
A Lack of Parasitic Reduction in the Obligate Parasitic Green Alga
Helicosporidium is a highly-adapted obligate parasite of animals. Its evolutionary origins were unclear for almost a century, but molecular analysis ultimately and surprisingly showed that it is a green alga, which means it has undergone an evolutionary transition from autotrophy to parasitism comparable to that of the malaria parasite Plasmodium and its relatives. Such transitions are often associated with the loss of biological functions that are no longer necessary in their novel environment and with the development of molecular mechanisms, sometimes quite sophisticated, to invade and take advantage of their hosts. Yet, very little is actually known about the early stages of the transition of a free-living organism to an obligate intracellular parasite. Here we sequenced the genome and transcriptome of Helicosporidium, and use it to show that the outcome of this transition is quite different from that of Plasmodium.
Published in the journal:
. PLoS Genet 10(5): e32767. doi:10.1371/journal.pgen.1004355
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004355
Summary
Helicosporidium is a highly-adapted obligate parasite of animals. Its evolutionary origins were unclear for almost a century, but molecular analysis ultimately and surprisingly showed that it is a green alga, which means it has undergone an evolutionary transition from autotrophy to parasitism comparable to that of the malaria parasite Plasmodium and its relatives. Such transitions are often associated with the loss of biological functions that are no longer necessary in their novel environment and with the development of molecular mechanisms, sometimes quite sophisticated, to invade and take advantage of their hosts. Yet, very little is actually known about the early stages of the transition of a free-living organism to an obligate intracellular parasite. Here we sequenced the genome and transcriptome of Helicosporidium, and use it to show that the outcome of this transition is quite different from that of Plasmodium.
Introduction
Helicosporidia are parasitic protists characterized by mature discoid cysts each containing a single filamentous and three ovoid cells [1], [2]. These parasites invade their invertebrate hosts per os and initiate their replicative stage within the digestive tract [3], [4]. The cysts, triggered by chemical changes in the gut, dehisce and release both the ovoid cells and filament cell. The ovoid cells remain in the gut lumen whereas the uncoiled and barbed filamentous cells penetrate the peritrophic membrane and become anchored to the host midgut cells. Over time the filamentous cells migrate through the midgut epithelium, breach the basement membrane, and invade the hemocoel. In the hemocoel the filament cells will transition to a vegetative stage that replicates by autosporulation; a select number at the four-cell stage will differentiate to the infectious cyst stage characterized by the three ovoid and a single filament cell [5], [6]. Unlike many parasites the vegetative cells of Helicosporidia can be cultured readily on defined media with limited nutrients, suggesting that despite being a parasitic species, they have retained a diverse slate of metabolic pathways allowing for saprobic growth.
The evolutionary origin of Helicosporidia remained uncertain for nearly 100 years since their initial description, although various characters were used to suggest some relationship with microsporidian, sporozoan, and myxosporidian parasites. Recently, however, ultrastructural observations surprisingly revealed that the vegetative state of Helicosporidium cells is similar to that of the achlorophylous trebouxiophyte green alga Prototheca [1], and subsequent phylogenetic inferences derived from actin/tubulin and plastid sequences strongly confirmed this affiliation [7], [8]. The discovery that Helicosporidia are trebouxiophycean green algae raises some interesting questions about the evolution of parasitism: within this single lineage are found free-living autotrophs like most other green algae, but also a variety of symbiotic species, opportunistic pathogens, and perhaps even obligate intracellular parasites, all of which diversified within a relatively narrow evolutionary timescale. The transformation from free-living to parasitic lifestyles often includes the shedding of metabolic functions that are no longer required as the parasite relies increasingly on its host for energy and nutrients [9]. The parasitic relationship may be opportunistic at first, but can switch to being obligate upon reaching a certain threshold of host-dependence, after which the formerly free-living organism can no longer revert to its previous lifestyle due to the ratchet-like nature of these losses. We do not often think of photosynthetic organisms with progenitors for parasitic ones, but a variety or parasitic lineages had at one time photosynthetic ancestors, including oomycetes, several dinoflagellates, and most famously the apicomplexan parasites such as the malaria parasite, Plasmodium (see [10], [11] and references therein). One of the first things to be lost in photosynthetic species is presumably their ability to harvest energy from light and fix carbon. Harnessing light from within large-bodied hosts is probably very difficult if not impossible, and the resulting metabolic deficit must lead to a significant shift in the balance between the host and parasite. Some of these lineages (e.g. oomycetes) probably evolved through a heterotrophic intermediate, but others possibly began their association with animals as phototrophs. How the transformation to parasites took place is of great interest, but unfortunately because it happened so long ago (around 1 bya for Plasmodium [12]) and is now so complete, the critical early stages have long been wiped away. Helicosporidia, in contrast, appear to have evolved from free-living autotrophs relatively recently [13], [14], and might therefore provide some interesting insights. Fossils records and molecular clock analyses suggest that Trebouxiophytes as a group arose in the early Neoproterozoic [13], from which the trebouxiophycean subgroup Chorellales later emerged around 100 million years ago (mya) [14]. Both Helicosporidium and the non-photosynthetic trebouxiophycean Prototheca arose from within the Chorellales [13], so the adaption to parasitism in Helicosporidia occurred less than 100 mya.
To specifically investigate how the metabolic and proteomic complexity of pathogenic Helicosporidia are distinguished from their free-living and symbiotic trebouxiophycean relatives, we sequenced the genome and transcriptome of Helicosporidium sp. ATCC50920, a parasite of the black fly Simulium jonesi [1], [15]. We show that the Helicosporidium genome is 2.5-fold smaller than genomes from the free-living and symbiotic trebouxiophytes, Coccomyxa subellipsoidea C-169 [16] and Chlorella variabilis NC64A [17], which are themselves extremely small for trebouxiophyte genomes. However, the reduction of the Helicosporidium genome is not tied to a massive reduction in metabolic functions: despite its small genome size and parasitic nature, it surprisingly still encodes all major metabolic pathways, with the exception of a small number specifically related to photosynthesis. Even here, the reduction is not complete: all genes relating to light harvesting and electron transport are missing, but the Helicosporidium carbon fixation pathway is nearly complete but for the lack of ribulose-1, 5-bisphosphate carboxylase/oxygenase (RuBisCO) and a pyruvate kinase. The smaller size of the Helicosporidium genome can be attributed to a greater degree of genome compaction (e.g. fewer and smaller introns, and smaller intergenic regions), and most significantly to a lower complexity of gene families, particularly those related to DNA packaging/replication pathways. We also show that the gene family complexity of other metabolic pathways has increased, in particular relating to chitin metabolism, which likely represented a key development in the ability of Helicosporidium to develop in the insect host. Overall, these results give our first view into the early stage in the transition from a free-living autotroph to an obligate pathogen.
Results
General features of the Helicosporidium draft genome
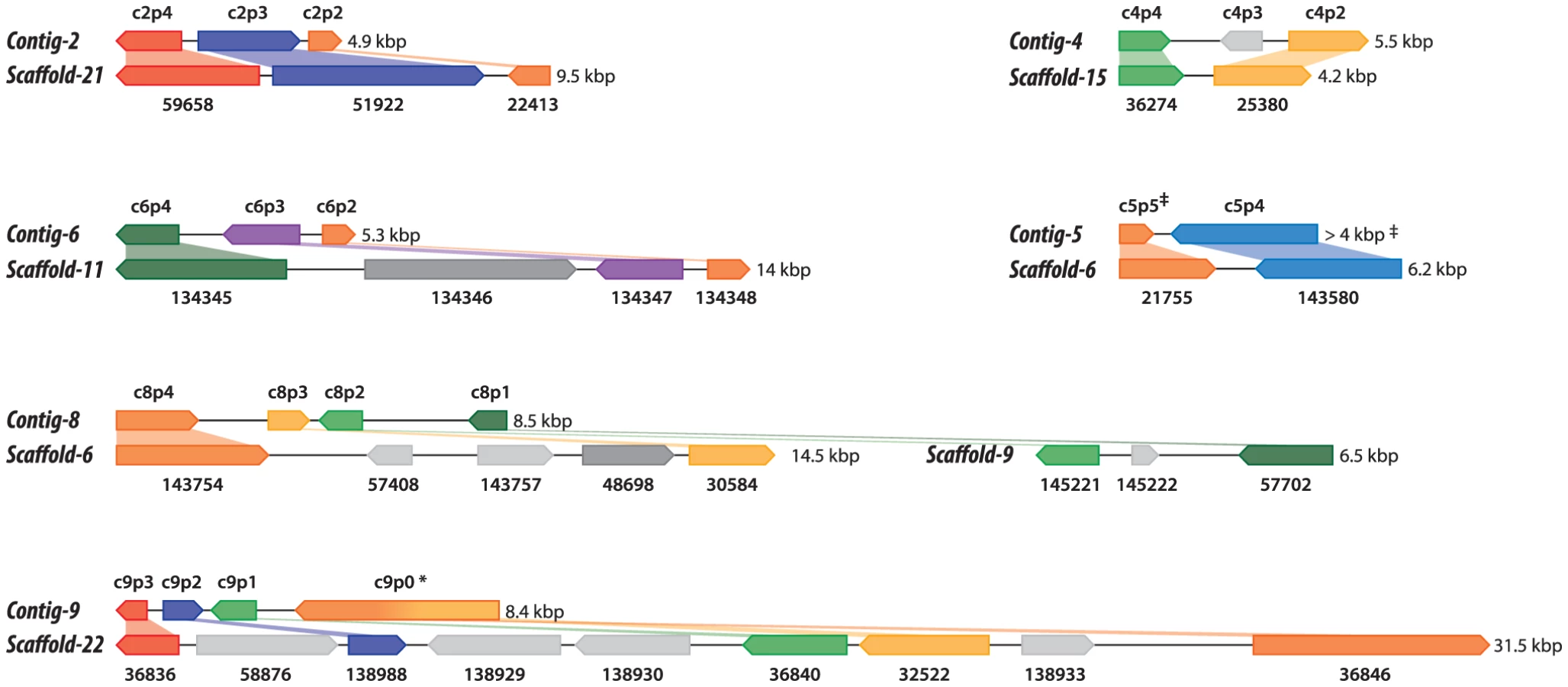
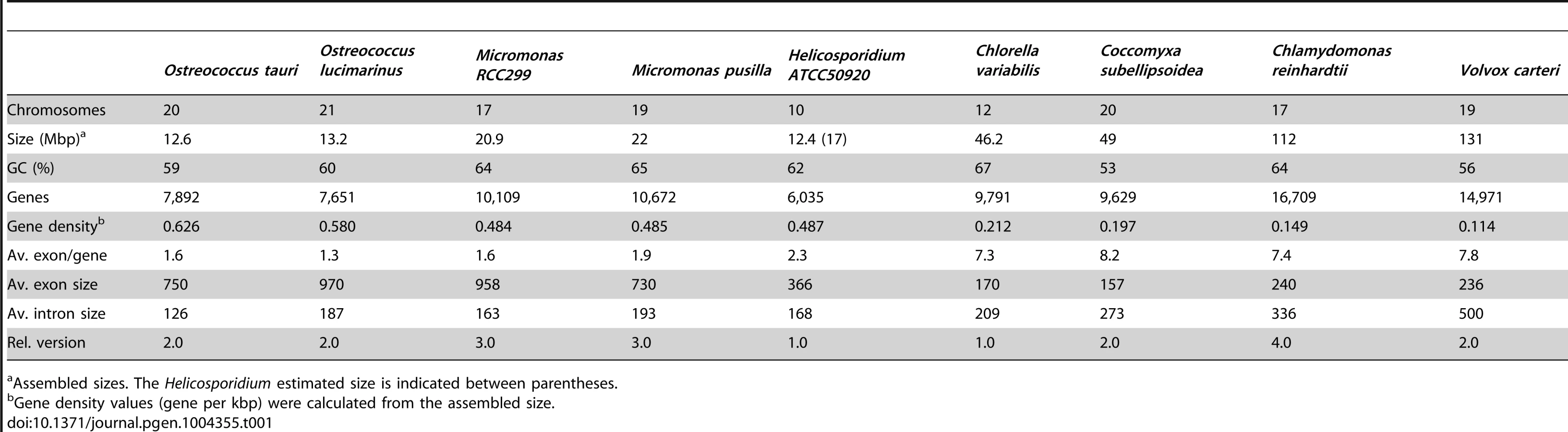
Shotgun Illumina reads of total DNA were assembled into 11,717 contigs totalling 13,684,556 bp (62.2% GC). Contamination filters suggest a maximum of 1% overall contamination, located in the smaller-sized contigs. Removal of the mitochondrial and plastid genomes and filtering of the small contigs resulted in 5,666 contigs of at least 500 bp in size (12,373,820 bp total; N50 3,036 bp, 61.7% GC), with an average coverage of 62× (Table 1). Based on the current data, we estimate the Helicosporidium genome at a maximum size of 17±0.5 Mbp. This corresponds well to a genome size estimate of 13 Mbp, derived from karyotype visualisation by clamped homogeneous electrical field (CHEF) electrophoresis [18]. A total of 6,035 protein-encoding genes were predicted among the assembled 12.4 Mbp, with an average of 2.3 exons (366 bp/exon) and 1.3 introns (168 bp/intron) per gene. Coding density in the Helicosporidium genome is high (0.487 gene/kb) compared to the free-living and symbiotic trebouxiophytes Coccomyxa (0.197 gene/kb) and Chlorella (0.212 gene/kb), but lower than that of the 12 Mbp genomes of the picoplanktonic prasinophycean green algae in the genus Ostreococcus (0.626 and 0.580 gene/kb; Table 1, Figure 1). Identifiable transposable elements are rare in the assembled Helicosporidium contigs, although micro- and minisatellites and regions of generally low complexity were found (Data S1). Comparing the genomic assemblies with the transcriptome revealed a total of 95.4% of the genes attributed to known metabolic pathways (Table S1) were found in both datasets, and only 3.6% were found exclusively in the transcriptome. This correlated well with the overall percentages of transcriptomic contigs mapping on the genomic ones (≥1000 bp; 92.3%, ≥1500 bp; 95.9%), suggesting that the total coding potential of Helicosporidium is well-represented in the draft genome. The Helicosporidium genome shares little gene order conservation with the other green algal genomes: only 30% of the genes located within its ten largest contigs are arrayed in syntenic clusters with those of Chlorella (Figure 2), with no apparent metabolic relationship between the genes present in these clusters.

A surprisingly near-complete metabolic profile
The Helicosporidium genome is small: it is approximately 2.5 times smaller than the two other complete trebouxiophyte genomes, Coccomyxa and Chlorella (Table 1, Figure 1), which are themselves at the extremely low end of the spectrum of estimated genome sizes in this lineage (Figure S1). But this small size is not a reflection of a severe reduction in metabolic potential. Indeed, the Helicosporidium genome encodes almost all of the major biological functions that are shared between the genomes of its trebouxiophycean relatives and that of the chlorophycean green alga Chlamydomonas reinhardtii (Table S1).
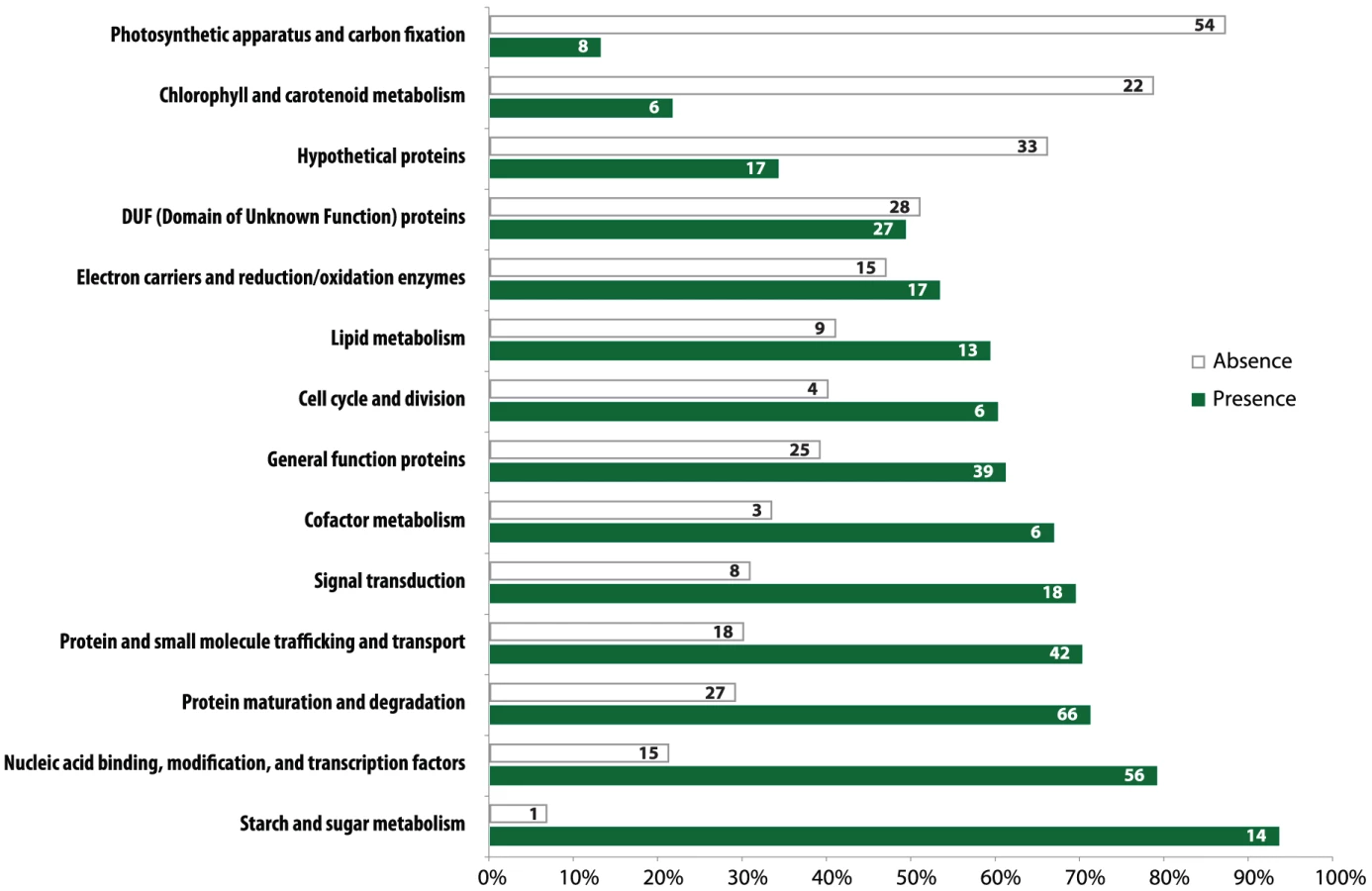
Gene loss in the Helicosporidium genome is significantly concentrated in photosynthesis-related pathways, and even here gene loss is surprisingly sparse given its non-photosynthetic, parasitic nature. The Helicosporidium genome encodes 56% of the plastid-targeted proteins predicted by the GreenCut2 database [19] (Figure 3), whereas both the photosynthetic Coccomyxa and Chlorella encode 96% of these proteins. The overall distribution of these losses in plastid metabolism is not random, but is concentrated on processes related to light-harvesting (Figure 4, Data S3, S4, S5, S6, S7, S8, S9, S10, S11). The heme synthesis branch of the tetrapyrrole pathway is complete in Helicosporidium, but the branch leading to the biogenesis of chlorophyll has been lost (Figures 4 and S2, Data S3, S4, S5, S6, S7, S8, S9, S10, S11). Similarly, Helicosporidium cannot synthesize carotenoids. It does not encode light-harvesting antenna proteins and photosystems I and II are completely absent, which parallels the loss of all photosynthesis-related genes in its plastid genome [20]. Surprisingly however, the Helicosporidium genome has retained an almost complete carbon fixation pathway despite lacking two major components rbcL/rbcS coding respectively for the large and small subunits of the ribulose-1,5-bisphosphate carboxylase oxygenase (RuBisCO) and ppdK, a pyruvate orthophosphate dikinase involved in pyruvate interconversions in the C4 pathway (Figure 4). Similarly, Helicosporidium has retained some proteins involved in electron transport and components of the F-type ATPase and cytochrome b6f (Figure 4). Starch and fatty acid metabolic pathways are more or less intact, as is the terpenoid biosynthesis pathway and its isoprenoid non-mevalonate MEP/DOXP synthesis branch. The SUF iron-sulfur cluster biosynthetic pathway is conserved as well, alongside its ISC/NIF mitochondrial counterpart [21], [22]. Not surprisingly the protein import and export systems are intact.
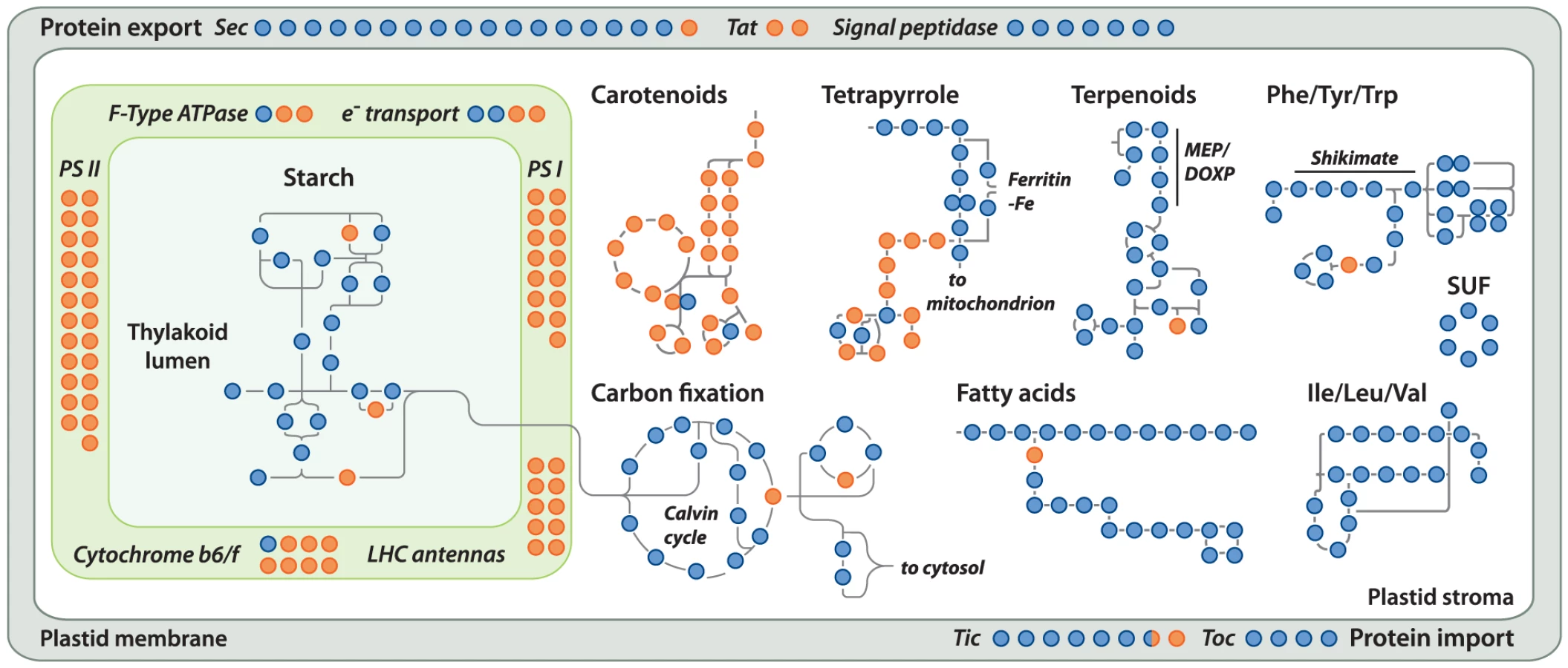
Outside the plastid, Helicosporidium metabolism shows little signs of significant reduction. The Helicosporidium genome encodes all proteins required for the biosynthesis of conventional aminoacyl-tRNAs, except selenocysteine. Despite its conservation across the green algae, we found no evidence for the presence of a selenocysteine synthase in the genomic and transcriptomic Helicosporidium datasets. The o-phosphoseryl-tRNA(sec) kinase also required for selenocystenyl-tRNA synthesis is missing too, however all other enzymes involved in the metabolism of selenocompounds are present. Helicosporidium appears incapable of endogenous RNA interference and, like the picoeukaryotes Ostreococcus tauri and Ostreococcus lucimarinus, lacks the genes coding for the Dicer and Argonaute proteins. These genes are found in single copies in the Chlorella and Coccomyxa genomes whereas three paralogous copies of DC1 and AGO1 are found in the Chlamydomonas genome [23]. In Chlamydomonas, this expanded set has been postulated to mediate the silencing of its numerous transposable elements [24]. The few losses observed in the remaining Helicosporidium pathways are either palliated by bypass enzymes or affect the synthesis or homeostasis of uncommon metabolites (Table S2).
The increased level of compaction and loss of photosynthetic genes in the Helicosporidium genome cannot explain its 2.5-fold reduction in genome size: other significant differences in gene content exist between the parasite and its free-living relatives. Given that Helicosporidium, Coccomyxa, and Chlorella were found to encode almost the same overall functional categories of genes in common with other green algae (Table S1), one possibility is that Helicosporidium possess fewer and/or smaller gene families. To investigate the complexity of gene families, we compared the Helicosporidium, Chlorella, and Coccomyxa predicted proteomes via an evolutionary gene network analysis [25]. A total of 100 connected components, excluding photosynthesis-related products, were found to exhibit a lower representation in Helicosporidium compared with its free-living relatives. These were manually curated into 9 functional categories based on annotation of the three gene sets (Tables S3 and S4, Figure 5). Interestingly, the functional categories where Helicosporidium has a reduced gene family complexity are for the most part not what are broadly defined as ‘operational’ genes where reduction that might be related to increased dependence on the host. Instead, the most drastic reductions in Helicosporidium are gene families in functional groups that are correlated with the size and complexity of the genome. The few exceptions are in amino acid and some other metabolic families, but most of the reduction relates to genes involved in chromosome packing, transcription, translation, post-translational modification, and protein turnover. Most surprisingly, Helicosporidium has not seen an increase in the complexity of transporter families, which might be expected of a parasite as host dependence grows; instead, this functional class is most reduced in Helicosporidium compared with Coccomyxa and Chlorella.

Gene family expansions: Chitinases
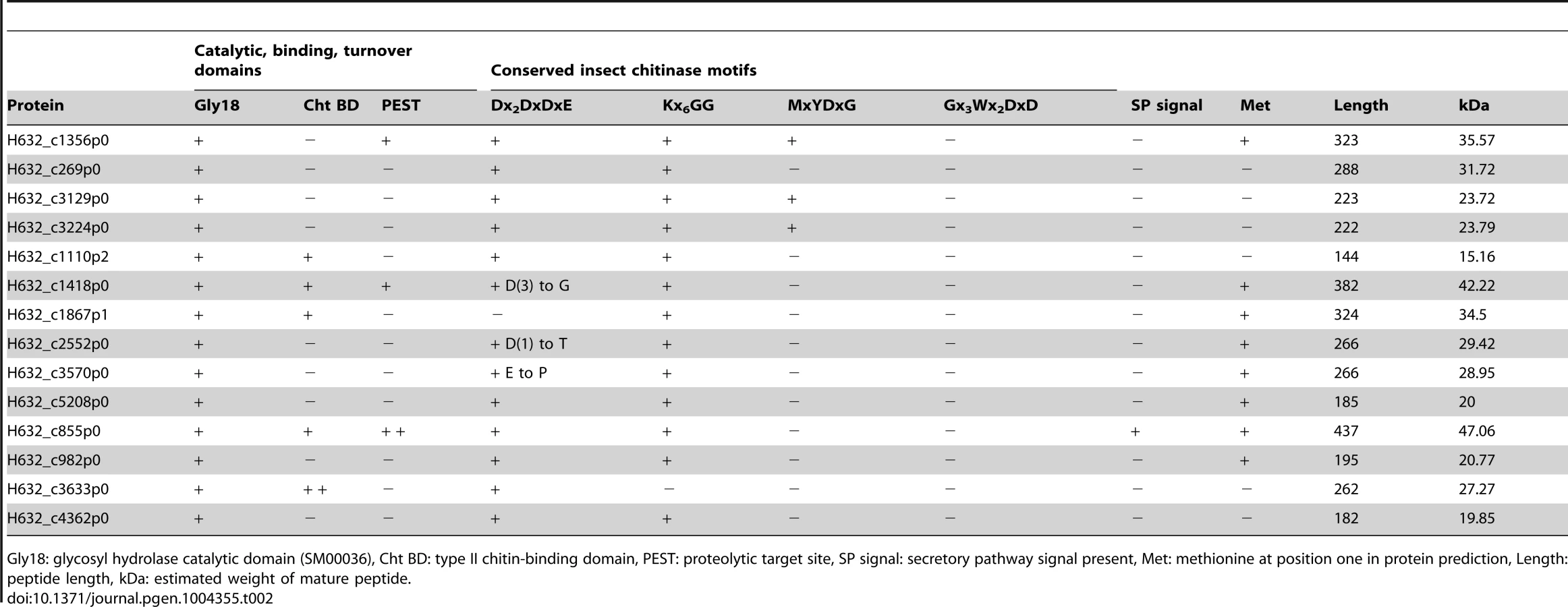
Taking an opposing view, to what gene families may have expanded due to the adaptation to parasitism, revealed a single obvious expansion of functional significance. A total of 14 genes with putative glycosyl hydrolase (GH) activity were identified throughout the Helicosporidium genome and were also found within its transcriptome (Table 2). All of these proteins appear to belong to the GH18 chitinase family, whereas plant chitinases normally come from the GH19 family. The Chlorella genome encodes two GH18 and one GH19 chitinase, and these are assumed to be involved in the remodelling of its cell wall, which has been experimentally demonstrated to contain chitin [17], [26]. The GH19 chitinase in Chlorella was acquired by horizontal gene transfer from a large DNA virus [17] and is not found in either the Helicosporidium or Coccomyxa genomes. Conversely, the Chlorella GH18 chitinases are found across the green algae. In Helicosporidium, the extra 12 copies appear to have been generated by recent duplication events. We found no evidence for the transfer of genetic material from insects in the Helicosporidium genome or transcriptome. A total of 13 of the 14 Helicosporidium chitinases contain the Dx2DxDxE/P motif essential for chitinolytic activity. Insect and bacterial chitinases with experimentally confirmed catalytic activity also contain one or more of three additional motifs: Kx6GG, MxYDx(x)G, and Gx3Wx2DxD [27]–[29]. Thirteen of the chitinases in Helicosporidium contain one or two of these additional motifs and demonstrate high conservation in their orientation (Table 2, Figure S3). However, in addition to H632_c1867p1 which appears to lack this domain, three show substitutions within the Dx2DxDxE motif and may be therefore be inactive.
Another class of proteins that might be expected to be relevant to the origin of parasitism in Helicosporidia are, unfortunately, the unidentified or ‘unique’ ORFs. To see whether these represented a high proportion of the genes in Helicosporidium, we identified predicted proteins of at least 100 amino acids that are not found in Coccomyxa and Chlorella, which resulted in 882 distinct proteins (Data S12). A small number of these proteins have clear or putative homologs in the Volvox and/or Chlamydomonas genomes but the vast majority are unique and have no known homologs. The few cases with homologues in Volvox and Chlamydomonas are sulfotransferases, glycosyltransferases or hydrolases with chitinase activity (as mentioned above), a 2-oxoglutarate Fe(II)-dependent oxygenase, and a cyclin. In contrast, the predicted proteins without known green algal homologs could not be assigned to any putative function in PFAM homology searches at an E-value cut-off of 1e-10. Five of these unknown proteins (H632_c233p3, H632_c338p0, H632_c531p0, H632_c1976p1, H632_c4072p0) display mid to low similarity with bacterial sequences of unknown function, but they are unambiguously encoded on contigs encoding clearly eukaryotic genes, and are therefore not bacterial contaminants. From the transcriptome, we can also conclude that the majority of these proteins are expressed, with 585 of the 882 found in transcriptome data at an E-value threshold of 1e-15.
Horizontal gene transfer from/to viruses
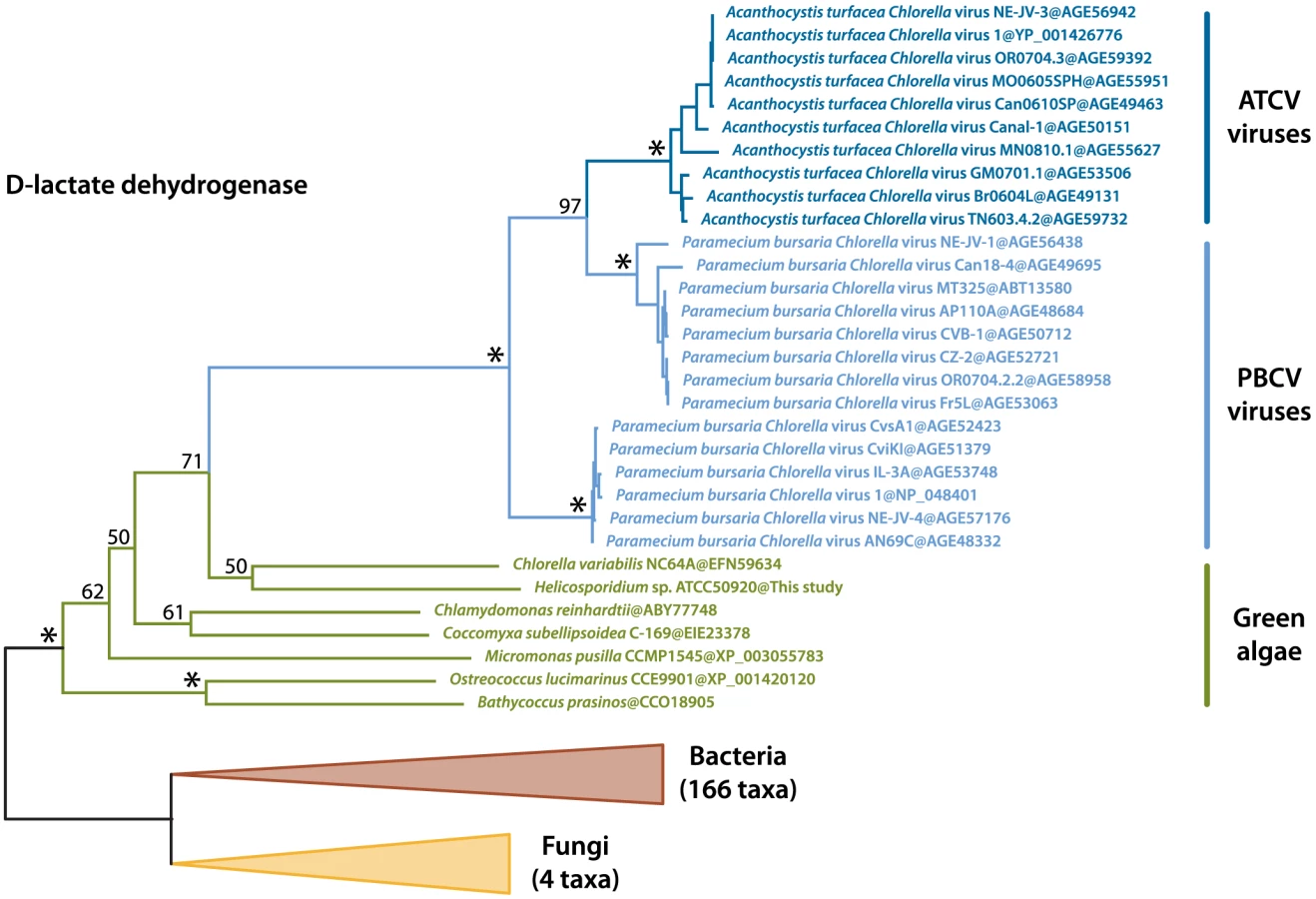
We identified two transcripts sharing a high identity with viral sequences (E-value threshold of 1e-40), the closest relatives being Paramecium bursaria Chlorella viruses (PBCV) and Acanthocystis turfacea Chlorella viruses (ATCV). These two transcripts were also identified in the Helicosporidium genomic contigs, and both are assembled with Helicosporidium nuclear genes and are therefore not the result of viral contamination. The first transcript (a374428r16) codes for a dUDP-D-glucose 4,6 dehydratase that is also found in the genomes of Chlorella, Coccomyxa and various other green algae, and has been reported as an example of host to virus horizontal gene transfer (HGT) [30], [31]. The second transcript (a28443r121) encodes a D-lactate-dehydrogenase and is also found across the green lineage. Phylogenetic analyses including the closest D-lactate-dehydrogenase sequences clusters the green algal sequences with homologues from nucleocytoplasmic large DNA viruses that infect them, with strong bootstrap support (Figure 6). Overall, this suggests D-lactate-dehydrogenase represents another case of host-virus HGT in the green algal lineage.
Discussion
A lack of loss in the evolution of parasitism
A recurring theme in the evolution of parasite genomes and parasitism in general is reduction. In parallel with the more constructive process of developing sometimes sophisticated mechanisms to invade and take advantage of their hosts, parasite evolution generally involves the selective pruning of biological functions that are no-longer mandatory to survival in the host. This reduction has occurred independently many times during the evolution of parasites, and is generally reflected in their genomes, streamlined sometimes solely by a loss of genes, but other times also by an overall shortening of coding and intergenic regions as well as a loss of introns, resulting in much smaller and compact genomes than those of their free-living relatives [9]. In very few cases have photosynthetic algae made this transition, but the famous exception is the apicomplexans, including the malaria parasite Plasmodium. Here the same process has taken place and although the plastid has been retained, it has been reduced to a cryptic form that lacks most of its ancestral metabolic pathways and has retained nothing whatsoever related to carbon metabolism.
Helicosporidium breaks from these trends in significant ways. With an almost perfect conservation of the core green algal metabolic pathways, its genome is small, but can hardly be considered reduced, which may reflect its relatively recent adaptation to parasitism. Particularly surprising is the retention of an almost complete pathway for carbon fixation. Helicosporidium has lost nearly all genes associated with light harvesting, photosystems, and chlorophyll biogenesis, so how it uses carbon fixation pathways is an interesting question. Carbohydrate storage, and in particular the use of starch could be the main driver behind the retention of carbon fixation genes. Successful parasites often sequester resources from their host [32] and converting simple sugar molecules to large starchy polymers polarizes the directionality of carbohydrate exchanges. Exactly how efficient are the Helicosporidium permeases at sugar uptake and other energy-related metabolites from the immediate environment is unclear; considering that this is one of the gene families of reduced complexity, but such sequestration may be a prerequisite for survival.
The coding density of the Helicosporidium genome is only marginally higher than its closest relatives, and almost on par with those of a number of free-living prasinophytes (Table 1; Figure 1). Green algal genome sizes range widely across lineages (Figure S1), and expansions as well as contractions are likely to have occurred several times independently. Accordingly, we cannot currently distinguish between a reduction of DNA packaging/replication/translation pathways in Helicosporidium, an expansion of these pathways in Chlorella/Coccomyxa, or a combination of both. A better phylogenetic framework as well a greater sampling depth of green algal nuclear genomes will be required to polarize the directionality of this event, but the reduced gene family complexity observed in picoprasinophytes [33]–[35] does argue in favour of an expansion from a lean ancestral state.
Despite its small size, the Helicosporidium genome does feature one small expansion. Chitinases are uniformly rare in green algae where genomic data are available, suggesting this gene family is ancestrally limited in the green algae (which fits with its likely function in remodelling a minor component of the cell wall in some species). The expansion of chitinases in Helicosporidium over its sister taxa Chlorella and Coccomyxa therefore likely represents a unique adaptation that directly resulted from or even contributed to its parasitic lifestyle in insect hosts. The Helicosporidium infection mechanism consists of the oral uptake of cysts, dehiscence in the midgut lumen, ingression through the midgut, and entry into the hemocoel where the organism multiplies. In a number of insect parasites, including the malarial parasite Plasmodium, the presence of exogenous chitinases from the GH18 family within the arthropod midgut tract is either mandatory for infection or associated with increased pathogenicity by allowing pathogens to pass through the peritrophic membrane [36], [37]. Disruption of chitinase activity with blocking agents interferes with the sporogonic development of malarial parasites, which is restored upon addition of exogenous chitinases [38]. One may speculate that the Helicosporidium chitinases serve multifunctional roles and operate at various stages in vivo. Potentially, chitinases sequestered in the cysts are activated by insect digestive proteases, and then digest the cyst wall to initiate the release of the invasive filament cell in the gut lumen. Alternatively, the chitinases released by the ovoid cells, may loosen the chitin matrix of the peritrophic membrane, allowing the ingress of the filamentous cell into the ectoperitrophic space. It is important to note that members of the GH18, in addition to binding to and digesting the chitin can also target various GlcNAc-containing glycans that comprise various exocellular matrices including insect basement membranes. Binding to such substrates may aid in the establishment of infection on the hemocoel associated tissues. Finally the production of these enzymes could soften the exoskeleton, which could play a role in the egress of the infectious cyst stage from diseased insects.
Materials and Methods
Tissue culture and DNA/RNA purification
Vegetative cells of the Helicosporidium sp. (ATCC50920) a parasite of the black fly Simulium jonesi [1], [15] was propagated in stationary cultures of sabouraud dextrose broth for five days at 27°C. Cells were harvested by centrifugation (5,000 rcf for 10 min) and pellets resuspended in a minimal volume sterile H2O and used for nucleic acid extraction. For genomic DNA preparation, a total of 2×109 cells were suspended in the yeast lysis buffer (Epicentre Biotechnologies, Madison WI) and homogenized with Bead Beater technology. The extracted nucleic acid phase was treated with DNase free RNase and subjected to chloroform phenol extraction, precipitated with ethanol, and suspended in TE buffer. A total of 14.1 µg high molecular weight DNA were recovered and submitted for sequencing. For total RNA extraction the resuspended harvested cells were immediately added to liquid N2 and ground with mortar and pestle to break the outer pellicles. The resulting frozen cell powders were processed initially with TRizol Reagent then processed with Purelink RNA Mini kit (Ambion). Column eluants were treated with RNase-free DNase and analysed with the 2100 Bioanalyzer (Agilent Technologies, Inc). Samples (10 µg) producing RIN values of 8.9 to 9.2 were selected for subsequent sequence analysis (see below).
DNA and RNA sequencing
Total DNA and RNA from Helicosporidium sp. ATCC 50920 (mitochondrial, plastid and nuclear) were sequenced by Fasteris SA (Plan-les-Ouates, Switzerland) using the Illumina platform. Two independent DNA sequencing runs were performed. In the first, 18,618,066 reads (54-bp paired ends; 323-bp inserts; average standard deviation, 19) totaling 1,005,375,564 bases were sequenced with the Illumina GA-IIx platform and the Chrysalis 36 cycles v 4.0 sequencing kit. In the second, 17,110,904 reads (51-bp paired ends; 241-bp inserts; average standard deviation, 64) totaling 872,656,104 bases were sequenced with the Illumina HiSeq 2000 platform and the TruSeq chemistry. Total RNA was sequenced using the Illumina directional mRNA-SEQ protocol with the Illumina HiSeq 2000 platform and the TruSeq chemistry. A total of 83,075,963 reads (100-bp single ends) were generated (8,307,596,300 bases total). Read quality for each Illumina data set was assessed with FastQC (version 0.10.1; Babraham Bioinformatics, Babraham Institute [http://www.bioinformatics.babraham.ac.uk]).
Genome assembly
Paired-end reads were assembled de novo with Ray [39] 2.0.0 rc8 using iterative k-mer values of 21 to 31 on 8 processing cores (2 Intel Xeon E5506 CPUs at 2.13 GHz) with a maximum RAM allowance of 96 Gb. The resulting contigs were filtered by size with sort_contigs.pl (Advanced Center for Genome Technology, University of Oklahoma [www.genome.ou.edu/informatics.html]), and contigs shorter than 500 bp were discarded. The contigs of at least 500 bp were conserved for downstream analyses. The 500+-bp contigs were used as canvas to generate a BLAST [40] database with MAKEBLASTDB from the NCBI BLAST 2.2.26 package, the mitochondrial and plastid contigs were identified by BLAST homology searches using the mitochondrial (GenBank accession number NC_017841, [41]) and plastid (GenBank accession number NC_008100, [20]) genomes as queries, and separated from the nuclear contigs. Putative contaminants were assessed by homology searches against the NCBI non-redundant database.
Transcriptome assembly
RNA-Seq reads were filtered using a sliding-window quality approach with Sickle (Bioinformatics Core, University of California, Davis [https://github.com/najoshi/sickle]) under the default parameters, and the overall read quality reassessed after filtering with FastQC. Illumina adapter sequences were then removed from the filtered sequences using custom Perl scripts, and PolyA-tails were removed from the reads with TrimEST from the EMBOSS [42] 6.4.0 package. The filtered transcriptome reads were assembled with Trinity's Inchworm module with a maximum RAM allowance of 90 Gb (–JM 90G) on 8 processing cores (2 Intel Xeon E5506 CPUs at 2.13 GHz). Contigs were filtered by size with sort_contigs.pl and contigs of at least 250 bp were selected for downstream analyses. Transcriptomic contigs were mapped on the genomic ones with GMAP version 2014-01-21 [43] using the default parameters.
Genome annotation
The nuclear contigs of at least 500-bp in length were sorted by size and renumbered incrementally using customs Perl scripts. Contigs were then processed with the Maker 2.11 annotation gauntlet [44], [45] using the Chlorella gene model as implemented in Augustus 2.5.5 [46]. The resulting GFF annotations files were processed, curated, and converted to GenBank annotations files using custom Perl scripts. Putative functions were assigned using homology searches against the PFAM database (E-value threshold of 1E-30; Table S5). Transposable elements were searched for with RepeatMasker [http://repeatmasker.org] using Repbase version 20130422 [47].
Genome size estimation
Illumina reads from the mitochondrial and plastid genome were first filtered out from the total dataset with bowtie 0.12.9 [48] using –un and –al the flags against indexes built from the organelle sequences. Filtered nuclear reads were then mapped with bowtie against the 5,666 contigs (≥500 bp) with the –S flag, and the coverage estimated from the SAM file with Tablet 1.12.12.05 [49] and the coveragestat.py python script. The genome size was then estimated using the following formula: [# of reads X read length]/coverage.
Pathways mapping and network analyses
KEGG metabolic pathway maps for the green algae Chlamydomonas reinhardtii, Volvox carteri, Ostreococcus tauri and Ostreococcus lucimarinus were retrieved from the KEGG pathway databases [50], [51], the proteins sorted accordingly, and then used as queries for homology searches against the Helicosporidium, Chlorella and Coccomyxa proteomic, genomic and transcriptomic datasets (the Chlorella and Coccomyxa data was retrieved from the JGI website). BLASTP and TBLASTN searches were performed using E-value thresholds of 1E-10 and 1E-05, respectively. Genes not found in searches against any of the three datasets were considered absent from the corresponding organism. Network analyses were performed according to [25]. Specifically, all possible edges were drawn between pairs of genes if their reciprocal BLASTP comparisons to one another met all of the following conditions: E-value<1E-10, minimal hit identity >20, at least 20% of the shortest gene's length had identical residues in the match, and the hit length >20 amino acids. The network was then filtered to include underrepresented Helicosporidium genes compared to Coccomyxa and Chlorella. Functional annotations for the genes comprising each connected component (GenBank, KOG, KEGG, Interpro [52], and Pfam) were used to characterize each connected component by its inferred biological function. Plastid-targeted proteins from GreenCut2's Table S2 [19] were extracted from the corresponding Chlamydomonas reinhardtii (version 3.1) and Arabidopsis thaliana (http://www.arabidopsis.org/) protein catalogs and converted to custom BLAST databases with MAKEBLASTDB from the NCBI BLAST package. Helicosporidium, Chlorella and Coccomyxa were searched independently against both GreenCut2 databases with BLASTP (proteins) and TBLASTN (genome and transcriptome) using E-value thresholds of 1E-10 and 1E-05, respectively.
Chitinase identification
Putative glycosyl hydrolases identified in the Helicosporidium genome were annotated for catalytic and chitin binding domains using SMART 7 [53] and endo-proteolytic sites often located within developmental insect chitinases were identified with ePESTfind [54]. The glycosyl hydrolase catalytic domains were annotated manually for the presence and orientation of key amino acid motifs. Secretory signal motifs were searched for with TargetP 1.1 [55] and PredAlgo [56].
Phylogenetic analyses
Amino acid sequences retrieved from GenBank were aligned with the L-INS-I algorithm from MAFFT 7.029b [57]. Phylogenetic models were selected with ProtTest 3.2 [58]. Maximum Likelihood phylogenetic reconstructions were performed with PHYML 3.0 [59] under the LG+Γ4+I model of amino acid substitution [60].
Data deposition
The Helicosporidium data was deposited at DDBJ/EMBL/GenBank under NCBI BioProject ID PRJNA188927 and accession AYPS00000000. The version described in this paper is version AYPS01000000. The predicted proteins and RNAs are also available in Data S13 and S14, respectively. All custom Perl scripts are available on GitHub (https://github.com/JFP-Laboratory).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BouciasDG, BecnelJJ, WhiteSE, BottM (2001) In vivo and in vitro development of the protist Helicosporidium sp. J Eukaryot Microbiol 48: 460–470.
2. TartarA (2013) The non-photosynthetic algae Helicosporidium spp.: Emergence of a novel group of insect pathogens. Insects 4: 375–391.
3. BläskeV-U, BouciasDG (2004) Influence of Helicosporidium spp. (Chlorophyta: Trebouxiophyceae) infection on the development and survival of three noctuid species. Environ Entomol 33: 54–61.
4. ConklinT, BläskeV-U, BecnelJJ, BouciasDG (2005) Infectivity of two isolates of Helicosporidium spp. (Chlorophyta: Trebouxiophyceae) in heterologous host insects. Florida Entomol 88: 431–440.
5. Bläske-LietzeV-U, BouciasDG (2005) Pathogenesis of Helicosporidium sp. (Chlorophyta: Trebouxiophyceae) in susceptible noctuid larvae. J Invertebr Pathol 90: 161–168.
6. Bläske-LietzeV-U, ShapiroAM, DentonJS, BottsM, BecnelJJ, et al. (2006) Development of the insect pathogenic alga Helicosporidium. J Eukaryot Microbiol 53: 165–176.
7. TartarA, BouciasDG, AdamsBJ, BecnelJJ (2002) Phylogenetic analysis identifies the invertebrate pathogen Helicosporidium sp. as a green alga (Chlorophyta). Int J Syst Evol Microbiol 52: 273–279.
8. TartarA, BouciasDG, BecnelJJ, AdamsBJ (2003) Comparison of plastid 16S rRNA (rrn16) genes from Helicosporidium spp.: evidence supporting the reclassification of Helicosporidia as green algae (Chlorophyta). Int J Syst Evol Microbiol 53: 1719–1723.
9. KeelingPJ, SlamovitsCH (2005) Causes and effects of nuclear genome reduction. Curr Opin Genet Dev 15: 601–608.
10. BlouinNA, LaneCE (2012) Red algal parasites: models for a life history evolution that leaves photosynthesis behind again and again. Bioessays 34: 226–235.
11. JanouškovecJ, HorákA, OborníkM, LukešJ, KeelingPJ (2010) A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc Natl Acad Sci USA 107: 10949–10954.
12. EscalanteAA, AyalaFJ (1995) Evolutionary origin of Plasmodium and other Apicomplexa based on rRNA genes. Proc Natl Acad Sci U S A 92: 5793–5797.
13. LeliaertF, SmithDR, MoreauH, HerronMD, VerbruggenH, et al. (2012) Phylogeny and molecular evolution of the green algae. CRC Crit Rev Plant Sci 31: 1–46.
14. De WeverA, LeliaertF, VerleyenE, VanormelingenP, Van der GuchtK, et al. (2009) Hidden levels of phylodiversity in Antarctic green algae: further evidence for the existence of glacial refugia. Proc Biol Sci 276: 3591–3599.
15. TartarA, BouciasDG (2004) The non-photosynthetic, pathogenic green alga Helicosporidium sp. has retained a modified, functional plastid genome. FEMS Microbiol Lett 233: 153–157.
16. BlancG, AgarkovaI, GrimwoodJ, KuoA, BrueggemanA, et al. (2012) The genome of the polar eukaryotic microalga Coccomyxa subellipsoidea reveals traits of cold adaptation. Genome Biol 13: R39.
17. BlancG, DuncanG, AgarkovaI, BorodovskyM, GurnonJ, et al. (2010) The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell 22: 2943–2955.
18. Tartar A (2004) Incertae sedis no more: The phylogenetic affinity of Helicosporidia University of Florida.
19. KarpowiczSJ, ProchnikSE, GrossmanAR, MerchantSS (2011) The GreenCut2 resource, a phylogenomically derived inventory of proteins specific to the plant lineage. J Biol Chem 286: 21427–21439.
20. De KoningAP, KeelingPJ (2006) The complete plastid genome sequence of the parasitic green alga Helicosporidium sp. is highly reduced and structured. BMC Biol 4: 12.
21. GodmanJ, BalkJ (2008) Genome analysis of Chlamydomonas reinhardtii reveals the existence of multiple, compartmentalized iron-sulfur protein assembly machineries of different evolutionary origins. Genetics 179: 59–68.
22. Dellibovi-RaghebTa, GisselbergJE, PriggeST (2013) Parasites FeS up: iron-sulfur cluster biogenesis in eukaryotic pathogens. PLoS Pathog 9: e1003227.
23. CeruttiH, MaX, MsanneJ, RepasT (2011) RNA-mediated silencing in Algae: biological roles and tools for analysis of gene function. Eukaryot Cell 10: 1164–1172.
24. Casas-MollanoJA, RohrJ, KimE-J, BalassaE, van DijkK, et al. (2008) Diversification of the core RNA interference machinery in Chlamydomonas reinhardtii and the role of DCL1 in transposon silencing. Genetics 179: 69–81.
25. HalaryS, McInerneyJO, LopezP, BaptesteE (2013) EGN: a wizard for construction of gene and genome similarity networks. BMC Evol Biol 13: 146.
26. KapaunE, ReisserW (1995) A chitin-like glycan in the cell wall of a Chlorella sp. (Chlorococcales, Chlorophyceae). 197: 577–582.
27. ZhuQ, ArakaneY, BeemanRW, KramerKJ, MuthukrishnanS (2008) Characterization of recombinant chitinase-like proteins of Drosophila melanogaster and Tribolium castaneum. Insect Biochem Mol Biol 38: 467–477.
28. KhajuriaC, BuschmanLL, ChenM-S, MuthukrishnanS, ZhuKY (2010) A gut-specific chitinase gene essential for regulation of chitin content of peritrophic matrix and growth of Ostrinia nubilalis larvae. Insect Biochem Mol Biol 40: 621–629.
29. WatanabeT, KoboriK, MiyashitaK, FujiinT, SakaiH, et al. (1993) Identification of glutamic acid 204 and aspartic acid 200 in chitinase A1 of Bacillus circulans WL-12 as essential residues for chitinase activity. J Biol Chem 268: 18567–18572.
30. Parakkottil ChothiM, DuncanGA, ArmirottiA, AbergelC, GurnonJR, et al. (2010) Identification of an L-rhamnose synthetic pathway in two nucleocytoplasmic large DNA viruses. J Virol 84: 8829–8838.
31. JeanniardA, DuniganDD, GurnonJR, Agarkova IV, KangM, et al. (2013) Towards defining the chloroviruses: a genomic journey through a genus of large DNA viruses. BMC Genomics 14: 158.
32. PombertJ-F, SelmanM, BurkiF, BardellFT, FarinelliL, et al. (2012) Gain and loss of multiple functionally related, horizontally transferred genes in the reduced genomes of two microsporidian parasites. Proc Natl Acad Sci U S A 109: 12638–12643.
33. DerelleE, FerrazC, RombautsS, RouzéP, WordenAZ, et al. (2006) Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci USA 103: 11647–11652.
34. WordenAZ, LeeJ, MockT, RouzéP, SimmonsMP, et al. (2009) Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science (80-) 324: 268–272.
35. PalenikB, GrimwoodJ, AertsA, RouzéP, SalamovA, et al. (2007) The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc Natl Acad Sci U S A 104: 7705–7710.
36. ShenZ, Jacobs-LorenaM (1997) Characterization of a novel gut-specific chitinase gene from the human malaria vector Anopheles gambiae. J Biol Chem 272: 28895–28900.
37. VinetzJM, ValenzuelaJG, SpechtCA, AravindL, LangerRC, et al. (2000) Chitinases of the avian malaria parasite Plasmodium gallinaceum, a class of enzymes necessary for parasite invasion of the mosquito midgut. J Biol Chem 275: 10331–10341.
38. ShahabuddinM, ToyoshimaT, AikawaM, KaslowDC (1993) Transmission-blocking activity of a chitinase inhibitor and activation of malarial parasite chitinase by mosquito protease. Proc Natl Acad Sci U S A 90: 4266–4270.
39. BoisvertS, LavioletteF, CorbeilJ (2010) Ray: Simultaneous assembly of reads from a mix of high-throughput sequencing technologies. J Comput Biol 17: 1519–1533.
40. AltschulSF, GishW, MillerW, MyersEW, LipmanDJ (1990) Basic Local Alignment Search Tool. J Mol Biol 215: 403–410.
41. PombertJ-F, KeelingPJ (2010) The mitochondrial genome of the entomoparasitic green alga Helicosporidium. PLoS One 5: e8954.
42. RiceP, LongdenI, BleasbyA (2000) EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet 16: 276–277.
43. WuTD, WatanabeCK (2005) GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 21: 1859–1875.
44. CantarelBL, KorfI, RobbSMC, ParraG, RossE, et al. (2008) MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18: 188–196.
45. HoltC, YandellM (2011) MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12: 491.
46. StankeM, DiekhansM, BaertschR, HausslerD (2008) Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24: 637–644.
47. JurkaJ, KapitonovVV, PavlicekA, KlonowskiP, KohanyO, et al. (2005) Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467.
48. LangmeadB, TrapnellC, PopM, SalzbergSL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25.
49. MilneI, BayerM, CardleL, ShawP, StephenG, et al. (2010) Tablet - next generation sequence assembly visualization. Bioinformatics 26: 401–402.
50. KanehisaM, GotoS, FurumichiM, TanabeM, HirakawaM (2010) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38: D355–360.
51. KanehisaM, GotoS, SatoY, FurumichiM, TanabeM (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40: D109–114.
52. QuevillonE, SilventoinenV, PillaiS, HarteN, MulderN, et al. (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33: W116–W120.
53. LetunicI, DoerksT, BorkP (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40: D302–D305.
54. RechsteinerM, RogersSW (1996) PEST sequences and regulation by proteolysis. Trends Biochem Sci 21: 267–271.
55. EmanuelssonO, NielsenH, BrunakS, von HeijneG (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300: 1005–1016.
56. TardifM, AtteiaA, SpechtM, CogneG, RollandN, et al. (2012) PredAlgo: a new subcellular localization prediction tool dedicated to green algae. Mol Biol Evol 29: 3625–3639.
57. KatohK, StandleyDM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780.
58. DarribaD, TaboadaGL, DoalloR, PosadaD (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27: 1164–1165.
59. GuindonS, DufayardJ-F, LefortV, AnisimovaM, HordijkW, et al. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59: 307–321.
60. LeSQ, GascuelO (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25: 1307–1320.
61. KapraunDF (2007) Nuclear DNA content estimates in green algal lineages: Chlorophyta and Streptophyta. Ann Bot 99: 677–701.
62. KapraunDF (2005) Nuclear DNA content estimates in multicellular green, red and brown algae: phylogenetic considerations. Ann Bot 95: 7–44.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 5
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
- Šanci na úspěšný průběh těhotenství snižují nevhodné hladiny progesteronu vznikající při umělém oplodnění
Nejčtenější v tomto čísle
- PINK1-Parkin Pathway Activity Is Regulated by Degradation of PINK1 in the Mitochondrial Matrix
- Phosphorylation of a WRKY Transcription Factor by MAPKs Is Required for Pollen Development and Function in
- Null Mutation in PGAP1 Impairing Gpi-Anchor Maturation in Patients with Intellectual Disability and Encephalopathy
- p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision