophage cytokine response to H. capsulatum. A previous study showed that Dectin-1 induces Raf-1 activation in a Syk-independent manner [26]. Activated Raf-1 antagonizes Syk-dependent non-canonical NF-κB activation by promoting inactive p65/RelB dimer formation [26]. Ligation of DC-SIGN activates Raf-1 which downregulates Borrelia burgdorferi- and TLR-induced TNF and IL-6 response by destabilizing their mRNAs and suppresses IL-12 response by impairing nucleosome remodeling at IL-12p35 promoter [43]. Together, these data suggest that activation of Raf-1 by ligation of PRRs negatively regulates and fine-tunes innate immune response. More studies are needed to demonstrate how Raf-1 negatively regulates H. capsulatum-induced TNF and IL-6 production.
There are only limited studies on the role of AP-1 in PRR crosstalk and in host defense against fungal infections. It has been shown that TLR2 and Dectin-1 cooperatively regulate zymosan-induced IL-10 production in DCs through an ERK-dependent, but AP-1-independent mechanism [44]. Other reports showed that Dectin-1 engagement in DCs triggers AP-1 activation, while curdlan stimulates a PLCγ2-dependent pathway, β-glucan on Aspergillus fumigatus activates a Syk-dependent pathway [22, 45, 46]. Our results demonstrate that CR3 acts in concert with Dectin-1 to activate both c-Jun and c-Fos. In addition, knocking down c-Jun and c-Fos in macrophages decreases H. capsulatum-induced TNF and IL-6 response, highlighting the role of AP-1 in host defense against fungal infections. Our MWA data show that PLCγ2 is activated by H. capsulatum. However, whether PLCγ2 is involved in CR3/Dectin-1-mediated AP-1 activation still remains to be determined. It is interesting to note that AP-1 activation is known to be associated with several disorders (ex. cancer and autoimmune diseases) by regulating genes involved in cell proliferation, angiogenesis and inflammation and inhibition of AP-1 activation is identified as a promising therapeutic strategy [47–49]. Our findings raise the possibility that administration of AP-1 inhibitor may increase susceptibility to fungal infections by suppressing proinflammatory cytokine production. Thus, AP-1inhibitor should be used with caution as a treatment modality for cancer and inflammatory disorder.
In addition to acting as a PRR to interact with pathogens, CR3 plays multiple roles in cellular processes including leukocyte extravasation, adhesion and chemotaxis [17]. This adds to the complexity of experimental design in addressing the role of CR3 in vivo. Intranasal or intraperitoneal infection of Itgam-/- mice with S. pneumonia results in infiltration of a large number of neutrophils to the infection sites [50, 51]. Neutrophil recruitment to the peritoneum is reduced in mice lacking CR3 after intraperitoneally challenge with C. albicans, yet accumulation of neutrophils in the kidney is comparable between WT and CR3-deficient mice intravenously infected with C. albicans [19, 52]. These studies demonstrated that abnormal neutrophil infiltration to the infection site should be considered as a factor influencing susceptibility in in vivo studies employing Itgam-/- mice. Indeed, we observed that unlike in WT mice, there is massive neutrophil infiltration to the lungs of Itgam-/- mice after intratracheal infection with H. capsulatum, a condition which may interfere the interaction between macrophages and the yeasts (S14A and S14B Fig). By contrast, the percentage and the number of neutrophils recruited to the spleen of Itgam-/- mice were commensurate with that in WT mice after intravenous inoculation of H. capsulatum. To focus on the roles of CR3 and Dectin-1 in macrophage interaction with H. capsulatum, we resorted to employ the disseminated histoplasmosis model by intravenous inoculation of the organism instead of pulmonary infection.
PRR signaling is known to act as a bridge that links innate and adaptive immunity. Signaling transduced by Dectin-1 can induce Th1 and Th17 responses [53] as well as priming cytotoxic T cells [54]. In addition, it has been reported that blockade of CR3 significantly reduces the Th1 and Th17 responses induced by A. fumigatus [55]. H. capsulatum infection triggers IFN-γ and IL-17 production by both CD4+ and CD8+ T cells, and both IFN-γ and IL-17 activate macrophages for inhibition of the replication of intracellular H. capsulatum [39, 41, 56, 57]. Mice deficient in IFN-γ or treated with neutralizing antibody against IFN-γ or IL-17 are more susceptible to H. capsulatum, showing the importance of IFN-γ and IL-17 in clearing this intracellular fungal pathogen [57–59]. However, whether and how PRRs participate in the development of H. capsulatum-induced IFN-γ and IL-17 response remains unclear. In this study, we show that both CR3 and Dectin-1 contribute to and function collaboratively in regulating TNF, IL-6, IL-17 and IFN-γ responses induced by H. capsulatum. TNF is a critical factor for the host to control H. capsulatum, and acts together with IFN-γ and IL-17 to provide protection [57, 60]. Although the role of IL-6 in histoplasomsis has not been well addressed, previous studies showed that the generation of H. capsulatum-induced IL-17 response is dependent on IL-6 and IL-6 deficiency leads to impairment of Th1 response in mice infected with C. albicans or A. fumigatus [57, 61, 62]. Our results also showed that, in addition to regulate macrophage TNF and IL-6 production, both CR3 and Dectin-1 are involved in IL-12 response in dendritic cells, suggesting the role of these receptors in regulating dendritic cells cannot be ignored. Moreover, our in vitro study showed that deficiency in either CR3 or Dectin-1 or both did not affect the intracellular growth of H. capsulatum in macrophages, strengthening the possibility that CR3 and Dectin-1 deficiency resulting in susceptibility to disseminated H. capsulatum infection is due to their roles in regulating IFN-γ and IL-17 responses. It is interesting to note that macrophages in the spleens of Itgam-/-Clec7a-/- mice were greatly diminished (almost exhausted) after infection with a lethal dose of H. capsulatum, presenting a picture that the macrophages are losing the battle to the fungal pathogen. While dendritic cells are major antigen-presenting cells and macrophage are the host, cytokine-producing cell and effector cell in infection by H. capsulatum, our study reveals that CR3 and Dectin-1 are of vital importance not only in their collaborative roles in macrophage cytokine production but also in instructing adaptive immune response against disseminated histoplasmosis.
In summary, we demonstrate for the first time the mechanism of receptor crosstalk between a member of the integrin family and CLR resulting in enhanced cytokine response. The collaboration between CR3 and Dectin-1 is through activation of Syk-JNK-AP-1 signaling pathway and dependent on formation of PRR clusters on lipid rafts. Our results also highlight the importance of CR3 and Dectin-1 in innate recognition that instructs antifungal adaptive immune response. Collectively, our findings provide a better understanding of the molecular mechanisms underlying the collaboration between CR3 and Dectin-1 and offer a valuable model for disentangling the intricacies of host-pathogen interactions.
Materials and Methods
Ethics statement
All animal experiments were undertaken in accordance with the Guidebook for the Care and Use of Laboratory Animals, 3rd Ed., 2007, published by The Chinese-Taipei Society of Laboratory Animal Sciences, approved by the Institutional Animal Care and Use Committee (IACUC, Permit number: 20130231) of National Taiwan University College of Medicine.
Fungus
Histoplasma capsulatum (Hc) strain 505 yeast cells were used in the whole study. Yeast cells were cultured at 37°C on brain heart infusion (BHI) agar supplemented with 1 mg/ml cysteine and 20 mg/ml glucose. Heat-killed yeast cells were prepared by treatment at 65°C for 2 h. To examine the surface expression of α-glucan and β-glucan, viable or heat-killed yeasts were fixed with 4% paraformaldehyde and stained with antibodies against α-(1,3)-glucan (Clone MOPC-104E) (Biolegend, San Diego, CA, USA) and β-(1,3)-glucan (Biosupplies, Parkville, Australia) followed by analysis with flow cytometry (BD FACSCanto II, BD Biosciences).
Mice
Itgam-/- (Stock number: 003991) and wild-type C57BL/6 (Stock number: 000664) mice were originally purchased from the Jackson Laboratories (Bar Harbor, ME, USA) and Clec7a-/- mice were generated by Dr. Gordon D. Brown [63]. Itgam-/-Clec7a-/- mice were generated by crossing Itgam-/- and Clec7a-/- mice. Mice heterozygous for a deletion in the Syk locus (Syk+/-) were obtained from Dr. Clifford Lowell (University of California, San Francisco, CA, USA) [64]. All strains used in this study were on C57BL/6 background. They were maintained and bred in the National Taiwan University College of Medicine Laboratory Animal Center (NTU CMLAC) or in the National Laboratory Animal Center (NLAC, Taiwan) under specific pathogen-free (SPF) conditions. In vivo infection experiments were performed following biosafety level 2 (BSL-2) guidelines.
Cells
Peritoneal macrophages were collected by lavage from mice at 4 days after peritoneal injection of 1 ml of 3% thioglycollate medium (Sigma-Aldrich, St Louis, MO, USA). Macrophages deficient in Syk were derived from fetal liver cells obtained from Syk-/- mouse embryos (E13.5-E15.5). Syk-/- embryos were separated from Syk+/+ and Syk+/- embryos by their exhibition of severe petechiae and confirmed by genotyping [65]. Single-cell suspensions from fetal liver tissues were cultured in 20% L929-cell conditioned medium for 7 days. Over 95% of the adherent cells were F4/80+ which were identified as fetal liver-derived macrophages (FLDMs).
Reagents and antibodies
Syk inhibitors SykI and BAY 61-3606, PI3K inhibitor LY294002, ERK inhibitor U0126, JNK inhibitor SP600125, and p38 inhibitor SB203580 were purchased from Calbiochem-Merck (Darmstadt, Germany). Raf-1 inhibitor GW5074, methyl-β-cyclodextrin (MβCD), and water-soluble cholesterol were obtained from Sigma-Aldrich.
Antibodies against phospho (p)-Zap-70 (Tyr319)/Syk (Tyr352), p-Akt (Tyr308), p-c-Raf (Ser338), p-ERK1/2 (Thr202/Tyr204), p-JNK (Thr183/Tyr185), JNK, p-p38 (Thr180/Tyr182), p-IKKα/β (Ser176/180), p-NF-κBp65 (Ser536), p-IκBα (Ser32), IκBα, p-c-Jun (Ser63), c-Jun, p-c-Fos (Ser32), and c-Fos were purchased from Cell Signaling (Beverly, MA, USA). Anti-Syk, anti-β-actin, HRP-conjugated anti-rabbit IgG, and HRP-conjugated anti-mouse IgG antibodies were purchased from GeneTex Inc. (Irvine, CA, USA). Blocking antibodies against CR3 (clone 5C6) and Dectin-1 (clone 2A11) were purchased from Serotec (Oxford, UK). Antibodies for cell surface staining, anti-CD11b (clone M1/70), anti-CD18 (clone GAME-46), and APC-conjugated anti-F4/80 antibody were obtained from eBioscience (San Diego, CA, USA), and anti-Dectin-1 (clone 218820) was purchased from R&D Systems (Minneapolis, MN, USA).
Stimulation with fungus and particulate ligands
Macrophages were stimulated with viable or heat-killed H. capsulatum yeasts (at a yeast-to-macrophage ratio of 20/1) or iC3b-caoted beads (at a bead-to-cell ration of 10/1) and 50 μg/ml depleted zymosan (InvivoGen, San Diego, CA, USA).The iC3b-coated beads were prepared as described previously [66]. Briefly, 2 × 108 of 3 μm Latex beads (Sigma-Aldrich) were incubated with PBS containing 20 μg/ml human IgM (Sigma-Aldrich) at 37°C for 60 min. Beads were washed with PBS then resuspended in freshly isolated mouse serum (diluted 1:1 in PBS) and incubated at 37°C for another 20 min. Beads were washed with Hank's balanced salt solution then resupspended in RPMI 1640 medium supplemented with 10% heat-inactivated FBS. During the process, the classical pathway of complement cascade was activated, resulting in C3b deposition on the surface of beads where it was rapidly and completely converted to iC3b [67].
Cytokine assays
Macrophages were stimulated with or without H. capsulatum or particulate ligands. Culture supernatants were collected after incubation at 37°C for different periods of time. The concentrations of TNF and IL-6 in the supernatants were quantified by enzyme-linked immunosorbent assay (ELISA) kit (eBioscience) following the manufacturer’s instructions.
Western blotting
Cells were lysed by PhosphoSafe Extraction Reagent cell lysis buffer (Novagen, Madison, WI, USA). Whole cell lysates were subjected to electrophoresis at 10% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE), and transferred to PVDF membrane. The membrane was incubated in buffer containing primary antibody against molecule of interest, followed by HRP-conjugated secondary antibody. The blot was developed by chemiluminescence using ECL solution (Millipore, Billerica, MA, USA). For normalization, the intensity of blots was quantified by ImageJ software (NIH, Bethesda, MD, USA).
Isolation of lipid raft fractions
Macrophages (3 × 107) were lysed with 0.5% Brij in TNE buffer [25 mM Tris (pH 7.5), 150 mM NaCl, 5 mM EDTA, protease inhibitors, 1 mM Na3VO4, and 1 mM NaF] and let stand on ice for 1 h. Lysates were then mixed with equal volume of 80% sucrose in TNE buffer and overlaid with 30% and 5% sucrose in the same buffer. The gradients were centrifuged at 40,000 × g in a SW55Ti rotor (Beckman Coulter, Fullerton, CA, USA) at 4°C for 18 h. Twelve fractions were collected and the proteins in the fractions were subjected to electrophoresis at 10% SDS-PAGE and Western blot analysis by using antibodies against CD11b (GeneTex), Dectin-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), Syk, p-Syk, and flotillin-1 (Cell Signaling).
Immunofluorescence staining and confocal microscopy
Macrophages were allowed to adhere on cover slide overnight and stimulated with heat-killed H. capsulatum yeasts. After stimulation, cells were fixed with 3% paraformaldehyde followed by permeabilization with 0.5% Triton X-100. Cells were blocked with PBS containing 5% heat-inactivated FBS and stained with Alexa Fluor 647-conjugated cholera toxin B (Invitrogen, Carlsbad, CA, USA), anti-p-Syk (Cell Signaling), anti-Dectin-1 (Serotec), and/or PE-conjugated anti-CD11b (eBioscience) antibodies. Cells were then stained with secondary Alexa Fluor-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA). Cell nuclei were stained with Hoechst 33258. The images were acquired with a Zeiss Axiovert 100TV confocal microscope (Carl Zeiss Inc., Jena, Germany) and analyzed by Zen software (Carl Zeiss Inc.) and ImageJ software (NIH).
Micro-Western Arrays (MWAs)
Peritoneal macrophages were stimulated with or without heat-killed H. capsulatum. Cells were lysed at different time points, and Micro-Western Arrays (MWAs) were performed to measure protein expression as previously described [68]. Blots were analyzed by Odyssey analysis software (Li-Cor Biosciences, USA). Heat maps were created by using PermutMatrix software (LIRMM).
siRNA transfection of macrophages
Peritoneal macrophages (1 × 106) were transfected with 30 pmol of control siRNA or siRNAs targeting c-Fos or c-Jun (Santa Cruz Biotechnology) using the Amaxa Nucleofector kit for mouse macrophages (Lonza, Basel, Switzerland) with a Nucleofector II electroporation device (Lonza). After transfection, cells were gently resuspended in RPMI 1640 medium supplemented with 20% heat-inactivated FBS and plated in 12-well tissue culture plate. Adherent cells were collected for cytokine assay and Western blot analysis 48 h later.
H. capsulatum infection, fungal burden and leukocyte populations in spleen
Mice were injected intravenously with low (2.5 × 105) or high (5 × 106) dose of H. capsulatum yeasts suspended in PRMI 1640 medium. For survival studies, mice were monitored for up to 30 days. For immunological studies, mice were killed on day 7 (low dose) or day 9 (high dose) after infection. To determine the fungal burden, spleens were weighted and homogenized in sterile RPMI 1640 medium. The homogenates were serially diluted and plated on glucose-pepton agar plates. Mycelial colonies were counted 10 days after incubation as described elsewhere [40]. To determine the percentage of leukocyte populations in spleen, splenocytes were stained for surface CD4, CD8, B220, F4/80, Ly6G and CD11c and analyzed by flow cytometry. All antibodies were purchased from eBioscience.
Ex vivo cytokine production and intracellular cytokine staining
To study cytokine production by splenocytes, single cell suspensions were prepared from the spleen. Five million splenocytes were cultured in RPMI 1640 complete medium containing 400 pg/ml of rIL-2 for 48 h. The concentrations of TNF, IL-6, IL-17A and IFN-γ in the culture supernatants were quantified by ELISA. To analyze intracellular IFN-γ, 1 × 106 splenocytes were cultured in RPMI 1640 complete medium for 24 h and monensin (2 μM, Sigma-Aldrich) was added at 6 h before harvest. Cells were stained with anti-CD4, anti-CD8 and anti-IFN-γ antibodies as described previously [40]. The percentage of IFN-γ-producing cells in the total CD4+ or CD8+ T cell populations was calculated by dividing the % of IFN-γ+CD4+ cells or IFN-γ+CD8+ by the % of CD4+ or CD8+ cells. All antibodies were purchased from eBioscience.
Statistics
The comparisons between multiple groups were analyzed with one-way ANOVA followed by Tukey post-hoc test or by Duncan post-hoc analysis using SPSS 22.0 statistical software (IBM, Armonk, NY, USA). The differences between two groups were tested by two-tailed t-test. Generalized Wilcoxon test was used to analyze mouse survival. Differences were considered significant at a P value of < 0.05.
Accession numbers
The accession numbers in the UniPortKB/SwissProt database of the proteins mentioned in this study are followed: CD11b, P05555; CD18, P11835; Dectin-1, Q6QLQ4; Syk, P43404; JNK, Q91Y86 (JNK1) and Q9WTU6 (JNK2); c-Fos, P01101; c-Jun, P05627; Raf-1, Q99N57; PLCγ2, Q8CIH5; Akt, P31750; ERK, Q63844 (ERK1) and P63085 (ERK2); p38, P47811; IKKα, Q60680; IKKβ, O88351; IκBα, Q9Z1E3; NF-κBp65, Q04207; PKCε, P16054; PKCη, P23298; PKCθ, Q02111; β-actin, P60710; GAPDH, P16858; flotillin-1, O08917; TNF, P06804; IL-6, P08505; IL-12p35, P43431; IL-12p40, P43432; IL-17A, Q62386; IFN-γ, P01580.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Brown GD, Denning DW, Levitz SM. Tackling human fungal infections. Science. 2012;336(6082):647. doi: 10.1126/science.1222236 22582229.
2. Gordon S. Pattern recognition receptors: doubling up for the innate immune response. Cell. 2002;111(7):927–30. 12507420.
3. Brown GD. Innate antifungal immunity: the key role of phagocytes. Annu Rev Immunol. 2011;29:1–21. doi: 10.1146/annurev-immunol-030409-101229 20936972.
4. Guimaraes AJ, de Cerqueira MD, Nosanchuk JD. Surface architecture of Histoplasma capsulatum. Front Microbiol. 2011;2:225. doi: 10.3389/fmicb.2011.00225 22121356.
5. Smits GJ, Kapteyn JC, van den Ende H, Klis FM. Cell wall dynamics in yeast. Curr Opin Microbiol. 1999;2(4):348–52. 10458981.
6. Hontelez S, Sanecka A, Netea MG, van Spriel AB, Adema GJ. Molecular view on PRR cross-talk in antifungal immunity. Cell Microbiol 2012;14(4):467–74. doi: 10.1111/j.1462-5822.2012.01748.x 22233321.
7. Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197(9):1107–17. doi: 10.1084/jem.20021787 12719479.
8. Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med. 2003;197(9):1119–24. doi: 10.1084/jem.20021890 12719478.
9. Dennehy KM, Ferwerda G, Faro-Trindade I, Pyz E, Willment JA, Taylor PR, et al. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol. 2008;38(2):500–6. doi: 10.1002/eji.200737741 18200499.
10. Ferwerda G, Meyer-Wentrup F, Kullberg B-J, Netea MG, Adema GJ. Dectin-1 synergizes with TLR2 and TLR4 for cytokine production in human primary monocytes and macrophages. Cell Microbiol. 2008;10(10):2058–66. doi: 10.1111/j.1462-5822.2008.01188.x 18549457.
11. Takahara K, Tokieda S, Nagaoka K, Takeda T, Kimura Y, Inaba K. C-type lectin SIGNR1 enhances cellular oxidative burst response against C. albicans in cooperation with Dectin-1. Eur J Immunol. 2011;41(5):1435–44. doi: 10.1002/eji.200940188 21400494.
12. Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Groß O, et al. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J Exp Med. 2009;206(9):2037–51. doi: 10.1084/jem.20082818 19703985.
13. Lin JS, Huang JH, Hung LY, Wu SY, Wu-Hsieh BA. Distinct roles of complement receptor 3, Dectin-1, and sialic acids in murine macrophage interaction with Histoplasma yeast. J Leukoc Biol. 2010;88(1):95–106. doi: 10.1189/jlb.1109717 20360401.
14. Arnaout MA. Structure and function of the leukocyte adhesion molecules CD11/CD18. Blood. 1990;75(5):1037–50. 1968349.
15. Vetvicka V, Thornton BP, Ross GD. Soluble beta-glucan polysaccharide binding to the lectin site of neutrophil or natural killer cell complement receptor type 3 (CD11b/CD18) generates a primed state of the receptor capable of mediating cytotoxicity of iC3b-opsonized target cells. J Clin Invest. 1996;98(1):50–61. doi: 10.1172/JCI118777 8690804.
16. Mayadas TN, Cullere X. Neutrophil β2 integrins: moderators of life or death decisions. Trends Immunol. 2005;26(7):388–95. doi: 10.1016/j.it.2005.05.002 15922663.
17. Abram CL, Lowell CA. The Ins and Outs of Leukocyte Integrin Signaling. Annu Rev Immunol. 2009;27(1):339–62. doi: 10.1146/annurev.immunol.021908.132554 19302044.
18. Harokopakis E, Hajishengallis G. Integrin activation by bacterial fimbriae through a pathway involving CD14, Toll-like receptor 2, and phosphatidylinositol-3-kinase. Eur J Immunol. 2005;35(4):1201–10. doi: 10.1002/eji.200425883 15739163.
19. Li X, Utomo A, Cullere X, Choi Myunghwan M, Milner Danny A Jr, Venkatesh D, et al. The β-Glucan receptor Dectin-1 activates the integrin Mac-1 in neutrophils via Vav protein signaling to promote Candida albicans clearance. Cell Host Microbe. 2011;10(6):603–15. doi: 10.1016/j.chom.2011.10.009 22177564.
20. Groves E, Dart AE, Covarelli V, Caron E. Molecular mechanisms of phagocytic uptake in mammalian cells. Cell Mol Life Sci. 2008;65(13):1957–76. doi: 10.1007/s00018-008-7578-4 18322649.
21. Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol. 2006;6(1):33–43. doi: 10.1038/nri1745 16341139.
22. Xu S, Huo J, Lee K- G, Kurosaki T, Lam K- P. Phospholipase Cγ2 is critical for Dectin-1-mediated Ca2+ flux and cytokine production in dendritic cells. J Biol Chem. 2009;284(11):7038–46. doi: 10.1074/jbc.M806650200 19136564.
23. Goodridge HS, Simmons RM, Underhill DM. Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J Immunol. 2007;178(5):3107–15. 17312158.
24. Gross O, Gewies A, Finger K, Schäfer M, Sparwasser T, Peschel C, et al. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature. 2006;442(7103):651–6. doi: 10.1038/nature04926 16862125.
25. Jia XM, Tang B, Zhu LL, Liu YH, Zhao XQ, Gorjestani S, et al. CARD9 mediates Dectin-1-induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J Exp Med. 2014;211(11):2307–21. doi: 10.1084/jem.20132349 25267792.
26. Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SCM, et al. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol. 2009;10(2):203–13. doi: 10.1038/ni.1692 19122653.
27. Goodridge HS, Shimada T, Wolf AJ, Hsu Y-MS, Becker CA, Lin X, et al. Differential use of CARD9 by Dectin-1 in macrophages and dendritic cells. J Immunol. 2009;182(2):1146–54. doi: 10.4049/jimmunol.182.2.1146 19124758.
28. Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106(7):2543–50. doi: 10.1182/blood-2005-03-1239 15956283.
29. Li B, Allendorf DJ, Hansen R, Marroquin J, Ding C, Cramer DE, et al. Yeast β-glucan amplifies phagocyte killing of iC3b-opsonized tumor cells via complement receptor 3-Syk-phosphatidylinositol 3-kinase pathway. J Immunol. 2006;177(3):1661–9. 16849475.
30. Pande G. The role of membrane lipids in regulation of integrin functions. Curr Opin Cell Biol 2000;12(5):569–74. 10978891.
31. Xu S, Huo J, Gunawan M, Su I-H, Lam K-P. Activated Dectin-1 localizes to lipid raft microdomains for signaling and activation of phagocytosis and cytokine production in dendritic cells. J Biol Chem. 2009;284(33):22005–11. doi: 10.1074/jbc.M109.009076 19525229.
32. Wu-Hsieh BA, Lee GS, Franco M, Hofman FM. Early activation of splenic macrophages by tumor necrosis factor alpha is important in determining the outcome of experimental histoplasmosis in mice. Infect Immun. 1992;60(10):4230–8. 1398934.
33. Rappleye CA, Eissenberg LG, Goldman WE. Histoplasma capsulatum α-(1,3)-glucan blocks innate immune recognition by the β-glucan receptor. Proc Natl Acad Sci U S A. 2007;104(4):1366–70. doi: 10.1073/pnas.0609848104 17227865.
34. Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 2005;24(6):1277–86. doi: 10.1038/sj.emboj.7600594 15729357.
35. Saijo S, Iwakura Y. Dectin-1 and Dectin-2 in innate immunity against fungi. Int Immunol. 2011;23(8):467–72. doi: 10.1093/intimm/dxr046 21677049.
36. Ratnoff WD, Pepple JM, Winkelstein JA. Activation of the alternative complement pathway by Histoplasma capsulatum. Infect Immun. 1980;30(1):147–9. 7439970.
37. Goodridge HS, Reyes CN, Becker CA, Katsumoto TR, Ma J, Wolf AJ, et al. Activation of the innate immune receptor Dectin-1 upon formation of a 'phagocytic synapse'. Nature. 2011;472(7344):471–5. doi: 10.1038/nature10071 21525931.
38. Inoue M, Moriwaki Y, Arikawa T, Chen YH, Oh YJ, Oliver T, et al. Cutting edge: critical role of intracellular osteopontin in antifungal innate immune responses. J Immunol. 2011;186(1):19–23. doi: 10.4049/jimmunol.1002735 21135164.
39. Wu-Hsieh BA, Howard DH. Inhibition of the intracellular growth of Histoplasma capsulatum by recombinant murine gamma interferon. Infect Immun. 1987;55(4):1014–6. 3104206.
40. Lin JS, Yang CW, Wang DW, Wu-Hsieh BA. Dendritic cells cross-present exogenous fungal antigens to stimulate a protective CD8 T cell response in infection by Histoplasma capsulatum. J Immunol. 2005;174(10):6282–91. 15879127.
41. Wu SY, Yu JS, Liu FT, Miaw SC, Wu-Hsieh BA. Galectin-3 negatively regulates dendritic cell production of IL-23/IL-17-axis cytokines in infection by Histoplasma capsulatum. J Immunol. 2013;190(7):3427–37. doi: 10.4049/jimmunol.1202122 23455499.
42. Subramanian Vignesh K, Landero Figueroa JA, Porollo A, Caruso JA, Deepe GS Jr. Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity. 2013;39(4):697–710. doi: 10.1016/j.immuni.2013.09.006 24138881.
43. Hovius JWR, de Jong MAWP, den Dunnen J, Litjens M, Fikrig E, van der Poll T, et al. Salp15 binding to DC-SIGN inhibits cytokine expression by impairing both nucleosome remodeling and mRNA stabilization. PLoS Pathog. 2008;4(2):e31. doi: 10.1371/journal.ppat.0040031 18282094.
44. Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest. 2006;116(4):916–28. doi: 10.1172/jci27203 16543948.
45. Tassi I, Cella M, Castro I, Gilfillan S, Khan WN, Colonna M. Requirement of phospholipase C-gamma2 (PLCgamma2) for Dectin-1-induced antigen presentation and induction of TH1/TH17 polarization. Eur J Immunol. 2009;39(5):1369–78. doi: 10.1002/eji.200839313 19404984.
46. Toyotome T, Adachi Y, Watanabe A, Ochiai E, Ohno N, Kamei K. Activator protein 1 is triggered by Aspergillus fumigatus beta-glucans surface-exposed during specific growth stages. Microb Pathog. 2008;44(2):141–50. doi: 10.1016/j.micpath.2007.08.015 17928189.
47. Palanki MS. Inhibitors of AP-1 and NF-kappa B mediated transcriptional activation: therapeutic potential in autoimmune diseases and structural diversity. Curr Med Chem. 2002;9(2):219–27. 11860356.
48. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002;4(5):E131–6. doi: 10.1038/ncb0502-e131 11988758.
49. Ye N, Ding Y, Wild C, Shen Q, Zhou J. Small molecule inhibitors targeting activator protein 1 (AP-1). J Med Chem. 2014;57(16):6930–48. doi: 10.1021/jm5004733 24831826.
50. Prince JE, Brayton CF, Fossett MC, Durand JA, Kaplan SL, Smith CW, et al. The differential roles of LFA-1 and Mac-1 in host defense against systemic infection with Streptococcus pneumoniae. J Immunol. 2001;166(12):7362–9. 11390487.
51. Rijneveld AW, de Vos AF, Florquin S, Verbeek JS, van der Poll T. CD11b limits bacterial outgrowth and dissemination during murine pneumococcal pneumonia. J Infect Dis. 2005;191(10):1755–60. doi: 10.1086/429633 15838804.
52. Soloviev DA, Jawhara S, Fonzi WA. Regulation of innate immune response to Candida albicans infections by alphaMbeta2-Pra1p interaction. Infect Immun. 2011;79(4):1546–58. doi: 10.1128/iai.00650-10 21245270.
53. LeibundGut-Landmann S, Grosz O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8(6):630–8. doi: 10.1038/ni1460 17450144.
54. Leibundgut-Landmann S, Osorio F, Brown GD, Reis e Sousa C. Stimulation of dendritic cells via the dectin-1/Syk pathway allows priming of cytotoxic T-cell responses. Blood. 2008;112(13):4971–80. doi: 10.1182/blood-2008-05-158469 18818389.
55. Gresnigt MS, Becker KL, Smeekens SP, Jacobs CW, Joosten LA, van der Meer JW, et al. Aspergillus fumigatus-induced IL-22 is not restricted to a specific Th cell subset and is dependent on complement receptor 3. J Immunol. 2013;190(11):5629–39. doi: 10.4049/jimmunol.1202601 23645883.
56. Lin JS, Wu-Hsieh BA. Functional T cells in primary immune response to histoplasmosis. Int Immunol. 2004;16(11):1663–73. doi: 10.1093/intimm/dxh168 15452021.
57. Deepe GS Jr., Gibbons RS. Interleukins 17 and 23 influence the host response to Histoplasma capsulatum. J Infec Dis. 2009;200(1):142–51. doi: 10.1086/599333 19469707.
58. Clemons KV, Darbonne WC, Curnutte JT, Sobel RA, Stevens DA. Experimental histoplasmosis in mice treated with anti-murine interferon-gamma antibody and in interferon-gamma gene knockout mice. Microbes Infect. 2000;2(9):997–1001. 10967280.
59. Allendoerfer R, Deepe GS Jr. Intrapulmonary response to Histoplasma capsulatum in gamma interferon knockout mice. Infect Immun. 1997;65(7):2564–9. 9199420.
60. Zhou P, Miller G, Seder RA. Factors involved in regulating primary and secondary immunity to infection with Histoplasma capsulatum: TNF-α plays a critical role in maintaining secondary immunity in the absence of IFN-γ. J Immunol. 1998;160(3):1359–68. 9570555.
61. Romani L, Mencacci A, Cenci E, Spaccapelo R, Toniatti C, Puccetti P, et al. Impaired neutrophil response and CD4+ T helper cell 1 development in interleukin 6-deficient mice infected with Candida albicans. J Exp Med. 1996;183(4):1345–55. 8666893.
62. Cenci E, Mencacci A, Casagrande A, Mosci P, Bistoni F, Romani L. Impaired antifungal effector activity but not inflammatory cell recruitment in interleukin-6-deficient mice with invasive pulmonary aspergillosis. J Infec Dis. 2001;184(5):610–7. doi: 10.1086/322793 11494166.
63. Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. 2007;8(1):31–8. doi: 10.1038/ni1408 17159984.
64. Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 1995;378(6554):303–6. doi: 10.1038/378303a0 7477353.
65. Yanagi S, Inatome R, Ding J, Kitaguchi H, Tybulewicz VLJ, Yamamura H. Syk expression in endothelial cells and their morphologic defects in embryonic Syk-deficient mice. Blood. 2001;98(9):2869–71. doi: 10.1182/blood.V98.9.2869 11675365.
66. Chow CW, Downey GP, Grinstein S. Measurements of phagocytosis and phagosomal maturation. Curr Protoc Cell Biol. 2004;Chapter 15:Unit 15.7. doi: 10.1002/0471143030.cb1507s22 18228442.
67. Newman SL, Mikus LK. Deposition of C3b and iC3b onto particulate activators of the human complement system. Quantitation with monoclonal antibodies to human C3. J Exp Med. 1985;161(6):1414–31. 2409200.
68. Ciaccio MF, Wagner JP, Chuu C-P, Lauffenburger DA, Jones RB. Systems analysis of EGF receptor signaling dynamics with microwestern arrays. Nat Methods. 2010;7(2):148–55. doi: 10.1038/nmeth.1418 20101245.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 7
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- RNA Virus Reassortment: An Evolutionary Mechanism for Host Jumps and Immune Evasion
- Activation of TLR2 and TLR6 by Dengue NS1 Protein and Its Implications in the Immunopathogenesis of Dengue Virus Infection
- N-acetylglucosamine Regulates Virulence Properties in Microbial Pathogens
- Characterization of a Prefusion-Specific Antibody That Recognizes a Quaternary, Cleavage-Dependent Epitope on the RSV Fusion Glycoprotein