
Noninfectious Retrovirus Particles Drive the / Dependent Neutralizing Antibody Response
Members of the APOBEC3 family of deoxycytidine deaminases counteract a broad range of retroviruses in vitro through an indirect mechanism that requires virion incorporation and inhibition of reverse transcription and/or hypermutation of minus strand transcripts in the next target cell. The selective advantage to the host of this indirect restriction mechanism remains unclear, but valuable insights may be gained by studying APOBEC3 function in vivo. Apobec3 was previously shown to encode Rfv3, a classical resistance gene that controls the recovery of mice from pathogenic Friend retrovirus (FV) infection by promoting a more potent neutralizing antibody (NAb) response. The underlying mechanism does not involve a direct effect of Apobec3 on B cell function. Here we show that while Apobec3 decreased titers of infectious virus during acute FV infection, plasma viral RNA loads were maintained, indicating substantial release of noninfectious particles in vivo. The lack of plasma virion infectivity was associated with a significant post-entry block during early reverse transcription rather than G-to-A hypermutation. The Apobec3-dependent NAb response correlated with IgG binding titers against native, but not detergent-lysed virions. These findings indicate that innate Apobec3 restriction promotes NAb responses by maintaining high concentrations of virions with native B cell epitopes, but in the context of low virion infectivity. Finally, Apobec3 restriction was found to be saturable in vivo, since increasing FV inoculum doses resulted in decreased Apobec3 inhibition. By analogy, maximizing the release of noninfectious particles by modulating APOBEC3 expression may improve humoral immunity against pathogenic human retroviral infections.
Published in the journal:
. PLoS Pathog 7(10): e32767. doi:10.1371/journal.ppat.1002284
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002284
Summary
Members of the APOBEC3 family of deoxycytidine deaminases counteract a broad range of retroviruses in vitro through an indirect mechanism that requires virion incorporation and inhibition of reverse transcription and/or hypermutation of minus strand transcripts in the next target cell. The selective advantage to the host of this indirect restriction mechanism remains unclear, but valuable insights may be gained by studying APOBEC3 function in vivo. Apobec3 was previously shown to encode Rfv3, a classical resistance gene that controls the recovery of mice from pathogenic Friend retrovirus (FV) infection by promoting a more potent neutralizing antibody (NAb) response. The underlying mechanism does not involve a direct effect of Apobec3 on B cell function. Here we show that while Apobec3 decreased titers of infectious virus during acute FV infection, plasma viral RNA loads were maintained, indicating substantial release of noninfectious particles in vivo. The lack of plasma virion infectivity was associated with a significant post-entry block during early reverse transcription rather than G-to-A hypermutation. The Apobec3-dependent NAb response correlated with IgG binding titers against native, but not detergent-lysed virions. These findings indicate that innate Apobec3 restriction promotes NAb responses by maintaining high concentrations of virions with native B cell epitopes, but in the context of low virion infectivity. Finally, Apobec3 restriction was found to be saturable in vivo, since increasing FV inoculum doses resulted in decreased Apobec3 inhibition. By analogy, maximizing the release of noninfectious particles by modulating APOBEC3 expression may improve humoral immunity against pathogenic human retroviral infections.
Introduction
Millions of years of co-evolution of retroviruses and mammalian hosts has led to the emergence of retroviral host restriction factors, some of which were discovered following major efforts to understand key steps in the HIV life cycle. Most of these genes, such as TRIM5α [1], Tetherin/Bst2 [2] and SAMHD1 [3], restrict retroviruses in the infected cell. In contrast, members of the APOBEC3 family of deoxycytidine deaminases are distinguished by their ability to inhibit retroviruses in the next target cell. In cell culture, co-transfection of APOBEC3 expression plasmids with retrovirus molecular clones does not decrease virus output, but the infectivity of the resulting virions is dramatically decreased [4]–[5]. These APOBEC3-containing virions are fusion-competent, but encounter post-entry blocks from early reverse transcription to integration [6]–[7], with G-to-A hypermutation of nascent reverse transcripts observed in most, but not all [8]–[11] retrovirus infections. Notably, while in vivo studies have largely focused on G-to-A hypermutation as a read-out of APOBEC3 function [12]–[14], the biological relevance of APOBEC3-mediated reduction of virion infectivity remains unclear. In fact, it is currently unknown whether high viral output with reduced infectivity can be detected in vivo, since multiple rounds of replication with Apobec3 restriction will ultimately result in decreased total virus titers [15]–[16].
Investigating the impact of APOBEC3 restriction in vivo is logistically difficult in humans due to potential redundancy in antiretroviral activities of seven human APOBEC3 members (APOBEC3A, B, C, D, F, G and H) (reviewed in [17]). Moreover, APOBEC3 activity is likely most relevant immediately following viral transmission, but such biological samples are very difficult to obtain from pathogenic human retrovirus infections. In contrast, mice encode a single Apobec3 gene (mA3) [4]–[5] that could be genetically disrupted. While mA3-deficient mice are physiologically normal [18], they proved more susceptible to pathogenic murine retroviral infections that include Mouse Mammary Tumor virus [8], Moloney Murine Leukemia Virus [19] and Friend retrovirus (FV) complex [20]–[21]. These studies provide a springboard for investigating the immunological impact of mA3 restriction in vivo.
FV causes severe splenomegaly and erythroleukemia in adult immunocompetent mice, and resistance and susceptibility to FV have been mapped to a variety of genes [22]–[23]. One of these genes, Recovery from Friend retrovirus gene 3 (Rfv3), influences the recovery of mice from viremia by promoting a more potent neutralizing antibody (NAb) response [24]–[25]. C57BL/6 (B6) mice recover from viremia, develop stronger NAb responses and are Rfv3 resistant, while BALB/c, A.BY and A/WySn strains have persistent viremia, develop weaker NAb responses and are Rfv3 susceptible. Our group and others demonstrated that the B6 mA3 gene acts as the classical Rfv3 resistance gene, promoting stronger NAb responses and facilitating recovery from FV viremia, infection, and disease in (B6×BALB/c)F1, (B6×A.BY)F1 and (B6×A/WySn)F1 mice [20], [26]–[27]. In addition, the B6 mA3 gene restricted acute FV replication in immune compartments [20]–[21], [27]–[28]. Acute FV inhibition was associated with significantly higher mA3 mRNA expression and splicing differences in Rfv3 resistant (B6) compared to Rfv3 susceptible strains (BALB/c, A.BY, A/WySn) [10], [21], [26], [29]. However, the mechanism through which B6 mA3 promotes FV-specific NAb responses remains unknown.
The APOBEC3 genes are evolutionarily related to Activation-Induced Deaminase, a B-cell specific enzyme that is critical for antibody affinity maturation and class-switching [30]. Thus, the identification of mA3 as Rfv3 led to the immediate hypothesis that mA3 may directly influence antibody development [20]. However, hapten immunization studies revealed that B6 mA3 influenced antibody affinity maturation only in the context of FV infection [28]. Thus, the underlying mechanism for the mA3/Rfv3 phenotype does not involve a direct effect of mA3 on B cell function. In fact, decreased immune dysfunction was found to be a critical component of how B6 mA3 promotes NAb responses [27]–[28]. We therefore hypothesized that mA3 influences NAb responses by promoting the release of noninfectious virions in vivo [28], driving NAb responses without eliciting pathology.
The mechanism for the mA3/Rfv3 phenotype may have implications for improving humoral immunity against human retroviruses, particularly against HIV-1. However, HIV-1 encodes Vif, which promotes the degradation of the human homologues APOBEC3G (hA3G) and APOBEC3F (hA3F) [31]–[34]. Surprisingly, despite the action of Vif, hA3G/hA3F-mediated G-to-A hypermutation was detected in HIV-1 sequences from clinical specimens [12]–[14]. These findings could in part be due to the emergence of defective Vif alleles [35]. However, high hA3G mRNA levels in primary cells or tissues were also associated with lower HIV-1 viral loads [36]–[37]. In rhesus macaques infected with SIV, rhesus macaque A3G (rhA3G) levels in colonic biopsies following mucosal vaccination also correlated with set-point plasma viral loads [38]. Thus, hA3G/hA3F may be induced to levels that saturate endogenous levels of Vif. This is consistent with in vitro observations that show inhibition of wild-type HIV-1 with increasing hA3G transfection levels [4]–[5]. Unfortunately, obtaining direct evidence that innate hA3G/hA3F restriction is saturable in humans is not feasible.
We therefore evaluated the in vivo antiviral activity and saturability of B6 mA3 in the context of FV infection of mice. The results provide long-sought insights into a fundamental APOBEC3 restriction phenotype that may have important implications for HIV-1 vaccine research.
Results
B6 mA3 promotes substantial release of noninfectious retroviral particles in vivo
(B6×BALB/c)F1 mice (genotype Rfv3r/s) are highly susceptible to FV infection but recover from plasma viremia by 28 days post-infection (dpi) due to the dominant, B6-encoded Rfv3 resistance gene [20], [26] (Figure S1A in Text S1). In contrast, the majority of (B6 mA3−/−×BALB/c)F1 mice (genotype Rfv3−/s) do not survive to 28 dpi, and those that survive display elevated plasma viremia, consistent with identity between Rfv3 and mA3 [20], [26] (Figure S1B in Text S1). We previously reported that the significant survival disadvantage of (B6 mA3−/−×BALB/c)F1 mice was linked to higher plasma viremia during acute infection (7 dpi), quantified on susceptible Mus dunni cells using an FV envelope-specific monoclonal antibody to detect foci of infectivity [39]. Thus, the Mus dunni assay measures infectious viremia. In contrast, a quantitative RT-PCR assay measuring total viral RNA copies (specifically, the F-MuLV helper virus component, as described in Materials and Methods) does not distinguish between infectious and noninfectious virions [26], [40]. To determine if B6 mA3 affected the relative amount of infectious virions released in vivo, we measured the ratio of virus titers obtained from both assays in plasma from (B6×BALB/c)F1 mice infected with 140 SFFU of FV (Figure 1).
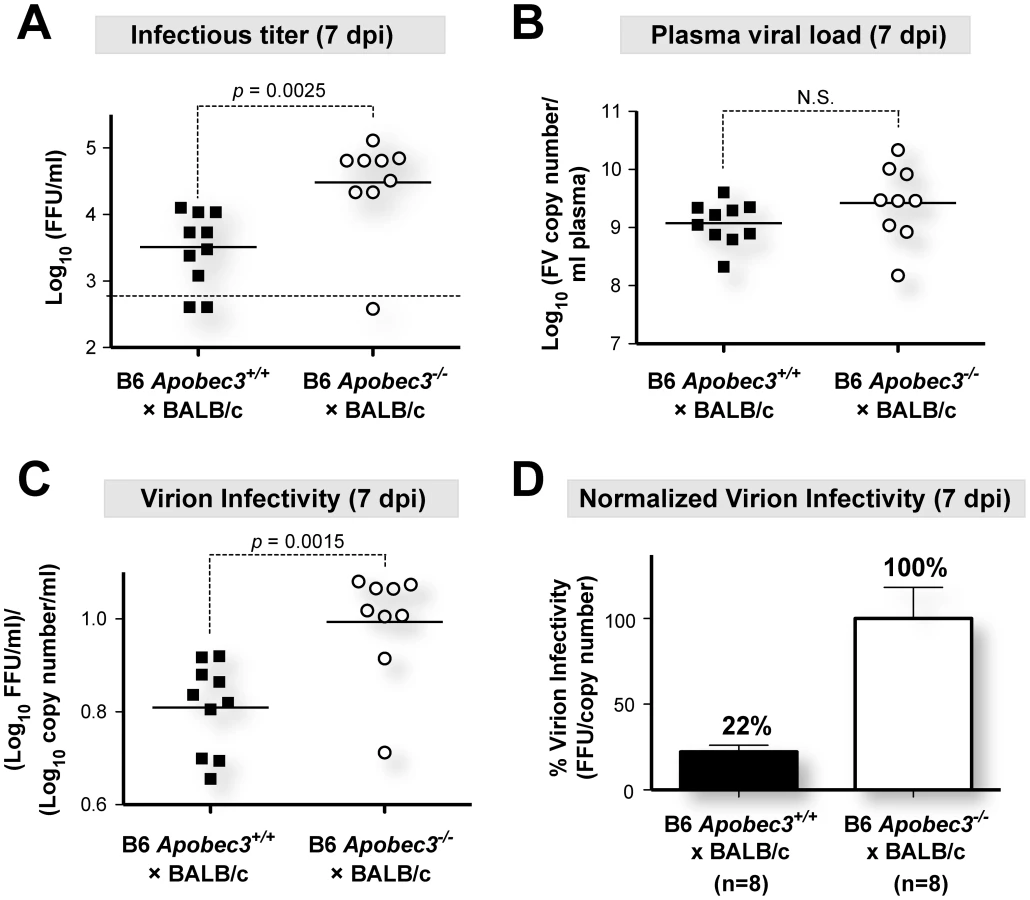
Interestingly, plasma samples from (B6 mA3+/+×BALB/c)F1 mice that had 10-fold lower infectious viremia titers at 7 dpi compared to (B6 mA3−/−×BALB/c)F1 mice (Figure 1A) had equivalent total viral RNA loads (Figure 1B). Thus, the fraction of infectious viral particles was significantly higher in mice without a functional B6 mA3 gene (Figure 1C). By setting the mean virion infectivity of (B6 mA3−/−×BALB/c)F1 mice to 100%, the relative infectivity of plasma virions from (B6 mA3+/+×BALB/c)F1 mice averaged only 22% (Figure 1D). Thus, B6 mA3 restriction resulted in approximately 5-fold higher levels of noninfectious virions at 7 dpi. Higher proportions of noninfectious particles were also observed in (B6 mA3+/+×A.BY)F1 versus (B6 mA3−/−×A.BY)F1 mice (Figure S2 in Text S1).
Minimal mA3-associated G-to-A substitutions in viral RNA and reverse transcripts from plasma virions isolated during acute infection
In the absence of HIV-1 Vif, hA3G and hA3F mediate high levels of G-to-A hypermutation in the minus strand of viral DNA, disrupting open reading frames and effectively inactivating HIV-1. In contrast, mA3 did not appear to induce G-to-A hypermutation against FV, MMTV and MLV [8]–[11], but these data were obtained either from cells infected in vitro or from bulk infected tissues. To investigate the mechanism of B6 mA3 restriction of plasma virions released during acute infection in vivo, we monitored FV sequence evolution in plasma from infected (B6 mA3+/+×BALB/c)F1 and (B6 mA3−/−×BALB/c)F1 mice (Figure 2A). Partial env sequences amplified from the FV inoculum stock phylogenetically clustered with each other (Fig. 2B, left panel), supporting their authenticity as reference sequences. Mutations that were already present in the inoculum quasispecies (Figure 2B, right panel) were then excluded from FV mutational analyses in infected mice.

Multiple FV env sequences were obtained from 7 dpi plasma of (B6 mA3+/+×BALB/c)F1 and (B6 mA3−/−×BALB/c)F1 mice. Relative to the FV inoculum sequences, the cumulative mutational load (Figure 2C), G-to-A substitutions (Figure 2C), and the continuity of the envelope open reading frames (Figure S3 in Text S1) in plasma viral RNA from (B6 mA3+/+×BALB/c)F1 and (B6 mA3−/−×BALB/c)F1 mice did not significantly differ from each other. Thus, the reduction in the infectivity of 7 dpi plasma virions from (B6 mA3+/+×BALB/c)F1 mice (Figure 1C) was likely not due to disproportionately mutated viral genomes.
Newly formed FV reverse transcripts in target cells following infection with plasma virions were next evaluated using approaches to bias for detection of G-to-A mutations [11], [41]–[42]. When compared to FV sequences from the inoculum and the corresponding 7 dpi plasma samples, similar mutation frequencies and total G-to-A substitutions were observed in (B6 mA3+/+×BALB/c)F1 and (B6 mA3−/−×BALB/c)F1 mice (Figure 2D). However, when these G-to-A substitutions were partitioned into mA3-associated dinucleotide preferences, the (B6 mA3+/+×BALB/c)F1 strain was associated with the detection of GG→AG and GA→AA mutations (Figure 2D). Thus, we could detect signatures of mA3-associated G-to-A mutations from reverse transcripts generated from acute B6 mA3+ plasma virions. However, even with techniques that significantly biased for the detection of such reverse transcripts, the G-to-A substitution frequency obtained for FV (0.14%) was at least 10-fold lower than that observed for HIV-1 ΔVif, which ranges from 1.3 to 6.5% [14]. Together, these findings suggest that mA3-mediated deamination plays a very minor role in restricting FV infection in vivo.
Acute plasma virions from (B6 mA3+/+×BALB/c)F1 mice are inhibited at the earliest stages of reverse transcription
The lack of mA3-associated G-to-A substitutions in reverse transcripts from acute plasma virions of (B6 mA3+/+×BALB/c)F1 mice argued in favor of a deamination-independent mechanism of mA3 inhibition. Human A3G can non-enzymatically impair an early step in HIV-1 reverse transcription involving the generation of strong stop DNA [7], [43]. Accordingly, we quantified the levels of newly formed early (R-U5 or strong-stop DNA) and late (R-gag) reverse transcripts following single-round infection of target cells with 7 dpi plasma FV virions (Figure 2A). These studies revealed a 9-fold decrease in early reverse transcripts from (B6 mA3+/+×BALB/c)F1 plasma virions compared to F1 mice lacking B6 mA3 (Figure 2E, left panel). No further decrease was observed in late reverse transcripts (Figure 2E, right panel). Thus, the post-entry block conferred by B6 mA3 on FV plasma virions occurred primarily during the earliest stages of reverse transcription.
Decreased cellular FV infection levels in (B6 mA3+/+×BALB/c)F1 mice
We previously reported that B6 mA3 decreased FV infection levels in target cells that include erythroblasts and B cells in (B6×A.BY)F1 mice at 7 dpi [28]. FV infected cells were quantified by flow cytometry using a Glyco-Gag specific monoclonal antibody, MAb 34 [44]. We now extend this observation to (B6×BALB/c)F1 mice. As shown in Table 1, the percentage of MAb 34+ cells was consistently lower in (B6 mA3+/+×BALB/c)F1 compared to (B6 mA3−/−×BALB/c)F1 strains in multiple cell subpopulations in the bone marrow and the spleen. Statistical significance was achieved with bone marrow erythroblasts and splenic B, T and dendritic cells. These findings revealed that at 7 dpi, B6 mA3 restriction of FV virion infectivity coincided with decreased FV infection of multiple target cells in vivo.
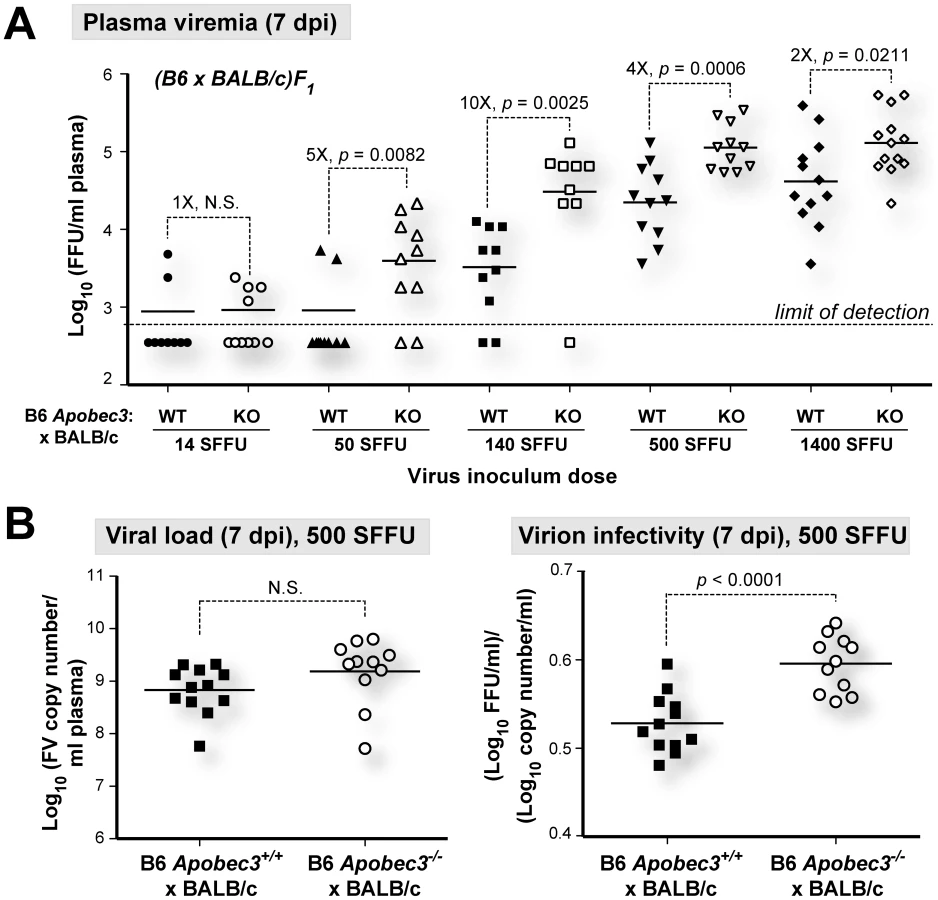
B6 mA3 is a saturable innate restriction factor in vivo
To test whether B6 mA3 restriction was saturable in vivo, we infected (B6 mA3+/+×BALB/c)F1 and (B6 mA3−/−×BALB/c)F1 mice with titrated doses of FV, and measured infectious plasma viremia at 7 dpi (Figure 3A). (B6 mA3−/−×BALB/c)F1 mice exhibited significantly higher 7 dpi infectious viremia compared to (B6 mA3+/+×BALB/c)F1 mice at all inoculum dosages, except at the lowest dose (14 SFFU), in which most titers were below the detection limit of the assay. The fold-difference in infectious viremia between (B6 mA3+/+×BALB/c)F1 and (B6 mA3−/−×BALB/c)F1 cohorts varied with the dose of the viral inoculum. The fold-difference in B6 mA3 restriction could not be accurately determined at 14 and 50 SFFU since several samples had infectious titers that were below the limit of detection. Maximum fold-difference in B6 mA3-mediated restriction was observed at 140 SFFU with 10-fold restriction (Figure 3A). At 500 SFFU and 1400 SFFU inoculum doses, there was only a 4-fold and 2-fold effect, respectively (Figure 3A). These results demonstrate that B6 mA3-mediated restriction was saturable in vivo. Notably, even at a higher viral dose that resulted in decreased B6 mA3 inhibition, similar viral RNA loads (Figure 3B; left panel) and higher noninfectious particle release (Figure 3B; right panel) were still observed in (B6 mA3+/+×BALB/c)F1 versus (B6 mA3−/−×BALB/c)F1 mice.
The B6 mA3-dependent NAb response correlates with IgG titers against intact virions
The B6 mA3 gene promoted NAb responses in (B6×A.BY)F1, (B6×A/WySn)F1 and pure B6 mice [20], [27]. However, measurements of NAb responses at 28 dpi in (B6×BALB/c)F1 mice were confounded by the increased mortality of (B6 mA3−/−×BALB/c)F1 mice [20], [26]. In contrast, infection with 10-fold lower dose (14 SFFU) resulted in all of the mice surviving to 28 dpi, providing an opportunity to revisit this question in this F1 strain (Figure 4A). As shown in Figure 4B, (B6 mA3+/+×BALB/c)F1 mice developed significantly stronger NAb responses than (B6 mA3−/−×BALB/c)F1 mice. Thus, the B6 mA3 gene promoted FV-specific NAb responses in four genetic backgrounds that include (B6×BALB/c)F1, (B6×A.BY)F1, (B6×A/WySn)F1 and B6 mice.
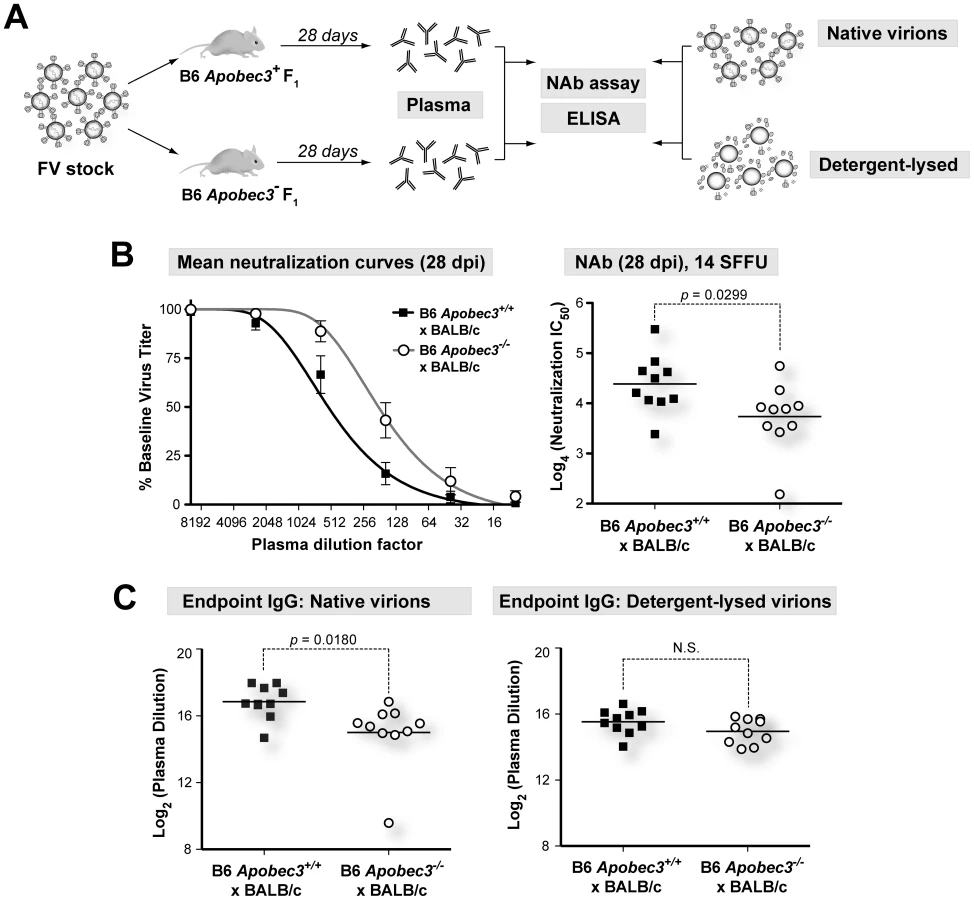
To determine which components of the 28 dpi plasma correlated with B6 mA3-dependent neutralization, we evaluated the binding titers of the major immunologlobulin isotypes in plasma, IgM and IgG. Native virions were bound to 96-well plates, and endpoint IgM and IgG titers were determined by indirect ELISA. Endpoint IgM titers of 28 dpi plasma from B6 mA3+ F1 and B6 mA3− F1 were not significantly different from each other (Figure S4 in Text S1). In contrast, endpoint IgG titers against native virions were significantly higher in (B6 mA3+/+×BALB/c)F1 compared to (B6 mA3−/−×BALB/c)F1 mice (Figure 4C, left panel). Notably, this difference was not detected if detergent lysed-virions were used (Figure 4C, right panel). Similar results were observed for IgG endpoint titers in (B6 mA3+/+×A.BY)F1 versus (B6 mA3−/−×A.BY)F1 mice (Figure S5 in Text S1). Thus, the B6 mA3-dependent antibody response was distinguished by an IgG response directed against intact virus particles.
Discussion
Mechanistic insights on the mA3/Rfv3 phenotype
The molecular identification of the classical resistance gene Rfv3 as mA3 solved a 30-year mystery in retrovirology [20], [26]–[27]. However, this discovery unlocked new questions, foremost of which is the mechanism for how mA3 promotes NAb responses. Recent studies suggested an indirect mechanism that linked the ability of B6 mA3 to restrict FV in vivo with a more vigorous B cell response [20]–[21], [27]–[28]. However, this finding seemed counterintuitive in light of studies on other viral infections, particularly HIV-1, which showed that viral antigen levels had to be preserved to maintain virus-specific antibody levels [45]–[47]. FV plasma viremia is routinely quantified using a focal infectivity assay that measures infectious virus [20], [39]. We therefore investigated whether this method underestimated the total numbers of FV particles in mice with functional Apobec3 activity.
Using a quantitative PCR assay for FV, B6 mA3+ F1 mice exhibited plasma viral RNA loads that were similar to B6 mA3-deficient F1 mice at 7 dpi. In other words, B6 mA3 activity led to no significant change in the physical numbers of virus particles. Instead, B6 mA3 activity reduced infectious virus titers, indicating the release of substantial levels of noninfectious FV particles. These B6 mA3-restricted plasma virions, which account for up to 80% of virions released during acute infection relative to B6 mA3− F1 mice, encounter a significant post-entry block in early reverse transcription in the next target cell, resulting in reduced FV infection in multiple cellular targets in vivo, including splenic B cells. Thus, high levels of B6 mA3-restricted FV particles likely drove the FV-specific B cell response that resulted in the development of potent NAbs (Figure 5).
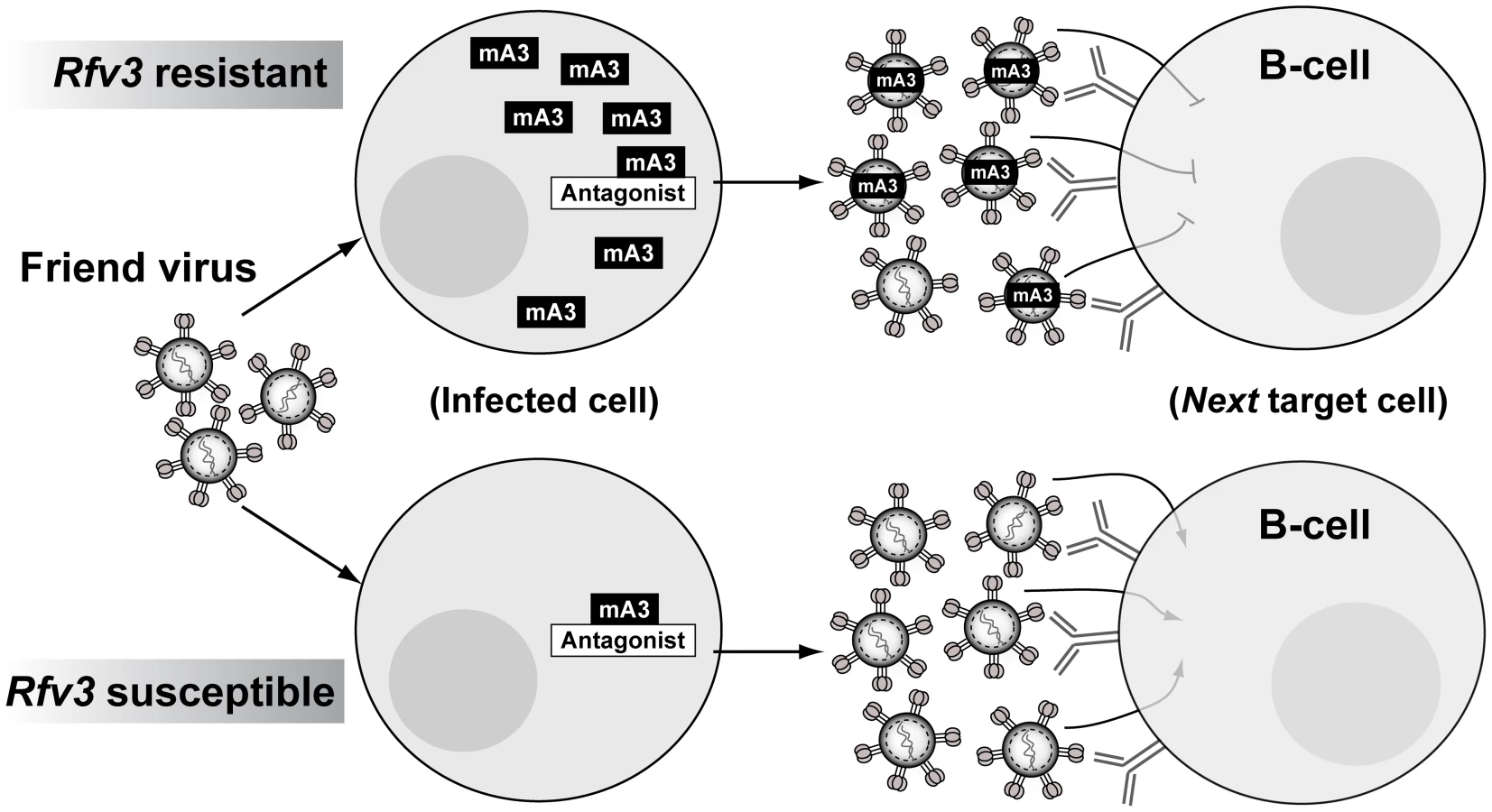
APOBEC3-restricted virions as B-cell immunogens
Native virions are potent inducers of humoral immunity. Repeating molecular patterns on virions may be particularly effective in cross-linking and activating B cell receptors, while viral nucleic acids could enhance B cell responses by activating Toll-like receptors [48]. The FV envelope glycoprotein is the primary target of NAbs [49]–[50], and is organized as a trimeric spike in the native virion, analogous to the HIV-1 envelope glycoprotein [51]–[52]. Detergent treatment disrupts retroviral trimers into monomeric subunits. In this study, we show that the more potent NAb response from B6 mA3+ F1 mice at 28 dpi correlated with significantly higher IgG binding titers against native, but not detergent-lysed virions. Thus, B6 mA3-restricted virions primed a more effective IgG response directed against native envelope trimers. Further studies are in progress to characterize the molecular attributes of this protective NAb response.
Novel insights on a fundamental APOBEC3 restriction phenotype
APOBEC3 is unique among known virus restriction factors due the circuitous nature of its inhibitory mechanism. Instead of simply restricting retroviruses in the infected cell, APOBEC3 evolved the ability to incorporate into budding virions and restrict intact virions in the next target cell [4]–[5]. This biological property of APOBEC3 is conserved from rodents (mA3) to humans (hA3G), suggesting an important evolutionary advantage. However, despite nearly 10 years since the discovery of this fundamental APOBEC3 phenotype [4]–[5], the benefits of next-round inhibition by APOBEC3 to the host remain mysterious. Our findings suggest that mA3 restriction functions as an innate mechanism that allow B cell epitopes to be presented in the context of native virions, subsequently driving the NAb response. This humoral immune response is characterized by high specificity and memory, attributes that could allow the host to effectively control the infection as well as prevent subsequent infections.
Implications for HIV-1 vaccine research
Although FV and HIV-1 infect different cell types and cause different diseases, functional similarities in APOBEC3 proteins from mice and humans suggest that concepts developed from the FV model may prove relevant to HIV-1 infection. Our current model on how mA3 promotes NAb responses implicates noninfectious virions as drivers of the Germinal Center (GC) cell response (Figure 5). These mA3 restricted virions retain immunogenicity but encounter a post-entry block in target cells that include B cells, reducing FV-induced immune dysfunction. While HIV-1 does not infect B cells, CD4+ T cells, the primary targets of HIV-1, also perform critical functions in the GC response, directly interacting with B cells to promote antigen-specific antibody development [53]. Thus, augmenting hA3G function during acute HIV-1 infection may preserve CD4+ T cell function in GCs and promote HIV-specific antibody development. In addition, the role of noninfectious virions in driving the mA3/Rfv3 phenotype further support the use of native virion mimics such as virus-like particles or stabilized trimers as base scaffolds for vaccine design [54]–[55], with the caveat that more sophisticated approaches are needed to elicit NAbs that could broadly neutralize HIV-1 strains from multiple subtypes.
The existence of a lentiviral A3G antagonist, Vif, provides an opportunity to experimentally test whether modulating A3G function can improve the lentivirus-specific humoral immune response. In the SIV model, mutating the Vif gene in SIV to attenuate its function [56] may allow for rhA3G to promote non-infectious virion release and improve humoral immunity in infected rhesus macaques. Similar studies on HIV-1 infection in humans are not possible, but therapeutic agents that block the Vif-hA3G interaction could prove useful. Unfortunately, compounds independently confirmed to specifically inhibit the Vif-hA3G interaction have yet to be identified. In this study, we provide the first evidence that innate mA3 restriction is saturable in vivo, possibly reflecting a delicate balance between the endogenous levels of mA3 and a putative mA3 antagonist encoded by FV, Glyco-Gag [57]. Thus, inducing hA3G levels to saturate, rather than disrupt, the interaction with Vif may be a viable alternative to promote hA3G activity (Figure 5). Notably, Interferon-alpha (IFN-α, a cytokine that could induce APOBEC3G expression in HIV-1 target cells in vitro [58]–[60], improved the kinetics of HIV-1 specific antibody development when clinically administered during acute HIV-1 infection in vivo [61]. Further studies on the link between IFN-α and APOBEC3 in vivo may provide critical insights on whether the saturability of APOBEC3 restriction can be exploited for therapy and vaccine development.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the University of Colorado Health Sciences Center Animal Care and Use Committee [Permit Number B-89709(10)1E]. All infections were performed under isoflurane anaesthesia, and all efforts were made to minimize suffering.
Mice
B6, BALB/c and A.BY mice were purchased from The Jackson Laboratory. B6 mA3 deficient mice were derived from the XN450 cell line [20] and backcrossed for 9 generations. Experimental groups consist primarily of (B6 mA3+/+×BALB/c)F1 versus (B6 mA3−/−×BALB/c)F1 mice. The rationale for an F1 transcomplementation approach is explained in more detail (Figure S1 in Text S1). Experiments were also performed in (B6 mA3+/+×A.BY)F1 versus (B6 mA3−/−×A.BY)F1 mice (Figures S2, S4 and S5 in Text S1). Note that B6, BALB/c and A.BY mice have a functional B cell Activating Factor Receptor (BAFF-R) and normal B cell maturation phenotype, as previously described [26]. These mice are also Fv1b and are therefore susceptible to B-tropic FV infection [62].
Description of FV inoculum stock
Mice were infected with FV complex derived from in vivo-passaged stocks originally used to describe Rfv3 [24], [63]. This B-tropic FV stock contains: replication-competent ecotropic Friend murine leukemia helper virus (F-MuLV); replication-defective spleen-focus forming virus (SFFV; Lilly-Steeves strain); lactate-dehydrogenase elevating virus (LDV). LDV is a ‘contaminant’ RNA virus that could enhance the pathogenicity of FV by delaying adaptive immune responses [64]–[65]. No polytropic or mink-cell focus-inducing viruses (MCFs) were detectable in the virus stock by focal immunofluorescence assay with antibodies Hy7 or mAb 516, which detect the vast majority of MCFs [66] (detection limit of 20/ml).
FV infection
SFFV titers in the FV stock were titered in BALB/c mice and expressed as spleen focus forming units (SFFU) per ml. (B6×BALB/c)F1 mice were infected intravenously via the retro-orbital route with 14 to 1400 SFFU (spleen focus forming units) in 300 µl RPMI and (B6×A.BY)F1 mice were infected with 1400 SFFU. All mice were >2 months old. Plasma samples were harvested at 7 or 28 days post-infection (dpi).
Infectious viremia titration
Infectious viremia titers were determined by serially diluting plasma into Mus dunni cells containing 4 µg/ml polybrene (Sigma; St Louis, MO). Infected cells were detected using a monoclonal antibody specific to F-MuLV gp70, MAb 720, as previously described [20], [39]. Briefly, F-MuLV gp70+ cells were detected following incubation with anti-mouse IgG conjugated to horseradish peroxidase and an insoluble substrate, 3-amino-9-ethylcarbazole (Sigma). Titers were expressed as log10 focus forming units (FFU) per ml of plasma.
Plasma viral load
Viral RNA copy numbers were quantified by real-time PCR (qPCR) as described [26], [40]. RNA from plasma (10 µl) was extracted using the RNAeasy kit (Qiagen; Valencia, CA), and used as template for one-step reverse transcription and PCR (Applied Biosystems; Carlsbad, CA) using FV-specific primers (FLVsense: 5′-GGACAGAAACTACCGCCCTG and FLVantisense: 5′-ACAACCTCAGACAACGAAGTAAGA) and probe (FLVprobe: FAM-TCGCCACCCAGCAGTTTCAGCAGC-TAMRA). Copy numbers were interpolated from an in-plate T7-transcribed RNA standard, and expressed as log10 copy number per ml of plasma.
Sequence alignments of the F-MuLV helper virus (GenBank Accession #Z11128), the Lilly-Steeves SFFV strain (V01552.1) and MCFs (L. H. Evans, unpublished data) revealed that the FV qPCR primers have low to no significant identity with SFFV or MCFs (data not shown). Furthermore, we tested whether the FV qPCR primers could detect any endogenous polytropic env sequences present at one copy per mouse genome. The FV qPCR assay consistently detects an input of 100 copies of cloned F-MuLV DNA. If the qPCR primers cross-react with endogenous MLV, then we should obtain a positive signal if >100 copies of uninfected genomic DNA are added into the reaction. An input of 100 ng genomic DNA (∼34,000 genomes) from uninfected B6, A.BY and BALB/c mice into the qPCR reaction yielded no detectable signals. In contrast, >105 copies were detected from 100 ng genomic DNA from FV infected B6 mice at 7 dpi (S. X. Li and M. L. Santiago, unpublished). Thus, we conclude that the qPCR assay is specific for the F-MuLV helper virus.
Sequence analysis of FV inoculum and plasma viral RNA
RNA was extracted from the FV inoculum and acute plasma using the RNAEasy kit (Qiagen) and reverse-transcribed using the RT2 cDNA synthesis kit (SA Biosciences; Frederick, MD). FV env sequences (849 bp) were obtained by amplifying with primers FV.f (ACTTATTCCAACCATACCTCT) and FV.r (TTTAGCTGGTGGTATTGTTGA) using the Phusion Hi-Fidelity PCR kit (Finnzymes; Woburn, MA). Amplicons were cloned using the TOPO cloning kit (Invitrogen; Carlsbad, CA). FV inoculum sequences were aligned with FB29 and PVC-211 (GenBank Z11128 and M93134) using ClustalX (http://www.clustal.org/). Phylogenetic trees were constructed using the neighbor joining method with 1000 subreplicates. PVC-111 was used as outgroup. Viral RNA sequences were compared with the FV inoculum consensus, excluding variations that were already detected in the inoculum. To quantify mutational loads, the total, G-to-A and mA3-associated mutational frequencies relative to the consensus were divided by the total number of base pairs, G nucleotides and GG/GA dinucleotides analyzed, respectively.
FV mutational analysis of newly formed reverse transcripts in target cells
To bias the detection of G-to-A mutations, we combined four approaches [11], [41]: (1) reverse transcripts were amplified following a single-round infection of Mus dunni cells, to enrich for potentially defective reverse transcripts; (2) a segment of env closest to the primer binding site was chosen, since this region may be present in single-stranded DNA form for longer duration and thereby more susceptible to deamination [67]; (3) Taq polymerase was utilized instead of Pfu polymerase, since Pfu polymerase activity is inhibited by deoxyuridines in DNA templates; and (4) a denaturation temperature of 88°C was used to enrich the detection of G-to-A hypermutated reverse transcripts, which should have a lower melting temperature. 7 dpi plasma samples (2.5 µl) from wild-type and B6 mA3− F1 mice were inoculated into Mus dunni cells in a 6-well plate containing 4 µg/ml polybrene. DNA was extracted after 2 days using the DNAeasy kit (Qiagen). PCR was performed with 10 ng DNA, 1× Sweet PCR mix (SA Biosciences), 2.5 mM dNTP, 1.25 pmol env primers FV.f and FV.r. Thermocycling conditions included a 95°C 15 min hot-start, followed by 30 cycles of denaturation at 88°C for 30 s, 55°C for 30 s and 72°C for 1.5 min. Amplicons were cloned using the TOPO-TA cloning kit (Invitrogen) and 4–8 clones were sequenced for each sample. Consensus from the FV inoculum and the corresponding plasma viral RNA sequences were used as reference for analysis of nucleotide substitutions using the HYPERMUT 2.0 software (hiv.lanl.gov).
Quantification of early and late reverse transcripts
DNA samples as described above were subjected to absolute quantifications for early (R-U5) and late (R-gag) FV transcripts, as well as a housekeeping gene, beta-actin, using a Taqman assay (Applied Biosystems; Foster City, CA) in a CFX96 real-time system (Bio-Rad; Hercules, CA). The primers and probes are listed as follows. Early reverse transcripts: R-U5.fwd, CTCCGATAGACTGAGTCG, R-U5.rev, AGACCCTCCCAAGGAACA, R-U5.probe FAM-CCCGTGTATCCAATAAATCCTCTTGC-TAMRA. PCR cycling conditions were 95°C 10 min followed by 40 cycles of 95°C for 15 sec and 55.7°C for 45 sec. The expected size of PCR product is 94 bp. Late reverse transcripts: R-U5.fwd and R-Gag.rev, TTCGACATCCTTCCAGTGGT and R-Gag.probe, FAM-CTGCAGCATCGTTCTGTGT-TAMRA. The PCR conditions for R-Gag were 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 61°C for 2 min 30 sec. The expected size of the R-Gag amplicon is 670 bp. Mouse beta-actin: Actin.fwd, GGCACCACACCTTCTACAATG, Actin.rev, GGGGTGTTGAAGGTCTCAAAC, and Actin.probe FAM-TGTGGCCCCTGAGGAGCACCC-TAMRA. PCR conditions included a hot-start for 95°C 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 50 sec. Absolute copy numbers were interpolated from a best-fit standard curve against a DNA standard.
Flow cytometry
Bone marrow and spleen cells (106 cells) were stained with MAb 34, an IgG2b monoclonal antibody specific for FV Glyco-Gag [44] for 30 min, then co-stained with: Ter119-FITC (clone TER-119), CD3-Alexa700 (17A2), (BD Biosciences; San Diego, CA); CD11c-PE-Cy7 (N418), (eBioscience; San Diego, CA); CD19-PerCP-Cy5.5 (6D5) (Biolegend; San Diego, CA) and anti-mouse IgG2b-APC (Columbia Biosciences; Columbia, MD). Isotype controls and cells from uninfected mice were used for gating. Cells were processed in an LSR-II flow cytometer (BD Biosciences), collecting up to 250,000 events per sample. Datasets were analyzed using the Flowjo software (Treestar; Ashland, OR).
Neutralizing antibody assay
Serial fourfold dilutions of heat-inactivated plasma were incubated with F-MuLV-N stock virus in the presence of guinea pig complement (Sigma). The antibody∶virus mixture was added into M. dunni cells and developed as in the plasma virus titrations [20]. The number of colonies were counted and compared to a no antibody control, which was set as 100%. Neutralization curves were constructed, and IC50 values were calculated based on a one-site sigmoidal fit using the Graphpad Prism software (Irvine, CA).
Virion preparation
Virion antigens were prepared from culture supernatants obtained from Mus dunni cells infected with F-MuLV-N in T-175 flasks in the presence of 4 µg/ml polybrene (Sigma). Cellular debris was pelleted at 1800× g for 5 min at 4°C, then the supernatants were passed through a 0.22 µm filter. The filtered supernatants were ultracentrifuged at 25,000× g for 2 hr at 4°C in SW28 Ultra-Clear tubes (Beckman Coulter; Brea, CA), and virion pellets were resuspended in 0.5 ml Tris Buffered Saline (TBS) containing 1× protease inhibitor cocktail (Calbiochem; La Jolla, CA) per tube and allowed to dissociate overnight at 4°C. In some preparations, the virion pellets were resuspended in TBS with 1% Empigen-BB (Sigma), a zwitterionic detergent that is commonly used to solubilize viral particles and liberate envelope monomers [68], [69]. Virions were aliquoted and total protein concentrations were determined using a BCA protein assay (Pierce; Rockford, IL).
Enzyme-linked immunosorbent assays (ELISAs)
ELISAs were performed at room temperature and 100 µl/well volumes unless otherwise indicated. Virions (200 ng per well) were coated into Immulon-4 HBX plates (Thermo Scientific Nunc; Rochester, NY) overnight at 4°C and blocked with SuperBlock (Pierce; Rockford, IL) for 2 hr. Serial 2-fold dilutions of plasma in Phosphate Buffered Saline (PBS) were added and incubated for 1 hr. After 6 washes with PBS with 0.05% Tween-20 (PBS-T), 1∶4000 biotinylated goat anti-mouse IgG (Southern Biotechnology; Birmingham, AL) was added and incubated for 1 hr. After 6 PBS-T washes, 1∶4000 streptavidin-conjugated horseradish peroxidase (Southern Biotechnology) was added and incubated for 30 min. Following 6 PBS-T washes, 100 µl of TMB substrate (BioFX Laboratories; Owings Mills, MD) was added per well and incubated in the dark for 15 min. The reaction was stopped with 0.3N sulfuric acid (Sigma), and absorbances were read at 450 nm in a Victor X5 plate reader (Perkin Elmer; Waltham, MA). The same procedures were followed for the IgM ELISAs, except that plasma samples were incubated overnight at 4°C, and 1∶10000 biotinylated goat anti-mouse IgM (Southern Biotechnology) was used. Endpoint titers were calculated by constructing one-site total best-fit curves using the Graphpad Prism software, and interpolating plasma concentrations that correspond to a cut-off value based on twice the mean absorbance background from wells with no plasma added. Samples from uninfected mice were also used as negative controls, and had absorbance values below the cut-off value (data not shown). Endpoint titers were expressed as log2 concentrations.
Statistical analyses
Differences between means were analyzed using a two-tailed Student's t test. The association between G-to-A mutations with B6 mA3 status was inferred by subjecting 2×2 contigency tables to a two-tailed Chi-square test. Differences with p values >0.05 were considered not statistically significant (N.S.).
Supporting Information
Zdroje
1. StremlauMOwensCMPerronMJKiesslingMAutissierP 2004 The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427 848 853
2. NeilSJZangTBieniaszPD 2008 Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451 425 430
3. LaguetteNSobhianBCasartelliNRingeardMChable-BessiaC 2011 SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474 654 657
4. SheehyAMGaddisNCChoiJDMalimMH 2002 Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418 646 650
5. MarianiRChenDSchrofelbauerBNavarroFKonigR 2003 Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114 21 31
6. MbisaJLBarrRThomasJAVandegraaffNDorweilerIJ 2007 HIV-1 cDNAs Produced in the Presence of APOBEC3G Exhibit Defects in Plus-Strand DNA Transfer and Integration. J Virol 81 7099 7110
7. BishopKNVermaMKimEYWolinskySMMalimMH 2008 APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog 4 e1000231
8. OkeomaCMLovsinNPeterlinBMRossSR 2007 APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 445 927 930
9. BrowneEPLittmanDR 2008 Species-specific restriction of apobec3-mediated hypermutation. J Virol 82 1305 1313
10. LangloisMAKemmerichKRadaCNeubergerMS 2009 The AKV murine leukemia virus is restricted and hypermutated by mouse APOBEC3. J Virol 83 11550 11559
11. PetitVGuetardDRenardMKerielASitbonM 2009 Murine APOBEC1 is a powerful mutator of retroviral and cellular RNA in vitro and in vivo. J Mol Biol 385 65 78
12. PaceCKellerJNolanDJamesIGaudieriS 2006 Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J Virol 80 9259 9269
13. KiefferTLKwonPNettlesREHanYRaySC 2005 G→A hypermutation in protease and reverse transcriptase regions of human immunodeficiency virus type 1 residing in resting CD4+ T cells in vivo. J Virol 79 1975 1980
14. KnoepfelSADi GiallonardoFDaumerMThielenAMetznerKJ 2011 In-depth analysis of G-to-A hypermutation rate in HIV-1 env DNA induced by endogenous APOBEC3 proteins using massively parallel sequencing. J Virol Methods 171 329 328
15. HacheGShindoKAlbinJSHarrisRS 2008 Evolution of HIV-1 isolates that use a novel Vif-independent mechanism to resist restriction by human APOBEC3G. Curr Biol 18 819 824
16. OkeomaCMHuegelALLingappaJFeldmanMDRossSR 2010 APOBEC3 Proteins Expressed in Mammary Epithelial Cells Are Packaged into Retroviruses and Can Restrict Transmission of Milk-Borne Virions. Cell Host Microbe 8 534 543
17. SantiagoMLGreeneWC 2008 The role of the Apobec3 family of cytidine deaminases in innate immunity, G-to-A hypermutation and evolution of retroviruses. DomingoEParrishCRHollandJJ Origin and Evolution of Viruses London, UK Academic Press 183 206
18. MiklMCWattINLuMReikWDaviesSL 2005 Mice deficient in APOBEC2 and APOBEC3. Mol Cell Biol 25 7270 7277
19. LowAOkeomaCMLovsinNde las HerasMTaylorTH 2009 Enhanced replication and pathogenesis of Moloney murine leukemia virus in mice defective in the murine APOBEC3 gene. Virology 385 455 463
20. SantiagoMLMontanoMBenitezRMesserRJYonemotoW 2008 Apobec3 encodes Rfv3, a gene influencing neutralizing antibody control of retrovirus infection. Science 321 1343 1346
21. TakedaETsuji-KawaharaSSakamotoMLangloisMANeubergerMS 2008 Mouse APOBEC3 restricts friend leukemia virus infection and pathogenesis in vivo. J Virol 82 10998 11008
22. ChesebroBMiyazawaMBrittWJ 1990 Host genetic control of spontaneous and induced immunity to Friend murine retrovirus infection. Annu Rev Immunol 8 477 499
23. HasenkrugKJDittmerU 2007 Immune control and prevention of chronic Friend retrovirus infection. Front Biosci 12 1544 1551
24. ChesebroBWehrlyK 1979 Identification of a non-H-2 gene (Rfv-3) influencing recovery from viremia and leukemia induced by Friend virus complex. Proc Natl Acad Sci U S A 76 425 429
25. DoigDChesebroB 1979 Anti-Friend virus antibody is associated with recovery from viremia and loss of viral leukemia cell-surface antigens in leukemic mice. Identification of Rfv-3 as a gene locus influencing antibody production. J Exp Med 150 10 19
26. SantiagoMLSmithDSBarrettBSMontanoMBenitezRL 2011 Persistent Friend virus replication and disease in Apobec3-deficient mice expressing functional B-cell-activating factor receptor. J Virol 85 189 199
27. Tsuji-KawaharaSChikaishiTTakedaEKatoMKinoshitaS 2010 Persistence of viremia and production of neutralizing antibodies differentially regulated by polymorphic APOBEC3 and BAFF-R loci in friend virus-infected mice. J Virol 84 6082 6095
28. SantiagoMLBenitezRLMontanoMHasenkrugKJGreeneWC 2010 Innate retroviral restriction by Apobec3 promotes antibody affinity maturation in vivo. J Immunol 185 1114 1123
29. OkeomaCMPetersenJRossSR 2009 Expression of murine APOBEC3 alleles in different mouse strains and their effect on mouse mammary tumor virus infection. J Virol 83 3029 3038
30. MuramatsuMKinoshitaKFagarasanSYamadaSShinkaiY 2000 Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102 553 563
31. StopakKde NoronhaCYonemotoWGreeneWC 2003 HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell 12 591 601
32. SheehyAMGaddisNCMalimMH 2003 The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med 9 1404 1407
33. MarinMRoseKMKozakSLKabatD 2003 HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med 9 1398 1403
34. ZhengYHIrwinDKurosuTTokunagaKSataT 2004 Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol 78 6073 6076
35. SimonVZennouVMurrayDHuangYHoDD 2005 Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog 1 e6
36. JinXBrooksAChenHBennettRReichmanR 2005 APOBEC3G/CEM15 (hA3G) mRNA levels associate inversely with human immunodeficiency virus viremia. J Virol 79 11513 11516
37. UlengaNKSarrADThakore-MeloniSSankaleJLEisenG 2008 Relationship between human immunodeficiency type 1 infection and expression of human APOBEC3G and APOBEC3F. J Infect Dis 198 486 492
38. SuiYZhuQGagnonSDzutsevATerabeM 2010 Innate and adaptive immune correlates of vaccine and adjuvant-induced control of mucosal transmission of SIV in macaques. Proc Natl Acad Sci U S A 107 9843 9848
39. RobertsonMNMiyazawaMMoriSCaugheyBEvansLH 1991 Production of monoclonal antibodies reactive with a denatured form of the Friend murine leukemia virus gp70 envelope protein: use in a focal infectivity assay, immunohistochemical studies, electron microscopy and western blotting. J Virol Methods 34 255 271
40. HeJYChengHJWangYFZhuYTLiGQ 2008 Development of a real-time quantitative reverse transcriptase PCR assay for detection of the Friend leukemia virus load in murine plasma. J Virol Methods 147 345 350
41. MahieuxRSuspeneRDelebecqueFHenryMSchwartzO 2005 Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J Gen Virol 86 2489 2494
42. SuspeneRGuetardDHenryMSommerPWain-HobsonS 2005 Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc Natl Acad Sci U S A 102 8321 8326
43. DoitshGCavroisMLassenKGZepedaOYangZ 2010 Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143 789 801
44. ChesebroBBrittWEvansLWehrlyKNishioJ 1983 Characterization of monoclonal antibodies reactive with murine leukemia viruses: use in analysis of strains of friend MCF and Friend ecotropic murine leukemia virus. Virology 127 134 148
45. VoltersvikPAlbrektsenGUlvestadEDyrhol-RiiseAMSorensenB 2003 Changes in immunoglobulin isotypes and immunoglobulin G (IgG) subclasses during highly active antiretroviral therapy: anti-p24 IgG1 closely parallels the biphasic decline in plasma viremia. J Acquir Immune Defic Syndr 34 358 367
46. MorrisLBinleyJMClasBABonhoefferSAstillTP 1998 HIV-1 antigen-specific and -nonspecific B cell responses are sensitive to combination antiretroviral therapy. J Exp Med 188 233 245
47. BinleyJMTrkolaAKetasTSchillerDClasB 2000 The effect of highly active antiretroviral therapy on binding and neutralizing antibody responses to human immunodeficiency virus type 1 infection. J Infect Dis 182 945 949
48. BachmannMFJenningsGT 2010 Vaccine delivery: a matter of size, geometry, kinetics and molecular patterns. Nat Rev Immunol 10 787 796
49. MesserRJDittmerUPetersonKEHasenkrugKJ 2004 Essential role for virus-neutralizing antibodies in sterilizing immunity against Friend retrovirus infection. Proc Natl Acad Sci U S A 101 12260 12265
50. BrittWJChesebroB 1983 Use of monoclonal anti-gp70 antibodies to mimic the effects of the Rfv-3 gene in mice with Friend virus-induced leukemia. J Immunol 130 2363 2367
51. YangXKurtevaSRenXLeeSSodroskiJ 2005 Stoichiometry of envelope glycoprotein trimers in the entry of human immunodeficiency virus type 1. J Virol 79 12132 12147
52. FassDHarrisonSCKimPS 1996 Retrovirus envelope domain at 1.7 angstrom resolution. Nat Struct Biol 3 465 469
53. ReinhardtRLLiangHELocksleyRM 2009 Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 10 385 393
54. ForsellMNSchiefWRWyattRT 2009 Immunogenicity of HIV-1 envelope glycoprotein oligomers. Curr Opin HIV AIDS 4 380 387
55. YoungKRMcBurneySPKarkhanisLURossTM 2006 Virus-like particles: designing an effective AIDS vaccine. Methods 40 98 117
56. SchmittKHillMSRuizACulleyNPinsonDM 2009 Mutations in the highly conserved SLQYLA motif of Vif in a simian-human immunodeficiency virus result in a less pathogenic virus and are associated with G-to-A mutations in the viral genome. Virology 383 362 372
57. KolokithasARosenkeKMalikFHendrickDSwansonL 2010 The glycosylated Gag protein of a murine leukemia virus inhibits the antiretroviral function of APOBEC3. J Virol 84 10933 10936
58. KoningFANewmanENKimEYKunstmanKJWolinskySM 2009 Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol 83 9474 9485
59. ChenKHuangJZhangCHuangSNunnariG 2006 Alpha interferon potently enhances the anti-human immunodeficiency virus type 1 activity of APOBEC3G in resting primary CD4 T cells. J Virol 80 7645 7657
60. PengGLeiKJJinWGreenwell-WildTWahlSM 2006 Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med 203 41 46
61. Adalid-PeraltaLGodotVColinCKrzysiekRTranT 2008 Stimulation of the primary anti-HIV antibody response by IFN-alpha in patients with acute HIV-1 infection. J Leukoc Biol 83 1060 1067
62. BestSLe TissierPTowersGStoyeJP 1996 Positional cloning of the mouse retrovirus restriction gene Fv1. Nature 382 826 829
63. LillyFSteevesRA 1973 B-tropic Friend virus: a host-range pseudotype of spleen focus-forming virus (SFFV). Virology 55 363 370
64. RobertsonSJAmmannCGMesserRJCarmodyABMyersL 2008 Suppression of acute anti-friend virus CD8+ T-cell responses by coinfection with lactate dehydrogenase-elevating virus. J Virol 82 408 418
65. MarquesRAntunesIEksmondUStoyeJHasenkrugK 2008 B lymphocyte activation by coinfection prevents immune control of friend virus infection. J Immunol 181 3432 3440
66. LavignonMWalkerJLPerrymanSMMalikFGKhanAS 1994 Characterization of epitopes defining two major subclasses of polytropic murine leukemia viruses (MuLVs) which are differentially expressed in mice infected with different ecotropic MuLVs. J Virol 68 5194 5203
67. YuQKonigRPillaiSChilesKKearneyM 2004 Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol 11 435 442
68. SandersRWVesanenMSchuelkeNMasterASchiffnerL 2002 Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J Virol 76 8875 8889
69. PantophletRWangMAguilar-SinoROBurtonDR 2009 The human immunodeficiency virus type 1 envelope spike of primary viruses can suppress antibody access to variable regions. J Virol 83 1649 1659
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 10
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- Severe Acute Respiratory Syndrome Coronavirus Envelope Protein Regulates Cell Stress Response and Apoptosis
- The SARS-Coronavirus-Host Interactome: Identification of Cyclophilins as Target for Pan-Coronavirus Inhibitors
- Biochemical and Structural Insights into the Mechanisms of SARS Coronavirus RNA Ribose 2′-O-Methylation by nsp16/nsp10 Protein Complex
- Evolutionarily Divergent, Unstable Filamentous Actin Is Essential for Gliding Motility in Apicomplexan Parasites