
Probing the Metabolic Network in Bloodstream-Form Using Untargeted Metabolomics with Stable Isotope Labelled Glucose
In this work we have followed the distribution of carbon derived from glucose in bloodstream form trypanosomes, the causative agent of African trypanosomiasis, revealing it to enter a diverse range of metabolites. The work involved using 13C-labelled glucose and following the fate of the labelled carbon with an LC-MS based metabolomics platform. Beyond glycolysis and the oxidative branch of the pentose phosphate pathway the label entered lipid biosynthesis both through glycerol 3-phosphate and also acetate. Glucose derived carbon also entered nucleotide synthesis through ribose and pyrimidine synthesis through oxaloacetate-derived aspartate. Appreciable quantities of the carboxylic acids succinate and malate were identified, although labelling patterns indicate they are not TCA cycle derived. Amino sugars and sugar nucleotides were also labelled as was inositol used in protein modification but not in inositol phospholipid headgroup production. We confirm active and essential oxaloacetate production in bloodstream form trypanosomes and show that phosphoenolpyruvate carboxykinase is essential to these parasites using RNA interference. The amount of glucose entering these metabolites is minor compared to the quantity that enters pyruvate excreted from the cell, but the observation that enzymes contributing to the metabolism of glucose beyond glycolysis can be essential offers potential new targets for chemotherapy against trypanosomiasis.
Published in the journal:
. PLoS Pathog 11(3): e32767. doi:10.1371/journal.ppat.1004689
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004689
Summary
In this work we have followed the distribution of carbon derived from glucose in bloodstream form trypanosomes, the causative agent of African trypanosomiasis, revealing it to enter a diverse range of metabolites. The work involved using 13C-labelled glucose and following the fate of the labelled carbon with an LC-MS based metabolomics platform. Beyond glycolysis and the oxidative branch of the pentose phosphate pathway the label entered lipid biosynthesis both through glycerol 3-phosphate and also acetate. Glucose derived carbon also entered nucleotide synthesis through ribose and pyrimidine synthesis through oxaloacetate-derived aspartate. Appreciable quantities of the carboxylic acids succinate and malate were identified, although labelling patterns indicate they are not TCA cycle derived. Amino sugars and sugar nucleotides were also labelled as was inositol used in protein modification but not in inositol phospholipid headgroup production. We confirm active and essential oxaloacetate production in bloodstream form trypanosomes and show that phosphoenolpyruvate carboxykinase is essential to these parasites using RNA interference. The amount of glucose entering these metabolites is minor compared to the quantity that enters pyruvate excreted from the cell, but the observation that enzymes contributing to the metabolism of glucose beyond glycolysis can be essential offers potential new targets for chemotherapy against trypanosomiasis.
Introduction
Trypanosoma brucei is a protozoan parasite, sub-species of which are responsible for human African trypanosomiasis (HAT) and animal African trypanosomiasis (Nagana) [1]. Current treatments for HAT are inadequate, and new preventative and therapeutic options are urgently required [2]. The parasite is transmitted between mammalian hosts by a tsetse fly vector. The physiological environments of mammalian blood and the tsetse midgut diverge significantly, and bloodstream form and insect form parasites reveal adaptive differences in biochemistry to allow optimised survival in these environments [3,4].
T. brucei possess glycosomes, peroxisome-derived organelles that contain enzymes required for glycolysis [5]. Within the tsetse fly, glucose is generally scarce with proline a key energy source [6]. Procyclic form (PCF) trypanosomes preferentially use proline as a source of carbon and energy [6,7,8], although in glucose-rich culture medium they preferentially utilize glucose through glycolysis [7,8]. For many years it has been widely accepted that bloodstream form (BSF) trypanosomes exhibit greatly reduced metabolic potential, where glycosomal glucose utilization through glycolysis is the sole energy source [4,5,9,10]. Under aerobic conditions pyruvate was considered the sole end-product, with the redox balance maintained by a mitochondrial alternative oxidase shunt acting to regenerate dihydroxyacetone phosphate (DHAP) from glycerol 3-phosphate [11]. Under anaerobiosis, equal quantities of pyruvate and glycerol are produced, where the reverse reaction of glycerol kinase (GK) generates glycerol from glycerol 3-phosphate [9]. Early studies on BSF trypanosomes isolated from rodents identified other secreted metabolites, including succinate, aspartate and alanine [12–14]. However, the fact that these products were present in small amounts and that stumpy form trypanosomes, a non-replicative form of the parasite pre-adapted for life in the tsetse fly [15,16], could contaminate the slender BSF preparations, led to these observations being considered of limited relevance in formulating models of a simplified pathway for glucose catabolism in the slender replicative form of T. brucei [9].
Evidence for a more diverse fate of glucose in BSF trypanosome metabolism has, however, emerged. For example, NMR analysis of glucose metabolism [17,18] confirmed glycerol and pyruvate as the major end products of glycolysis, while also detecting significant amounts of alanine. The conversion of pyruvate to alanine may even be essential to BSF trypanosomes, since it was not possible to knockout the alanine aminotransferase (AAT) gene [19], although significant reduction of its transcript by RNAi was possible [19]. An oxidative branch of the pentose phosphate pathway (PPP) is operative [20], and 6-phosphogluconate dehydrogenase is essential to these forms [21,22]. Glucose-derived ribose was also shown to be incorporated into cellular nucleotides in BSF trypanosomes [23,24], and inositol derived from glucose is incorporated into the glycosylphosphatidyl inositol anchors attached to the abundant variant surface glycoprotein coat of these cells [25,26]. Recently it was shown that pyruvate dehydrogenase (PDH) becomes essential if slender bloodstream forms are deprived of threonine as a source of acetate [27]; in this situation PDH is needed to generate acetate from glucose metabolism.
The systematic application of reverse genetics to PCF trypanosomes combined with NMR analyses of end products of metabolism has demonstrated the importance of succinate fermentation pathways in both the glycosome and mitochondrion, through a route independent of the classical TCA cycle [28–30]. A glycosomal succinate shunt converts phosphoenolpyruvate (PEP) produced in the cytosol to oxaloacetate using PEP carboxykinase (PEPCK) following glycosomal reuptake. PEP can also be converted to pyruvate, which enters the mitochondrion to feed the PDH complex for production of acetyl-CoA, which is converted by the mitochondrial acetate:succinate CoA-transferase (ASCT) and acetyl-CoA thioesterase (ACH) to produce acetate [30,31].
The advent of metabolomics technology [32–34] allows an unbiased analysis of metabolism across diverse pathways in T. brucei. Stable-isotope labelling permits direct detection of active metabolic pathways within a live cell system [35–39] and offers a direct route to follow the distribution of atoms from a precursor substrate through the metabolic network. We have previously applied the approach to PCF trypanosomes [38], and here extend the methodology to make a comprehensive assessment of the distribution of glucose-derived carbon through the BSF trypanosome.
Results
Extensive glucose utilization for anabolic processes in bloodstream form T. brucei
BSF T. brucei were fed a 50:50 mixture of U-12C and U-13C-glucose as the major carbon source for one hour and 24 hours. Glycolytic intermediates were rapidly labelled, as expected as a result of high flux through the glycolytic pathway [10], with complete (50%) labelling of the major end-product, pyruvate. Significant labelling was observed in metabolites of many other pathways, indicating extensive anabolic utilization of glucose. In total, over 150 metabolites were detected with glucose-derived carbon labelling, including sugar phosphates, sugar nucleotides, lipids and secondary metabolites derived from glycolytic intermediates and end-products (S1 Table). Whilst excreted pyruvate accounts for the majority of catabolised glucose [10], glucose-derived pyruvate also serves as a major source of alanine and acetyl-CoA within the cell. Furthermore, metabolism of glucose through the oxidative branch of the PPP provides ribose 5-phosphate, as evidenced by its being 50% U-13C-labelled, which in turn is utilized for nucleotide biosynthesis. Of significant interest was the labelling of numerous carboxylic acids including malate, fumarate and succinate, and extensive labelling in aspartate and various pyrimidines. In all, these results demonstrate extensive utilization of glucose for anabolic purposes in bloodstream form T. brucei (Fig 1).
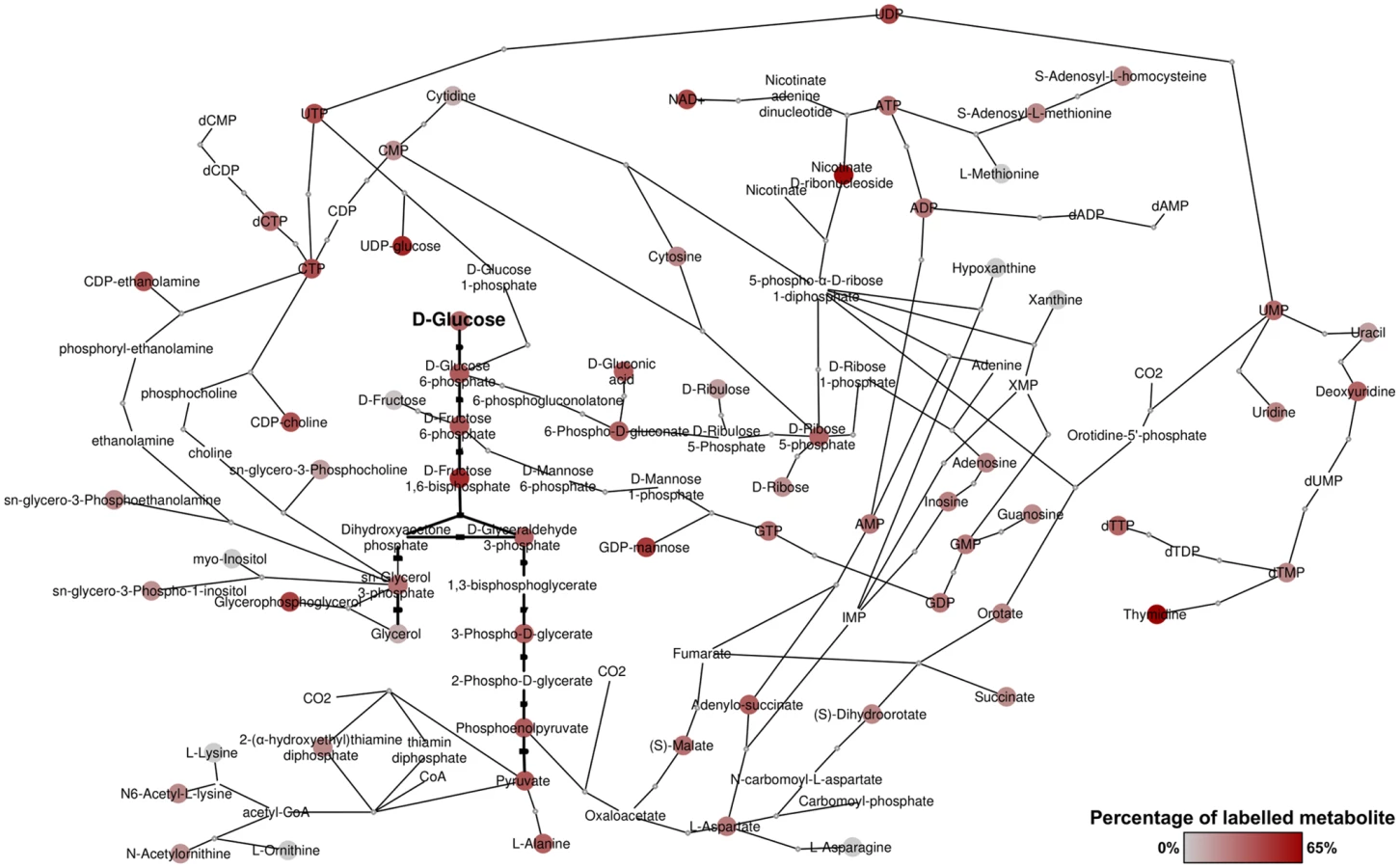
Revisiting glycolysis in bloodstream form T. brucei
The glycolytic pathway is highly active in T. brucei, which explains the 6-carbon labelling in glucose 6-phosphate (G6P), fructose 6-phosphate (F6P) and fructose-1,6-bisphosphate (FBP), and the 3-carbon labelling in the triose phosphate intermediates of lower glycolysis (Fig 2).
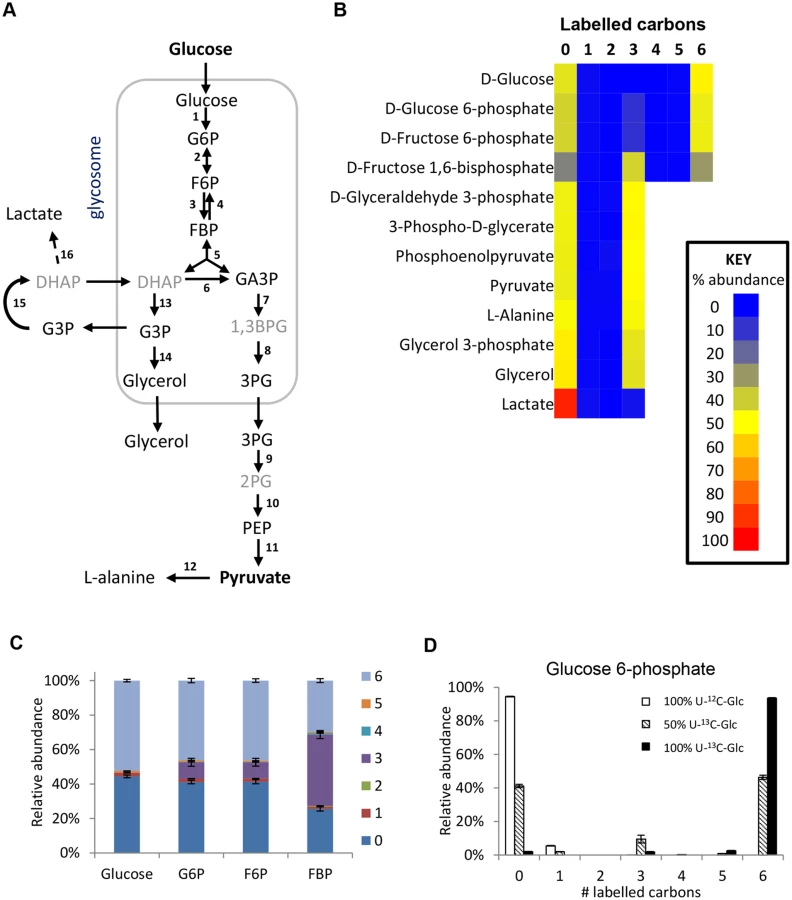
Interestingly, additional 3-carbon labelled isotopologues of hexose phosphates were observed. These isotopologues represent 10% of the intracellular G6P and F6P, and 40% of the FBP. These metabolites were identified by exact mass and retention time using authentic standards, although we cannot absolutely rule out other metabolites of the same mass and retention time (i.e. unanticipated hexose phosphate isomers) co-eluting with these metabolites. These findings are inconsistent with unidirectional glucose flow through the Embden-Meyerhof-Parnas glycolytic pathway. Labelling studies with 100% U-13C-glucose produced very little 3-labelled hexose phosphate (~2% of the hexose phosphate is 3-labelled even when only ~97% of the glucose is labelled inside the cells), indicating that all carbons in the hexose phosphates are originally derived from glucose (Fig 2d). Considering this lack of another source and the absence of other reactions that could produce 3-labelled hexose-phosphates, the 3-labelled hexose-phosphates most likely derive from the 3-labelled fructose bisphosphate (possibly via the reverse phosphofructokinase reaction although a contribution from fructose bisphosphatase cannot be ruled out). Gluconeogenesis from amino acids or other carbon sources is not identified, consistent with previous observations [20].
The high percentage of 3-carbon labelled FBP when using the 50:50 mix of U-13C-glucose and U-12C-glucose seems counter-intuitive, given the very strong net forward flux through glycolysis and the high concentration of FBP. However, our computational analysis shows that it is not impossible. The aldolase reaction is reversible (the net aldolase flux = forward flux—reverse flux). The observed labelling would require that the part of the FBP pool created by the “reverse” aldolase reaction, working in the direction of FBP production, would represent 85% (in HMI medium) or 97% (in CMM medium) of the total FBP pool (see S1 Text for detailed explanations). These large contributions by the reverse reaction are possible under certain conditions, in spite of the net flux strongly favouring the forward direction. Simulations using a previously published collection of models which are described in detail in [40] show that under the classical assumption that glycosomes are impermeable, reverse aldolase fluxes can vary widely with plausible kinetic parameter values, most frequently with between 0 and 60% of the FBP pool coming from the “reverse” aldolase reaction (i.e. between 0 and 29% being potentially 3-labelled). However, if the glycosome is semi-permeable to metabolites smaller than FBP itself (model 3 in ref 40) due to non-selective pore-forming channels in the glycosomal membrane [41], high reverse aldolase flux, accounting for 60–99% of the FBP pool is favoured (S1 Text). This would be in excellent agreement with the observed labelling pattern.
In addition to pyruvate, its transamination product alanine was produced, probably via AAT, an enzyme previously shown to be essential to BSF trypanosomes [19]. Very low levels of labelled lactate were also detected, with all three carbons labelled. No lactate dehydrogenase has been identified in T. brucei, although L-lactate production has been shown to occur through an unusual methylglyoxal detoxification pathway in trypanosomes [42], which may correspond to the metabolite we find of the corresponding mass.
A number of potential pyruvate-derived conjugated metabolites were observed, although their significance is unknown. For example, an abundant metabolite with either 3C or 6C labelling corresponding to the molecular formula C6H8O6, was putatively identified as parapyruvate [43]. Pyruvate also appeared to form adducts with basic amino acids to produce metabolites with mass, labelling and predicted retention times consistent with carboxyethyl-L-arginine and carboxyethyl-L-ornithine (S1 Table). Another abundant novel 3-carbon labelled metabolite was detected with a molecular formula of C6H9NO4S, a likely condensation product of pyruvate and cysteine, while C7H11NO5 may correspond to the formation of a pyruvate-threonine adduct. These metabolites were more abundant in cells grown in HMI11 compared to CMM, consistent with the greater abundance of L-cysteine and L-threonine in the richer medium. In the absence of authentic standards, however, further analysis will be required to confirm the structure and physiological relevance of these compounds.
Pentose phosphate pathway
The pentose phosphate pathway intermediates (6-phosphogluconate and ribose 5-phosphate) are fully labelled, indicating a linear oxidative PPP derived from glucose 6-phosphate (Fig 3). The complete (5-carbon) labelling of ribose in purine nucleotides confirms that this pathway is the major source of ribose 5-phosphate for nucleotide synthesis (Fig 3e). This contrasts with the situation in PCF T. brucei, which also possess an active non-oxidative branch of the PPP [20] and show the characteristic 2-, 3- and 5-carbon ribose 5-phosphate labelling as a consequence of the transaldolase and transketolase (TKT) reactions that combine fully labelled and fully unlabelled precursors [38]. The lack of these isotopologues thus confirms the lack of TKT in BSFs [20,44].
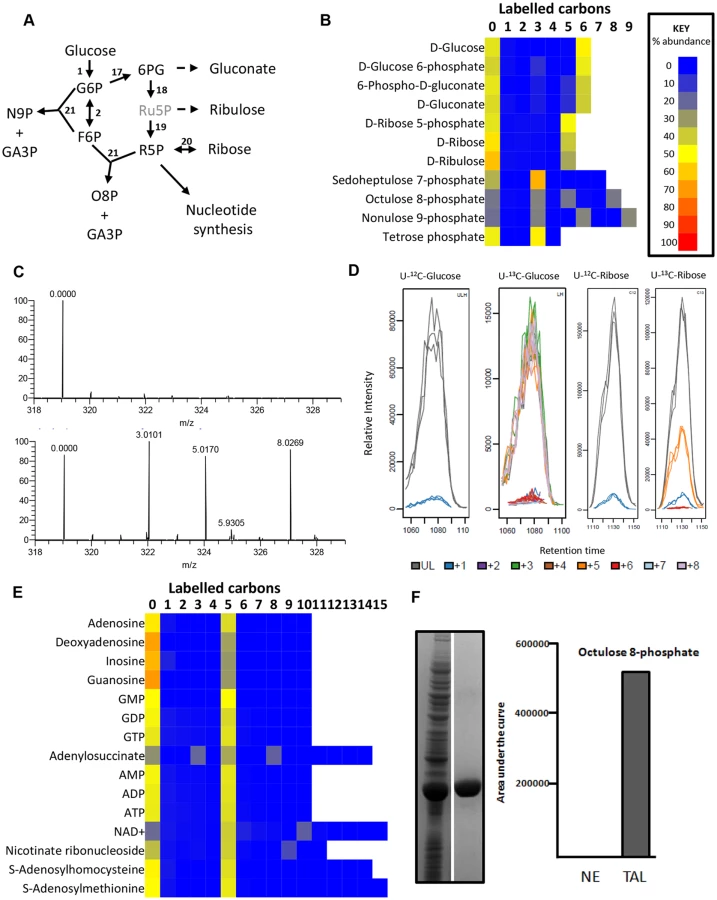
Two high molecular weight sugar phosphates, octulose 8-phosphate (O8P) and nonulose 9-phosphate (N9P) were detected and putatively identified by accurate mass and predicted retention time (as authentic standards are not available). The labelling patterns (Fig 3c-d) indicate production by transaldolase activity from a three carbon unit donated from an aldose donor to either a pentose phosphate (to produce O8P) or hexose phosphate (to produce N9P). Interestingly, BSF T. brucei have previously been shown to possess transaldolase activity [20], in spite of the absence of TKT. The absence of 2C- and 6C- labelled O8P in BSFs, while they are present in PCFs [38], indicates that a PPP intermediate is the precursor for this metabolite. We expressed and purified the gene encoding T. brucei transaldolase (Tb927.8.5600) in Escherichia coli. When the enzyme was mixed with ribose 5-phosphate and F6P as substrates, mass spectrometry revealed that O8P was produced, confirming the ability of this enzyme to carry out this reaction (Fig 3f). Moreover, cells were labelled with U-13C-ribose which resulted in extensive labelling of ribose 5-phosphate (produced by ribokinase), and significant 5-carbon labelling in O8P (Fig 3d).
The detection of metabolites with formulas consistent with sedoheptulose 7-phosphate (C7H15O10P) and erythrose 4-phosphate (C4H9O7P) was surprising in a cell without TKT activity. However, the 50% U-13C-glucose labelling reveals only the 3-carbon labelled isotopologue of each, confirming that these are not products of TKT. Further investigation found that the abundance of the C7H15O10P peak was very low in all samples, and the retention time of C4H9O7P was inconsistent with the authentic standard for erythrose 4-phosphate. More work is therefore required to elucidate the identity and source of these metabolites, and to confirm that a non-canonical pathway involved in sugar phosphate remodelling exists in trypanosomes.
Nucleotide salvage
The lack of uniform labelling in purine bases, nucleosides and nucleotides confirms the requirement for exogenous purine bases in trypanosomatids which lack purine biosynthetic pathways (Fig 3e). However, glucose-derived ribose produces 5-carbon labelled purine nucleotides, as purine salvage pathways are highly active [45]. Transporters for the efficient uptake of purine nucleosides have been characterized, and guanosine and inosine appear to be the major purine sources in in vitro culture, even in the presence of excess hypoxanthine [46]. These labelling data also confirm that nucleoside hydrolases [47] that form purine bases are the primary route for nucleotide salvage from imported nucleosides and that glucose-derived ribose 5-phosphate is added through purine phosphoribosyltransferase [48]. The almost complete labelling of the ribose moiety of adenosine nucleotides indicates minimal purine salvage from exogenous adenosine by adenosine kinase under our in vitro growth conditions, in spite of its high activity in these cells [49].
Intracellular synthesis of various cofactors, including NAD+ and S-adenosyl methionine (AdoMet), was also confirmed by incorporation of labelled ribose into the nucleotide subunits. AdoMet is an important precursor in polyamine synthesis, resulting in production of 5’methylthioadenosine (MTA). The 5-carbon labelling in these two metabolites confirms that the labelling is in the ribose moiety. However, the absence of labelling in methionine precludes activity of a methionine salvage pathway as previously proposed [50]. Methionine uptake from serum [51] apparently fulfils all methionine requirements of BSF parasites. The absence of labelling in α-ketomethylthiobutyrate (KMTB), the last intermediate in the methionine salvage pathway, further confirms the lack of function in this proposed pathway. KMTB probably arises instead from transamination of methionine, and it was previously shown to be excreted from parasites along with other amino acid-derived keto acids [46].
Carboxylic acid derivatives of catabolised glucose
An active TCA cycle is reportedly not operative in BSF T. brucei, and the observation that <1% of detected 2-oxoglutarate contains label from glucose corroborates this (S1 Table). A small amount of acetyl-coA and oxaloacetate were, nevertheless, directed into citrate, as seen/observed by the 2-, 3- and 5-carbon labelling in excess of the natural abundance of 13C by a total of approximately 1% in cells grown in HMI11, and 5% in CMM-grown cells (S2 Table). The relevance of this low level citrate production is not known. PCF T. brucei do not use the classical citrate—malate shuttle, instead utilizing a unique acetate shuttle to provide acetyl-CoA to the cytosol [31].
Isotope analysis of intracellular metabolites from cells incubated with 50% U-13C-labelled glucose reveals three-carbon labelled isotopologues of succinate and malate, representing 35% (70% when corrected for 50% labelled glucose precursor) of the intracellular malate (Fig 4) and 26% of succinate (52% corrected). This labelling pattern is consistent with the production of succinate via a succinate fermentation pathway. The first enzyme of this pathway, PEPCK, is found in BSF trypanosomes, albeit at a lower level than PCF [52,53]. Although oxaloacetate is not detectable on this analytical platform, the observation of the 3-carbon labelled isotopologue in aspartate (a transamination product of oxaloacetate) supports the production of oxaloacetate from PEP. The total labelling in aspartate (62% corrected) is comparable to malate (70% corrected), demonstrating that intracellular aspartate is primarily synthesized from glucose-derived oxaloacetate, rather than being taken up from the surrounding medium, which contains aspartate at 100 μM [46]. BSF trypanosomes failed to demonstrate appreciable aspartate transport capability [54]. T. brucei encodes three separate isoforms of malate dehydrogenase (MDH), localized to the mitochondrion, glycosomes and the cytosol, respectively [55]. The glycosomal isoform is reportedly absent from the BSF, while the cytosolic isoform is present at higher levels in BSF than procyclics [55]. Additionally a small amount of the mitochondrial isoform is reportedly present [55–57]. The succinate fermentation pathway therefore might involve oxaloacetate produced in the glycosome via PEPCK which might re-enter the cytosol and possibly the mitochondrion for further metabolism. Further work, systematically removing or knocking down expression of different isoforms, followed by metabolic profiling, will be required to deconvolute the sources of these partially reduced products of glucose catabolism in the BSF trypanosome.
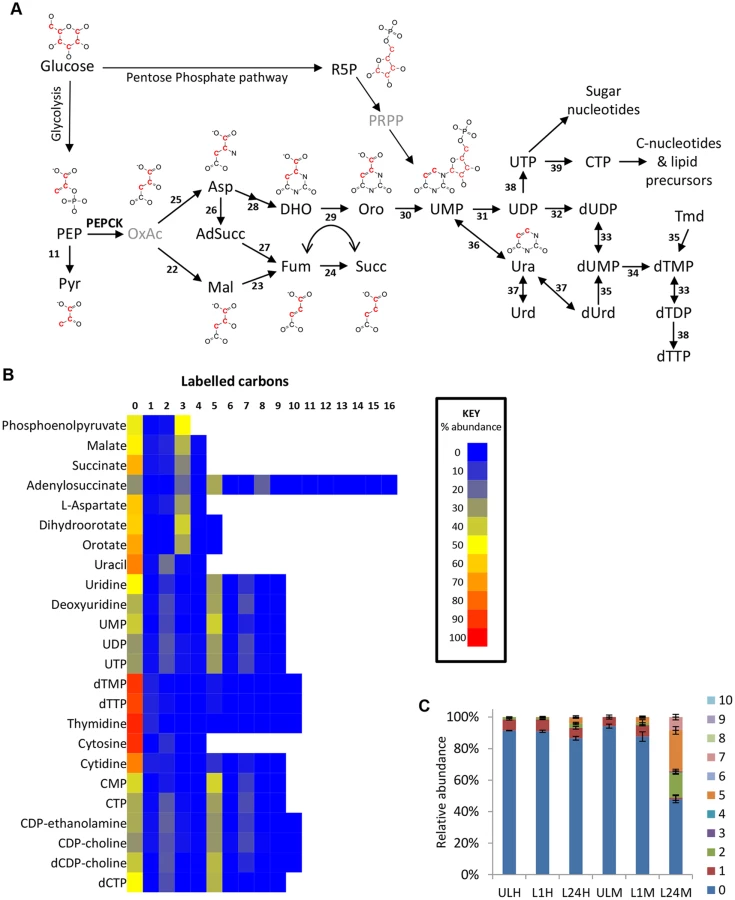
Aspartate use in nucleotide biosynthesis
Oxaloacetate-derived aspartate is a key precursor in pyrimidine synthesis, being converted to orotate via dihydroorotate. Dihydroorotate and orotate were detected with isotope enrichment equivalent to aspartate, ~35% 3-labelled carbons (70% corrected), confirming de novo synthesis of pyrimidines in this way. Uracil labelling was slightly lower (44% corrected), suggesting some uptake of uracil from the medium. However, higher levels of 2-labelling in UMP and UDP suggest that de novo synthesized orotate is the major source of pyrimidines under these conditions (Fig 4). Labelling of uridine and cytidine nucleotides was not significantly different between the two different culture media used in this study, HMI11 and CMM. However, significant differences were observed in thymidine nucleotides. dTTP and dTMP abundance in CMM-grown cells was only half that observed for HMI11, which contains 20 μM added thymidine. The isotope labelling patterns of dTTP and dTMP in CMM matched those of UMP and the other pyrimidine nucleotides, indicating complete synthesis from de novo synthesized pyrimidines by thymidylate synthetase in thymidine-poor media [58,59]. dTMP and dTTP in HMI11-grown cells were less than 10% labelled, confirming a preference for thymidine salvage by the action of thymidine kinase when exogenous thymidine is available (Fig 4c).
Aspartate is also an important intermediate in the purine salvage pathway, in which the formation of adenylosuccinate and release of fumarate results in nitrogen transfer from aspartate to IMP, producing AMP. Adenylosuccinate was detected with 3-carbon labelling, which confirms the role of glucose-derived aspartate in the purine salvage pathway (Fig 3e).
PEPCK is essential to BSF T. brucei
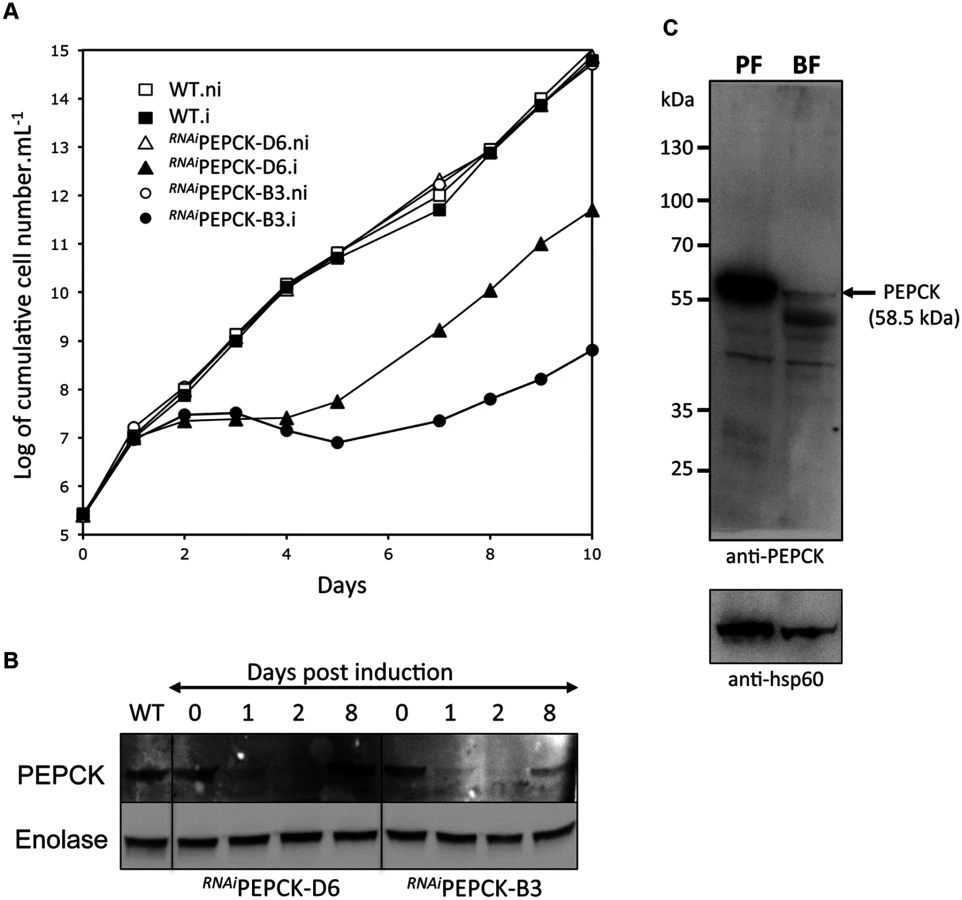
The discovery that glucose enters amino acid and nucleotide pathways via oxaloacetate indicates that PEPCK might play an unexpectedly important role in BSF trypanosomes. Indeed, a genome-wide RNAi screen indicated the gene could be essential [60]. To test this further, western blot analysis was performed with anti-PEPCK immune serum, showing that PEPCK is expressed in the BSF, albeit at much lower abundance than in the PCF (Fig 5). Two cell lines (RNAiPEPCK-B3 and RNAiPEPCK-D6) were then generated to investigate the role of PEPCK in BSF T. brucei by RNAi down-regulation of pepck gene expression. Both cell lines showed a growth arrest two days after tetracycline induction. A phenotypic reversion 5 days post-induction correlated with a re-expression of PEPCK (Fig 5).
RNAiPEPCK labelled metabolomics
In order to measure the metabolic role of PEPCK, the production of succinate from glucose in BSF trypanosomes was measured using 1H-NMR, as previously described for PCF T. brucei [61]. The parental BSF 427 strain was incubated for 5 hours in PBS/NaHCO3 medium containing 4 mM D-glucose as the only carbon source and the incubation medium was analyzed by 1H-NMR spectroscopy. In addition to pyruvate, detectable amounts of succinate were quantitatively measured, at levels ~2.7% of the excreted pyruvate (Fig 6a and Table 1). The amount of excreted succinate was decreased 4.3 fold in the RNAiPEPCK-B3 mutant after two days of induction compared to the wild-type cells (47 ±17 versus 201 ±89 nmol/h/mg of protein). Succinate secretion increased five days after induction (79 ±18 nmol/h/mg of protein) (Fig 6a and Table 1), as a consequence of pepck re-expression (Fig 5).
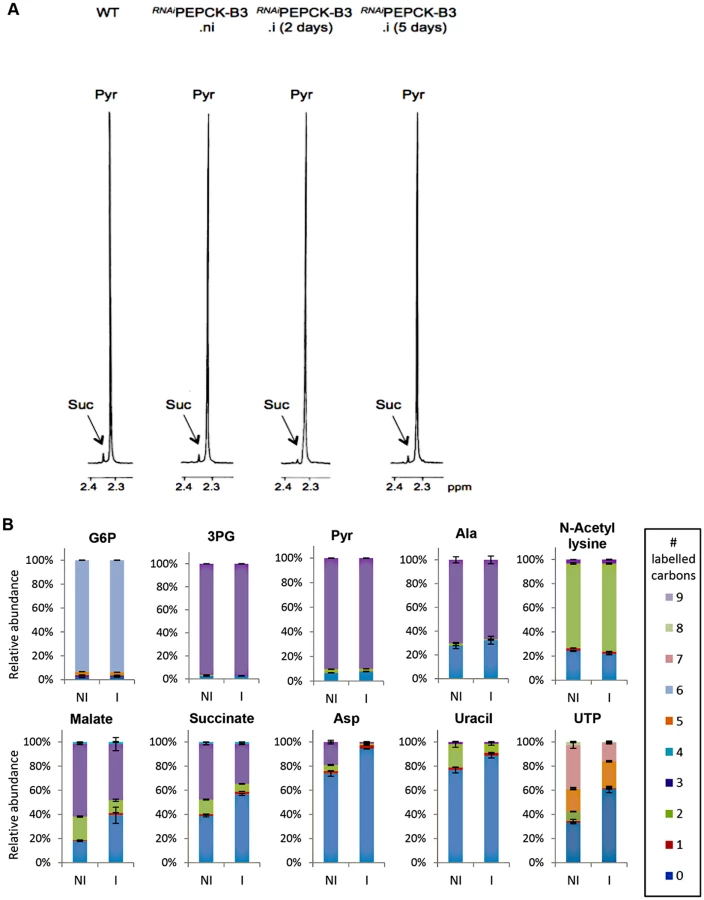
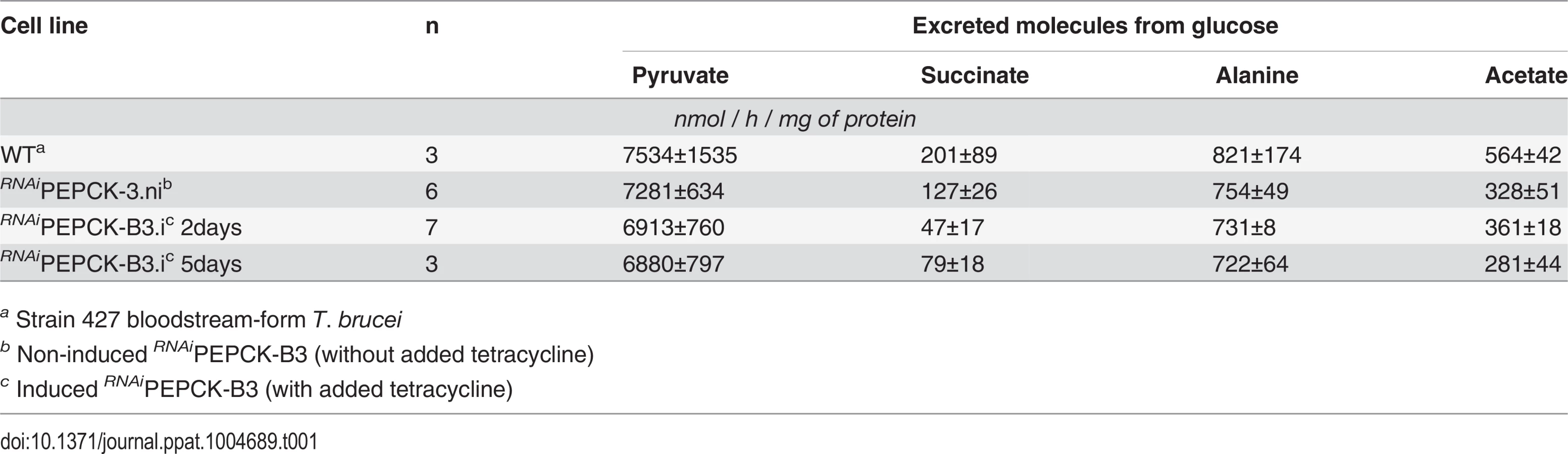
To confirm reduction of succinate production in the RNAiPEPCK-B3 tetracycline-induced cell line (at 2 days), we determined incorporation of 13C-labelling from U-13C-glucose into intracellular metabolites by LC-MS. A reduction of 13C3 incorporation into malate (24 ± 10%) and succinate (29 ± 2%) was observed in the induced RNAiPEPCK-B3 cell line compared to the non-induced cells (Fig 6b). Labelling in other products of PEPCK-derived oxaloacetate was also significantly reduced, including aspartate (15-fold), uracil (3-fold) and UTP (2-fold). In contrast, 13C incorporation into metabolites produced upstream of the PEPCK step (G6P, 3PG and pyruvate), and those in other pathways derived from glycolytic intermediates (alanine, acetyl-CoA, and ribose 5-phosphate) were not significantly altered (Fig 6b).
Acetate production in BSF T. brucei
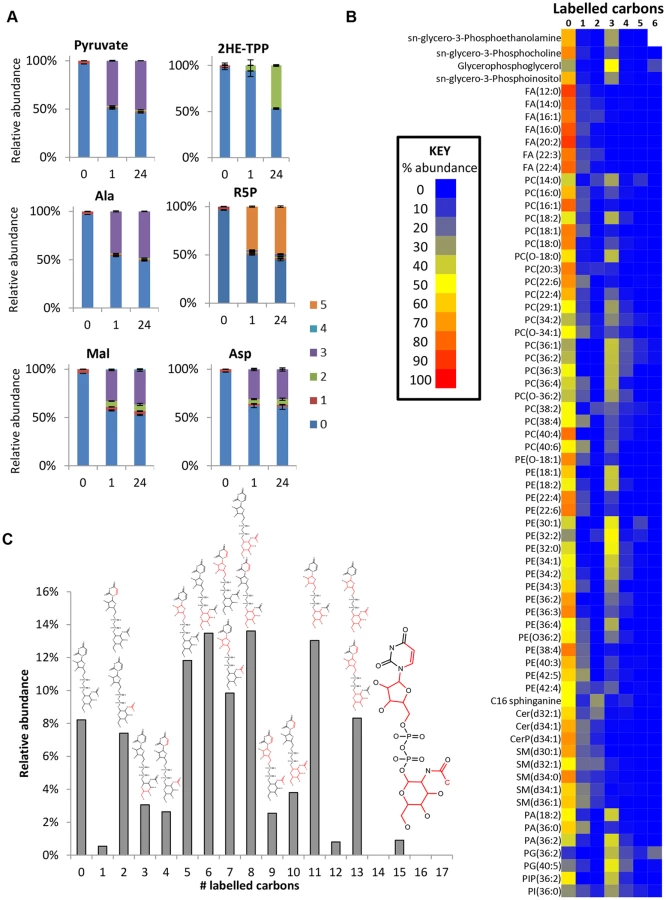
In addition to its secretion and conversion to alanine, pyruvate is a major source of acetyl-CoA in BSF trypanosomes. Although the intracellular concentration of acetyl-CoA was below the limit of detection for our analytical method, evidence of its production is provided by 2-hydroxyethyl thiamine pyrophosphate (Fig 7a), the intermediate in pyruvate’s conversion to acetyl-CoA by PDH. The presence of two labelled carbons indicates formation directly from glucose-derived pyruvate. Labelling was minimal after one hour, but complete labelling was observed at 24 hours, suggesting that flux through this pathway is significantly slower than through most other pathways (Fig 7a). It has recently been shown [27] that PDH knockdown is conditionally lethal in BSF T. brucei. Both threonine, via a threonine dehydrogenase (TDH) pathway, and glucose, via PDH, can provide acetate and the latter route becomes essential when threonine is absent, or redundant due to loss of TDH [27]. Furthermore, acetylated metabolites including acetylcarnitine, acetyllysine, acetylornithine and acetylglutamine were all detected with 2 labelled carbons from glucose (S1 Table). The unlabelled proportion of these metabolites probably relates to threonine’s key role in acetate provision [27].
Glucose contributions to fatty acid and lipid biosynthesis
De novo fatty acid synthesis was detected in BSF T. brucei by incorporation of two labelled carbon units in free fatty acids and phospholipids (Fig 7b). However, the very low level of labelling in fatty acids suggests that de novo fatty acid synthesis is minor compared to salvage mechanisms, and a high level of lysophospholipid uptake may represent the principal source of fatty acids in these cells [62] with elongation accounting for modelling fatty acids of different chain length [63].
The additional 3-carbon labelling observed in glycerophospholipids demonstrates that lipid salvage utilizes glycolysis-derived glycerol 3-phosphate for synthesis of phospholipid head-groups. Despite the requirement for salvage of host (or culture medium) lipids for the provision of choline, and most fatty acids, around half of the glycerol phosphate in lipid head-groups was derived from glycolysis (ranging from 8 to 100% corrected labelling for detected PC (phosphatidylcholine) and PE (phosphatidylethanolamine) lipids; Fig 7 and S1 Table, the variability relating to the fact that the relative contributions of the salvage and de novo synthesis pathways are not uniform across lipid species.
Nucleotide sugars and glycoconjugate precursors
Several other labelled metabolites were detected (S1 Table) indicating that glucose enters amino sugars (N-acetyl-D-glucosamine and N-acetyl-D-glucosamine 6-P), sugar nucleotides (UDP-Glc, or UDP-Gal GDP-man and UDP-GlcNAC; Fig 7c) and low-level labelling in other sugars (tentatively identified as myo-inositol, fructose, glucuronate and arabinonate, although accurate identification of sugars is difficult using accurate mass-based metabolomics, as many isomers are theoretically possible for most carbohydrate metabolites; hence, additional orthogonal approaches are required to confidently identify these structures. Nothwithstanding their abundance is clearly low compared to glucose and fructose the two primary hexoses. Mannose is absent in its free form, its generation occurring only when nucleotide conjugated and in phosphorylated form [24]). Targeted studies of sugar nucleotide biosynthesis have already demonstrated de novo biosynthesis of UDP-Glc [64] and GDP-Man [24] from glucose, and analysis of inositol metabolism demonstrated de novo synthesis of inositol for protein glycosylation, in addition to inositol salvage for lipid biosynthesis [25,26]. Data from the current study demonstrated less than 10% labelling in inositol, and confirmed that the inositol moiety of the lipid headgroup glycerol-phosphoinositol was unlabelled (i.e. not formed by de novo synthesis from glucose, consistent with targeted analyses conducted elsewhere to investigate the origin of inositol in T. brucei [25,26]).
Discussion
The introduction of metabolomic technologies, particularly in conjunction with stable isotope tracing has, in recent years, transformed our ability to analyse metabolism in biological systems. For example, isotopologue studies into Leishmania [65], Plasmodium [36,37] and Toxoplasma [35] have identified novel pathways and resolved long-standing questions of metabolism. The African trypanosome, Trypanosoma brucei, is the causative agent of human African trypanosomiasis, a neglected tropical disease of sub-Saharan Africa, for which new drugs are urgently needed. The slender bloodstream form of these parasites has generally been considered to have a highly streamlined glucose catabolic pathway [3–5,9,10]. The localization of the first seven enzymes of the glycolytic pathway to membrane-bounded peroxisome-like organelles known as glycosomes [5] has been proposed to be instrumental to the regulation of flux through glycolysis by providing an environment in which ATP/ADP and NAD/NADH can be balanced [9,10]. This arrangement has enabled the production of a well-defined dynamic mathematical model of glycolysis [9,10] and the models have been able to predict several important phenomena, such as the contribution of different enzymes to the overall control of the pathway. Recent iterations have included the addition of parameters that explicitly take into account uncertainty related to the system [66] and also the activity of the pentose phosphate pathway [22] and the presence of permeability pores that allow free diffusion of several glycolytic intermediates between the cytosol and glycosome [40].
Here we have taken the systematic route to analyse the full extent of glucose metabolism in long slender BSF trypanosomes.
Extensive pyruvate production was observed, consistent with existing models of glycolytic flux for energy production. However, the 3-carbon labelled isotopologues observed in glycolytic hexose phosphates reveals previously unreported complexity in this pathway. The lack of transketolase [20,42] indicates that a canonical non-oxidative pentose phosphate cycle is missing from these cells and thus is not the source of 3-labelled hexose phosphates. The role of transaldolase in BSFs is uncertain, and transaldolase-mediated production of 3-labelled fructose 6-phosphate (from F6P and GA3P) could be hypothesized, although this appears to be a redundant reaction. Alternatively, aldolase [67], acting to combine either U-13C or U-12C DHAP with U-13C or U-12C GA3P would produce the relative proportions of 0-, 3- and 6-labelled fructose bisphosphate as identified here. The further production of 3-labelled F6P (and G6P) from FBP would require fructose-1,6-bisphosphatase activity, and both transcriptomic [68] and proteomic [69] data indicate that this enzyme is present in BSF T. brucei. A family of models, simulating flux through glycolysis whilst taking into account our uncertainties about the system and its distribution between glycosome and cytosol [40], reveals that if metabolites smaller than FBP are able to exchange between compartments while FBP is not, then a significant proportion of the total FBP in the cell can indeed arise through the condensation of GA3P and DHAP. The data, therefore, supports the proposed role of pores creating a semi-permeable glycosome [41]. Further work is required to fully investigate the intermediate fluxes in glycolytic metabolism and to determine the temporal and spatial aspects of metabolism that give rise to these unexpected observations.
At this point we cannot rule out definitively the production of a compound with the mass and retention time of FBP as an artefact of the experimental system. The same might be true of other exotic, unexpected species identified in this study. However, a non-enzymatic aldol condensation of triose phosphates is highly unlikely, given that non-enzymatic condensations and additions generally require extreme pH conditions (<1 or > 9). To confirm this, DHAP and GA3P were mixed in CMM medium followed by extraction in the conditions used to extract our cellular lysates and storage for seven days at -80°C, and no FBP or other potential condensation or addition products were observed by LC-MS. We therefore consider it likely that many of the metabolites that we describe here are bona fide products of trypanosome metabolism, and the incorporation of carbon-13 label confirms the endogenous production of these metabolites (compared to the unlabelled metabolites primarily acquired from the serum in the growth media), although their physiological function, or indeed relevance remains unknown. Metabolites can be produced either enzymatically or chemically in cells as inevitable by-products of metabolism, methylglyoxal being an example already discussed here [42].
Apart from the identification of numerous unexpected metabolites and pathways operating in BSF trypanosomes, our experiments also rule out the operation of other pathways previously assumed to be functional. For example, a rapid deamination of aromatic amino acids was believed to contribute to a methionine cycle involved in provision of decarboxylated S-adenosylmethionine used in polyamine synthesis [50]. Here, however, we demonstrate that carbons from glucose do not enter methionine, which is provided exclusively through cellular uptake instead [51]. This means that the pathways of methylthioadenosine detoxification remain to be elucidated in the BSF trypanosome.
While the majority of glucose flows to secreted pyruvate under aerobic culture conditions, we show that glucose metabolism is pervasive in the BSF parasites. In addition to pyruvate and glycerol, various other metabolites are secreted, including alanine, the transaminated product of pyruvate. This reaction appears to be essential since the alanine transaminase gene could not be knocked out of the T. brucei genome [19], although knockdown by RNAi was possible. Succinate was also secreted. The succinate produced in BSF trypanosomes is primarily labelled in three carbons, which indicates its derivation from glycolytic phosphoenolpyruvate, rather than from a canonical TCA cycle, which would produce primarily 2-carbon and 4-carbon labelled isotopologues. Analysis of the phosphoproteome of BSFs identified glycosomal NADH-dependent fumarate dehydrogenase [70]. However, it is not possible to distinguish between glycosomal, cytosolic or potentially mitochondrial derivation of these metabolites, and systematic metabolite profiling following gene knockout or knockdown of various enzymes will be necessary to deconvolute these pathways.
Although it has been suggested that glucose makes negligible contribution to anabolic processes in BSF trypanosomes [10], numerous isolated studies have revealed that glucose serves as a precursor in processes including sugar nucleotide and glycoconjugate synthesis [23], inositol production [25,26] and recently acetate production [27]. Glucose also enters pyrimidine biosynthesis via oxaloacetate and lipid metabolism via glycerol 3-phosphate as well as acetate production. RNAi knockdown of PEPCK confirmed this activity as being essential in BSF trypanosomes. The same enzyme was not essential to PCFs, although its contribution is major in this parasitic insect form [71]. We recently demonstrated that loss of bound phosphate from the glycolytic pathway in the glycosome via the PPP requires a mechanism to retain the bound phosphate balance [22]. PEPCK offers an obvious means to restore glycosomal ATP, and this link to the PPP might explain the unexpectedly essential nature of this enzyme to the BSF trypanosome.
Although the untargeted mass spectrometry platform we use here is intrinsically unable to quantify the total flux of glucose carbon into the various pathways discussed here, it is clearly the case that the overwhelming majority of glucose is catabolised to pyruvate (Table 1). The total amount glucose entering these other pathways would be in the order of just a few percent of the total consumed, in agreement with the measurements of Haanstra et al [10]. Although only a small minority of the total glucose consumption, however, glucose is the major carbon source for the production of many of these extra-glycolytic metabolites, and this flux is clearly important given the fact that several enzymes, including PEPCK shown here, turn out to be essential to the metabolism of these parasites.
The identification of novel essential enzymes in the BSF trypanosome may have important implications for chemotherapy. Glycolysis has long been considered an attractive target given the absolute dependency on glucose for energy and carbon provision to these cells [72]; our results show that additional pathways around the periphery of glycolysis might be equally promising.
Materials and Methods
Parasite culture
T. brucei brucei bloodstream form (s427) was cultured in vitro in HMI11 [73] and CMM [44] supplemented with 10% fetal calf serum (FCS) Gold (PAA, Piscataway, NJ) at 37°C, 5% CO2. Initial 5 ml cultures in 25 ml vented flasks (Corning) were grown to a maximum density of 4 × 106 cells/ml and subcultured by 1-in-100 or 1-in-1,000 dilution every 2 or 3 days, respectively. A hemocytometer (Neubauer) was used for cell counts.
Labelling experiments and sample preparation
Two days prior to the extraction, 2 × 104 cells/ml were seeded in 30 ml of HMI11 and CMM. Growth media was then replaced with media containing 50% of 13C-labelled glucose (12.5 mM of 12C-glucose + 12.5 mM of 13C-glucose for HMI11, and 5 mM of 12C-glucose and 5 mM of 13C-glucose for CMM culture) at 24 h and 1 h prior to extraction. All samples were extracted at the same time atequivalent cell densities of approximately 2 ×106 cells/ml. To yield 5 ×107 cells, appropriate volumes of cell culture were harvested and quenched in EtOH/dry ice bath as previously described [74]. Cells were centrifuged at 1250 ×g for 10 min and spent media removed; cells were washed with pre-cooled phosphate buffered saline (PBS) at 4°C. After centrifugation at 1250 ×g for 5 min, the supernatant was removed and metabolites extracted by adding 100 μl of (ice cold) chloroform/methanol/water (1:3:1) and mixed vigorously for 1 h at 4°C. The extraction mixtures were centrifuged at 13,000 ×g for 10 min and the supernatants collected and stored at -80°C prior to analysis. In order to assess instrument performance, one pooled quality control (QC) sample was prepared by mixing an equal volume of all the samples. Independent biological replicates were prepared on different days.
LC-MS methodologies
Hydrophilic interaction liquid chromatography (HILIC) was carried out on a Dionex UltiMate 3000 RSLC system (Thermo Fisher Scientific, Hemel Hempstead, UK) using a ZIC-pHILIC column (150 mm × 4.6 mm, 5 μm column, Merck Sequant) as previously described [74]. Briefly, the column was maintained at 30°C and samples were eluted with a linear gradient (20 mM ammonium carbonate in water, A and acetonitrile, B) over 46 min at a flow rate of 0.3 ml/min as follows: 80% B to 20% B at 30 min, to 5% B at 32 min and held to 39 min for washing, to 80% B at 40 min and held to 46 min for re-equilibration. The injection volume was 10 μl and samples were maintained at 4°C prior to injection. For the MS analysis, a Thermo Orbitrap Exactive (Thermo Fisher Scientific) was operated in polarity switching mode and the MS settings were as follows: resolution 50,000, AGC 106, m/z range 70–1400, sheath gas 40, auxiliary gas 5, sweep gas 1, probe temperature 150°C, and capillary temperature 275°C. For positive mode ionisation: source voltage +4.5 kV, capillary voltage +50 V, tube voltage +70 kV, skimmer voltage +20 V. For negative mode ionisation: source voltage-3.5 kV, capillary voltage-50 V, tube voltage-70 V, skimmer voltage-20 V. Mass calibration was performed for each polarity immediately prior to each analysis batch. The calibration mass range was extended to cover small metabolites by inclusion of low-mass contaminants with the standard Thermo calmix masses (below m/z 1400), C2H6NO2 for positive ion electrospray ionisation (PIESI) mode (m/z 76.0393) and C3H5O3 for negative ion electospray ionisation (NIESI) mode (m/z 89.0244). To enhance calibration stability, lock-mass correction was also applied to each analytical run using these ubiquitous low-mass contaminants.
Data processing and analysis
Raw LC-MS data were processed with XCMS for untargeted peak detection [75], and mzMatch.R [76] was employed for peak matching and annotation of related peaks. Tentative metabolite identification was carried out by IDEOM using the default parameters [77]. Metabolite identification was performed by matching accurate masses and retention times of authentic standards (MSI confidence level 1, i.e. an annotated compound matched to a standard with two orthogonal approaches), but when standards were not available, predicted retention times were calculated by a previously validated model [78] (MSI confidence level 2, i.e. a putatively annotated compound based on its exact mass determination). Published literature and pathway/genome databases were considered to improve annotation in cases where isomers could not be differentiated based on accurate mass and retention time. Metadata to support the identification of each metabolite is available in the IDEOM file for each study (S3 Table). Metabolites included in the manuscript were manually annotated using authentic standards where available. However, note that many of the metabolite names given in the Ideom file are generated automatically as the software provides a best match to database entries of the given mass and formula. In the absence of additional information these must be considered as putatively-annotated hits; the confidence score in the column adjacent to that hit serves as a guide to this. Clearly it is beyond the scope of any study to provide authenticated annotations to many hundreds of detected compounds, but the full datasets are included in the spirit of open access data.
The mzmatch-ISO software [39] was used to extract all isotopologue abundances from all identified and putative annotated metabolites. Raw data are available in the associated IDEOM files (S3 Table) and chromatograms are in separate pdf files (S1–S2 Figs). Labelled metabolites are given in S4 Table and S5 Table. Data analysis and interpretation was based on the 24 hour time-point, at approximate steady state, unless otherwise stated.
NMR experiments
T. brucei bloodstream form cells (2.5 x107) were collected by centrifugation at 1,400 g for 10 min, washed with PBS containing 4 mM glucose and incubated for 5 h at 37°C in 2.5 ml of incubation buffer (PBS supplemented with 5 g/l NaHCO3, pH 7.4) containing 4 mM glucose. The integrity of the cells during the incubation was checked by microscopic observation. The supernatant was collected and 50 μl of malate solution in D2O (20 mM) was added as internal reference. 1H-NMR spectra were performed at 125.77 MHz on a Bruker DPX500 spectrometer equipped with a 5 mm broadband probe head. Measurements were recorded at 25°C with an ERETIC method. This method provides an electronically synthesized reference signal [79]. Acquisition conditions were as follows: 90° flip angle, 5,000 Hz spectral width, 32 K memory size, and 9.3 sec total recycle time. Measurements were performed with 256 scans for a total time close to 40 min. Before each experiment, phase of ERETIC peak was precisely adjusted. Resonances of obtained spectra were integrated and results were expressed relative to ERETIC peak integration.
Inhibition of pepck gene expression by RNAi
Inhibition of pepck gene expression by RNAi in the 427 strain was performed by expression of stem-loop “sense/anti-sense” RNA molecules of the targeted sequences introduced in the pHD1334 expression vector. First, the pLew-PEPCK-SAS plasmid was constructed to target a 481 bp fragment of the pepck gene (from position 22 bp to 503 bp). Briefly, a PCR-amplified 569 bp fragment, containing the antisense pepck sequence with restriction sites added to the primers was inserted into the HindIII and BamHI restriction sites of the pLew100 plasmid. Then a PCR-amplified fragment containing the sense pepck sequence (505 bp) was inserted upstream of the anti-sense sequence, using HindIII and XhoI restriction sites (XhoI was introduced at the 3’-extremity of the antisense PCR fragment). The resulting plasmid (pLew-PEPCK-SAS) contains a sense and antisense version of the targeted gene fragment, separated by a 52 bp fragment, under the control of the PARP promoter linked to a prokaryotic tetracycline (Tet) operator. Then the PEPCK-SAS HindIII-BamHI cassette extracted from the pLew-PEPCK-SAS plasmid was inserted in HindIII-BamHI digested pHD1334 vector. The RNAiPEPCK cell lines were produced by transfecting the 427 “double marker” (Hyg-Neo) monomorphic cell line with the pHD-PEPCK-SAS plasmid. Transfected cells were selected in IMDM medium containing, hygromycin (5 μg/ml), neomycin (2.5 μg/ml) and phleomycin (2.5 μg/ml). Induction of double-stranded RNA expression was performed by addition of 10 μg/ml tetracycline.
Cloning and expression of the T. brucei transaldolase gene
A gene annotated as transaldolase in the T. brucei genome database Tb427.08.5600, was amplified with the following primers 5’-GCCATATGAATCAACTGGAGAGCCT-3’ and 5’-GCCTCGAGTTACAAAAGTGTAGCGTGA-3’. The PCR product was first cloned into the vector pGEM-T easy and then transferred to the pET28a(+) expression vector using the introduced Nde1 and Xho1 restriction sites. The resultant plasmid was sequenced to confirm fidelity and transformed into E. coli strain BL21 (DE3) for expression at 37°C or 4 hours after induction with IPTG. Expressed protein was observed by SDS protein gel electrophoresis and purified using Ni2+ chelate chromatography (Fig 2f). The ability of transaldolase to produce octulose 8-phosphate was determined by mixing ribose 5-phosphate (5 mM) and fructose 6-phosphate (5 mM) in Tris-HCl (pH 7.2) and adding transaldolase (1.2 mg/mL) then incubating at 20°C for 2 hrs. Alternatively the reaction was set up with no enzyme. The presence of octulose 8-phosphate was determined by direct infusion of post-reaction mix into the Exactive mass spectrometer (Fig 2f).
Western blot analyses
Total protein extracts of wild-type or mutant BSF of T. brucei (5 x106 cells) were size-fractionated by SDS-PAGE (10%) and immunoblotted on Immobilon-P filters (Millipore) [80]. Immunodetection was performed as described [81,82] using primary antibodies, rat anti-T. brucei PEPCK (diluted 1:1000; gift from T. Seebeck, Bern, Switzerland), rabbit anti- T. brucei enolase (diluted 1:1,000, gift from P. Michels, Edinburgh, UK) and mouse anti-HSP60 (diluted 1:10 000) [83], and as secondary antibodies, anti-rat, anti-rabbit or anti-mouse IgG conjugated to horseradish peroxidase (BioRad, 1:5,000 dilution). Revelation was performed using the SuperSignal West Pico Chemiluminescent Substrate as described by the manufacturer (Thermo Fisher Scientific). Alternatively, for quantitative analyses, revelation was performed using the Luminata Crescendo Western HRP Substrate (Millipore). Images were acquired and analyzed with a KODAK Image Station 4000 MM and quantitative analyses were performed with the KODAK MI application.
Supporting Information
Zdroje
1. Brun R, Blum J, Chappuis F, Burri C (2010) Human African trypanosomiasis. Lancet 375:148–159. doi: 10.1016/S0140-6736(09)60829-1 19833383
2. Barrett MP (2010) Potential new drugs for human African trypanosomiasis: some progress at last. Curr Opin Infect Dis 23:603–608. doi: 10.1097/QCO.0b013e32833f9fd0 20844428
3. Besteiro S, Barrett MP, Rivière L, Bringaud F (2005) Energy generation in insect stages of Trypanosoma brucei: metabolism in flux. Trends Parasitol 21:185–191. 15780841
4. Bringaud F, Rivière L, Coustou V (2006) Energy metabolism of trypanosomatids: adaptation to available carbon sources. Mol Biochem Parasitol 149:1–9. 16682088
5. Opperdoes FR, Borst P (1977) Localization of nine glycolytic enzymes in a microbody-like organelle in Trypanosoma brucei: the glycosome. FEBS Lett 80:360–364. 142663
6. Bursell E (1963) Aspects of the metabolism of amino acids in the tsetse fly, Glossina (Diptera) J Insect Physiol 9:439–452.
7. Lamour N, Rivière L, Coustou V, Coombs GH, Barrett MP, et al. (2005) Proline metabolism in procyclic Trypanosoma brucei is down-regulated in the presence of glucose. J Biol Chem 280:11902–11910. 15665328
8. Coustou V, Biran M, Breton M, Guegan F, Rivière L, et al. (2008) Glucose-induced remodeling of intermediary and energy metabolism in procyclic Trypanosoma brucei. J Biol Chem 283:16342–16354. doi: 10.1074/jbc.M709592200 18430732
9. Bakker BM, Michels PA, Opperdoes FR, Westerhoff HV (1997) Glycolysis in bloodstream form Trypanosoma brucei can be understood in terms of the kinetics of the glycolytic enzymes. J Biol Chem 272:3207–3215. 9013556
10. Haanstra JR, van Tuijl A, van Dam J, van Winden W, Tielens AG, et al. (2012) Proliferating bloodstream-form Trypanosoma brucei use a negligible part of consumed glucose for anabolic processes. Int J Parasitol 42:667–673. doi: 10.1016/j.ijpara.2012.04.009 22580731
11. Opperdoes FR, Borst P, Bakker S, Leene W (1977) Localization of glycerol-3-phosphate oxidase in the mitochondrion and particulate NAD+-linked glycerol-3-phosphate dehydrogenase in the microbodies of the bloodstream form to Trypanosoma brucei. Eur J Biochem 76:29–39. 142010
12. Ryley JF (1962) Studies on the metabolism of the protozoa. 9. Comparative metabolism of blood-stream and culture forms of Trypanosoma rhodesiense. Biochem J 85:211–223. 13983265
13. Grant PT, Fulton JD (1957) The catabolism of glucose by strains of Trypanosoma rhodesiense. Biochem J 66:242–250. 13445679
14. Fulton JD, Stevens TS (1945) The glucose metabolism in vitro of Trypanosoma rhodesiense. Biochem J 39:317–320. 16747912
15. Rico E, Rojas F, Mony BM, Szoor B, Macgregor P, et al. (2013) Bloodstream form pre-adaptation to the tsetse fly in Trypanosoma brucei. Front Cell Infect Microbiol 3:78. doi: 10.3389/fcimb.2013.00078 24294594
16. Brown RC, Evans DA, Vickerman K (1972) Developmental changes in ultrastructure and physiology of Trypanosoma brucei. Trans R Soc Trop Med Hyg 66:336–337. 5048840
17. Mackenzie NE, Hall JE, Seed JR, Scott AI (1982) Carbon-13 nuclear-magnetic-resonance studies of glucose catabolism by Trypanosoma brucei gambiense. Eur J Biochem 121:657–661. 7056262
18. Hall JE, Mackenzie NE, Mansfield JM, McCloskey DE, Scott AI (1988) 13C-NMR analysis of alanine metabolism by isolated perfused livers from C3HeB/FeJ mice infected with African trypanosomes. Comp Biochem Physiol B 89:679–685. 3378406
19. Spitznagel D, Ebikeme C, Biran M, Nic a' Bháird N, Bringaud F, et al. (2009) Alanine aminotransferase of Trypanosoma brucei—a key role in proline metabolism in procyclic life forms. FEBS J 276:7187–7199. doi: 10.1111/j.1742-4658.2009.07432.x 19895576
20. Cronín CN, Nolan DP, Voorheis HP (1989) The enzymes of the classical pentose phosphate pathway display differential activities in procyclic and bloodstream forms of Trypanosoma brucei. FEBS Lett 244:26–30. 2924907
21. Barrett MP, Le Page RW (1993) A 6-phosphogluconate dehydrogenase gene from Trypanosoma brucei. Mol Biochem Parasitol 57:89–99. 8426618
22. Kerkhoven EJ, Achcar F, Alibu VP, Burchmore RJ, Gilbert IH, et al. (2013) Handling uncertainty in dynamic models: the pentose phosphate pathway in Trypanosoma brucei. PLoS Comput Biol 9:e1003371. doi: 10.1371/journal.pcbi.1003371 24339766
23. Turnock DC, Ferguson MA. (2007) Sugar nucleotide pools of Trypanosoma brucei, Trypanosoma cruzi, and Leishmania major. Eukaryot Cell 6:1450–1463. 17557881
24. Kuettel S, Wadum MC, Güther ML, Mariño K, Riemer C, et al. (2012) The de novo and salvage pathways of GDP-mannose biosynthesis are both sufficient for the growth of bloodstream-form Trypanosoma brucei. Mol Microbiol 84:340–351. doi: 10.1111/j.1365-2958.2012.08026.x 22375793
25. Martin KL, Smith TK (2006) The glycosylphosphatidylinositol (GPI) biosynthetic pathway of bloodstream-form Trypanosoma brucei is dependent on the de novo synthesis of inositol. Mol Microbiol 61:89–105. 16824097
26. Gonzalez-Salgado A, Steinmann ME, Greganova E, Rauch M, Mäser P, et al. (2012) myo-Inositol uptake is essential for bulk inositol phospholipid but not glycosylphosphatidylinositol synthesis in Trypanosoma brucei. J Biol Chem 287:13313–13323. doi: 10.1074/jbc.M112.344812 22351763
27. Mazet M, Morand P, Biran M, Bouyssou G, Courtois P, et al. (2013) Revisiting the central metabolism of the bloodstream forms of Trypanosoma brucei: production of acetate in the mitochondrion is essential for parasite viability. PLoS Negl Trop Dis 7:e2587. doi: 10.1371/journal.pntd.0002587 24367711
28. van Weelden SW, van Hellemond JJ, Opperdoes FR, Tielens AG (2005) New functions for parts of the Krebs cycle in procyclic Trypanosoma brucei, a cycle not operating as a cycle. J Biol Chem 280:12451–12460. 15647263
29. Coustou V, Biran M, Besteiro S, Rivière L, Baltz T, et al.(2006) Fumarate is an essential intermediary metabolite produced by the procyclic Trypanosoma brucei. J Biol Chem 281:26832–26846. 16857679
30. Millerioux Y, Morand P, Biran M, Mazet M, Moreau P, et al. (2012) ATP synthesis-coupled and-uncoupled acetate production from acetyl-CoA by mitochondrial acetate:succinate CoA-transferase and acetyl-CoA thioesterase in Trypanosoma. J Biol Chem 287:17186–17197. doi: 10.1074/jbc.M112.355404 22474284
31. Rivière L, Moreau P, Allmann S, Hahn M, Biran M, et al. (2009) Acetate produced in the mitochondrion is the essential precursor for lipid biosynthesis in procyclic trypanosomes. Proc Natl Acad Sci U S A 106:12694–12699. doi: 10.1073/pnas.0903355106 19625628
32. Creek DJ, Anderson J, McConville MJ, Barrett MP (2012) Metabolomic analysis of trypanosomatid protozoa. Mol Biochem Parasitol 181:73–84. doi: 10.1016/j.molbiopara.2011.10.003 22027026
33. Breitling R, Vitkup D, Barrett MP (2008) New surveyor tools for charting microbial metabolic maps. Nat Rev Microbiol 6:156–161. 18026122
34. Breitling R, Pitt AR, Barrett MP (2006) Precision mapping of the metabolome. Trends Biotechnol 24:543–548. 17064801
35. MacRae JI, Sheiner L, Nahid A, Tonkin C, Striepen B, et al. (2012) Mitochondrial metabolism of glucose and glutamine is required for intracellular growth of Toxoplasma gondii. Cell Host Microbe 12:682–692. doi: 10.1016/j.chom.2012.09.013 23159057
36. MacRae JI, Dixon MW, Dearnley MK, Chua HH, Chambers JM, et al. (2013) Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol 11:67. doi: 10.1186/1741-7007-11-67 23763941
37. Storm J, Sethia S, Blackburn GJ, Chokkathukalam A, Watson DG, et al. (2014) Phosphoenolpyruvate carboxylase identified as a key enzyme in erythrocytic Plasmodium falciparum carbon metabolism. PLoS Pathog 10:e1003876. doi: 10.1371/journal.ppat.1003876 24453970
38. Creek DJ, Chokkathukalam A, Jankevics A, Burgess KE, Breitling R, et al. (2012) Stable isotope-assisted metabolomics for network-wide metabolic pathway elucidation. Anal Chem 84:8442–8447. doi: 10.1021/ac3018795 22946681
39. Chokkathukalam A, Jankevics A, Creek DJ, Achcar F, Barrett MP, Breitling R (2013) mzMatch-ISO: an R tool for the annotation and relative quantification of isotope-labelled mass spectrometry data. Bioinformatics. 29:281–283. doi: 10.1093/bioinformatics/bts674 23162054
40. Achcar F, Barrett MP, Breitling R (2013) Explicit consideration of topological and parameter uncertainty gives new insights into a well-established model of glycolysis. FEBS J 280:4640–4651. doi: 10.1111/febs.12436 23865459
41. Gualdron-López M, Vapola MH, Miinalainen IJ, Hiltunen JK, Michels PA, Antonenkov VD (2012) Channel-forming activities in the glycosomal fraction from the bloodstream form of Trypanosoma brucei. PLoS One 7:e34530. doi: 10.1371/journal.pone.0034530 22506025
42. Greig N, Wyllie S, Patterson S, Fairlamb AH (2009) A comparative study of methylglyoxal metabolism in trypanosomatids. FEBS J 276:376–386. doi: 10.1111/j.1742-4658.2008.06788.x 19076214
43. Montgomery CM, Webb JL (1956) Metabolic studies on heart mitochondria. II. The inhibitory action of parapyruvate on the tricarboxylic acid cycle. J Biol Chem 221:359–368. 13345825
44. Stoffel SA, Alibu VP, Hubert J, Ebikeme C, Portais JC et al. (2011) Transketolase in Trypanosoma brucei. Mol Biochem Parasitol 179:1–7. doi: 10.1016/j.molbiopara.2011.04.006 21570429
45. Fish WR, Looker DL, Marr JJ, Berens RL (1982) Purine metabolism in the bloodstream forms of Trypanosoma gambiense and Trypanosoma rhodesiense. Biochim Biophys Acta 719:223–231. 6817814
46. Creek DJ, Nijagal B, Kim DH, Rojas F, Matthews KR, et al. (2013) Metabolomics guides rational development of a simplified cell culture medium for drug screening against Trypanosoma brucei. Antimicrob Agents Chemother 57:2768–2779. doi: 10.1128/AAC.00044-13 23571546
47. Parkin DW (1996) Purine-specific nucleoside N-ribohydrolase from Trypanosoma brucei brucei. Purification, specificity, and kinetic mechanism. J Biol Chem 271:21713–21719. 8702965
48. Davies MJ, Ross AM, Gutteridge WE (1983) The enzymes of purine salvage in Trypanosoma cruzi, Trypanosoma brucei and Leishmania mexicana. Parasitology 87:211–217. 6316234
49. Vodnala M, Fijolek A, Rofougaran R, Mosimann M, Mäser P, Hofer A (2008) Adenosine kinase mediates high affinity adenosine salvage in Trypanosoma brucei J Biol Chem 283:5380–5388. doi: 10.1074/jbc.M705603200 18167353
50. Berger BJ, Dai WW, Wang H, Stark RE, Cerami A (1996) Aromatic amino acid transamination and methionine recycling in trypanosomatids. Proc Natl Acad Sci USA 93: 4126–4130. 8633027
51. Hasne MP, Barrett MP (2000) Transport of methionine in Trypanosoma brucei brucei. Mol Biochem Parasitol 111:299–307. 11163438
52. Durieux PO, Schütz P, Brun R, Köhler P (1991) Alterations in Krebs cycle enzyme activities and carbohydrate catabolism in two strains of Trypanosoma brucei during in vitro differentiation of their bloodstream to procyclic stages. Mol Biochem Parasitol 45:19–27. 1904988
53. Hart DT, Misset O, Edwards SW, Opperdoes FR (1984) A comparison of the glycosomes (microbodies) isolated from Trypanosoma brucei bloodstream form and cultured procyclic trypomastigotes. Mol Biochem Parasitol 12:25–35. 6749187
54. Hasne, MP (2001) Amino acid transporters in Trypanosoma brucei brucei. University of Glasgow PhD thesis.
55. Aranda A, Maugeri D, Uttaro AD, Opperdoes F, Cazzulo JJ, et al.(2006) The malate dehydrogenase isoforms from Trypanosoma brucei: subcellular localization and differential expression in bloodstream and procyclic forms. Int J Parasitol 36:295–307. 16321390
56. Opperdoes FR, Borst P, Spits H (1977) Particle-bound enzymes in the bloodstream form of Trypanosoma brucei. Eur J Biochem 76:21–28. 195809
57. Opperdoes FR, Borst P, Bakker S, Leene W (1977) Localization of glycerol-3-phosphate oxidase in the mitochondrion and particulate NAD+-linked glycerol-3-phosphate dehydrogenase in the microbodies of the bloodstream form to Trypanosoma brucei. Eur J Biochem 76:29–39. 142010
58. Ali JA, Tagoe DN, Munday JC, Donachie A, Morrison LJ, de Koning HP (2013) Pyrimidine biosynthesis is not an essential function for Trypanosoma brucei bloodstream forms. PLoS One 8:e58034. doi: 10.1371/journal.pone.0058034 23505454
59. Ali JA, Creek DJ, Burgess K, Allison HC, Field MC, et al. (2013) Pyrimidine salvage in Trypanosoma brucei bloodstream forms and the trypanocidal action of halogenated pyrimidines. Mol Pharmacol 83:439–453. doi: 10.1124/mol.112.082321 23188714
60. Alsford S, Turner DJ, Obado SO, Sanchez-Flores A, Glover L, et al. (2011) High-throughput phenotyping using parallel sequencing of RNA interference targets in the African trypanosome. Genome Res 21:915–924. doi: 10.1101/gr.115089.110 21363968
61. Coustou V, Besteiro S, Biran M, Diolez P, Bouchaud V et al. (2003) ATP generation in the Trypanosoma brucei procyclic form: cytosolic substrate level is essential, but not oxidative phosphorylation. J Biol Chem 278:49625–49635. 14506274
62. Bowes AE, Samad AH, Jiang P, Weaver B, Mellors A (1993) The acquisition of lysophosphatidylcholine by African trypanosomes. J Biol Chem 268:13885–13892. 8314756
63. Lee SH, Stephens JL, Paul KS, Englund PT (2006) Fatty acid synthesis by elongases in trypanosomes. Cell 126:691–699. 16923389
64. Mariño K, Güther ML, Wernimont AK, Amani M, Hui R, et al. (2010) Identification, subcellular localization, biochemical properties, and high-resolution crystal structure of Trypanosoma brucei UDP-glucose pyrophosphorylase. Glycobiology 20:1619–1630. doi: 10.1093/glycob/cwq115 20724435
65. Saunders EC, Ng WW, Chambers JM, Ng M, Naderer T, et al. (2011) Isotopomer profiling of Leishmania mexicana promastigotes reveals important roles for succinate fermentation and aspartate uptake in tricarboxylic acid cycle (TCA) anaplerosis, glutamate synthesis, and growth. J Biol Chem 286:27706–27717. doi: 10.1074/jbc.M110.213553 21636575
66. Achcar F, Kerkhoven EJ, SilicoTryp Consortium, Bakker BM, Barrett MP, et al.(2012) Dynamic modelling under uncertainty: the case of Trypanosoma brucei energy metabolism. PLoS Comput Biol 8:e1002352. doi: 10.1371/journal.pcbi.1002352 22379410
67. Hannaert V, Saavedra E, Duffieux F, Szikora JP, Rigden DJ et al.(2003) Plant-like traits associated with metabolism of Trypanosoma parasites. Proc Natl Acad Sci USA 100:1067–1071. 12552132
68. Jensen BC, Sivam D, Kifer CT, Myler PJ, Parsons M (2009) Widespread variation in transcript abundance within and across developmental stages of Trypanosoma brucei. BMC Genomics 10:482. doi: 10.1186/1471-2164-10-482 19840382
69. Gunasekera K, Wüthrich D, Braga-Lagache S, Heller M, Ochsenreiter T (2012) Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genomics 13:556. doi: 10.1186/1471-2164-13-556 23067041
70. Nett IR, Martin DM, Miranda-Saavedra D, Lamont D, Barber JD, et al. (2009) The phosphoproteome of bloodstream form Trypanosoma brucei, causative agent of African sleeping sickness. Mol Cell Proteomics 8:1527–1538. doi: 10.1074/mcp.M800556-MCP200 19346560
71. Millerioux Y, Ebikeme C, Biran M, Morand P, Bouyssou G, et al. (2013) The threonine degradation pathway of the Trypanosoma brucei procyclic form: the main carbon source for lipid biosynthesis is under metabolic control. Mol Microbiol 90:114–29. doi: 10.1111/mmi.12351 23899193
72. Verlinde CL, Hannaert V, Blonski C, Willson M, Périé JJ, et al. (2001) Glycolysis as a target for the design of new anti-trypanosome drugs. Drug Resist Updat 4:50–65. 11512153
73. Hirumi H, Hirumi K (1989) Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J Parasitol 75:985–989. 2614608
74. t’Kindt R, Jankevics A, Scheltema R, Zheng L, Watson DG (2010). Towards an unbiased metabolic profiling of protozoan parasites: optimisation of a Leishmania sampling protocol for HILIC-orbitrap analysis. Anal Bioanal Chem 398:2059–2069. doi: 10.1007/s00216-010-4139-0 20824428
75. Tautenhahn R, Bottcher C, Neumann S (2008). Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics 9:504. doi: 10.1186/1471-2105-9-504 19040729
76. Scheltema RA, Jankevics A, Jansen RC, Swertz MA, Breitling R (2011) PeakML/mzMatch: A File Format, Java Library, R Library, and Tool-Chain for Mass Spectrometry Data Analysis. Anal Chem 83:2786–2793. doi: 10.1021/ac2000994 21401061
77. Creek DJ, Jankevics A, Burgess KEV, Breitling R, Barrett MP (2012) IDEOM: An Excel interface for analysis of LC-MS based metabolomics data. Bioinformatics 28:1048–1049. doi: 10.1093/bioinformatics/bts069 22308147
78. Creek DJ, Jankevics A, Breitling R, Watson DG, Barrett MP, et al. (2011) Toward Global Metabolomics Analysis with Hydrophilic Interaction Liquid Chromatography—Mass Spectrometry: Improved Metabolite Identification by Retention Time Prediction. Anal Chem 83:8703–8710. doi: 10.1021/ac2021823 21928819
79. Akoka S, Barantin L, Trierweiler M (1999) Concentration Measurement by Proton NMR Using the ERETIC Method. Anal Chem 71:2554–2557. doi: 10.1021/ac981422i 21662801
80. Harlow E, Lane D (1988) Antibodies. A laboratory manual. Cold Spring Harbor Laboratory Press.
81. Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press.
82. Denise H, Giroud C, Barrett MP, Baltz T (1999) Affinity chromatography using trypanocidal arsenical drugs identifies a specific interaction between glycerol-3-phosphate dehydrogenase from Trypanosoma brucei and Cymelarsan. Eur J Biochem 259:339–346. 9914512
83. Bringaud F, Peyruchaud S, Baltz D, Giroud C, Simpson L, Baltz T (1995) Molecular characterization of the mitochondrial heat shock protein 60 gene from Trypanosoma brucei. Mol Biochem Parasitol 74:119–123. 8719252
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 3
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling
- Antimicrobial-Induced DNA Damage and Genomic Instability in Microbial Pathogens
- Attenuation of Tick-Borne Encephalitis Virus Using Large-Scale Random Codon Re-encoding
- BILBO1 Is a Scaffold Protein of the Flagellar Pocket Collar in the Pathogen