
Non-redundant and Redundant Roles of Cytomegalovirus gH/gL Complexes in Host Organ Entry and Intra-tissue Spread
The role of viral glycoprotein entry complexes in viral tropism in vivo is a question central to understanding virus pathogenesis and transmission for any virus. Studies were limited by the difficulty in distinguishing between viral entry into first-hit target cells and subsequent cell-to-cell spread within tissues. Employing the murine cytomegalovirus entry complex gH/gL/gO as a paradigm for a generally applicable strategy to dissect these two events experimentally, we used a gO-transcomplemented ΔgO mutant for providing the complex exclusively for the initial cell entry step. In immunocompromised mice as a model for recipients of hematopoietic cell transplantation, our studies revealed an irreplaceable role for gH/gL/gO in initiating infection in host organs relevant to pathogenesis, whereas subsequent spread within tissues and infection of the salivary glands, the site relevant to virus host-to-host transmission, are double-secured by the entry complexes gH/gL/gO and gH/gL/MCK-2. As an important consequence, interventional strategies targeting only gO might be efficient in preventing organ manifestations after a primary viremia, whereas both gH/gL complexes need to be targeted for preventing intra-tissue spread of virus reactivated from latency within tissues as well as for preventing the salivary gland route of host-to-host transmission.
Published in the journal:
. PLoS Pathog 11(2): e32767. doi:10.1371/journal.ppat.1004640
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004640
Summary
The role of viral glycoprotein entry complexes in viral tropism in vivo is a question central to understanding virus pathogenesis and transmission for any virus. Studies were limited by the difficulty in distinguishing between viral entry into first-hit target cells and subsequent cell-to-cell spread within tissues. Employing the murine cytomegalovirus entry complex gH/gL/gO as a paradigm for a generally applicable strategy to dissect these two events experimentally, we used a gO-transcomplemented ΔgO mutant for providing the complex exclusively for the initial cell entry step. In immunocompromised mice as a model for recipients of hematopoietic cell transplantation, our studies revealed an irreplaceable role for gH/gL/gO in initiating infection in host organs relevant to pathogenesis, whereas subsequent spread within tissues and infection of the salivary glands, the site relevant to virus host-to-host transmission, are double-secured by the entry complexes gH/gL/gO and gH/gL/MCK-2. As an important consequence, interventional strategies targeting only gO might be efficient in preventing organ manifestations after a primary viremia, whereas both gH/gL complexes need to be targeted for preventing intra-tissue spread of virus reactivated from latency within tissues as well as for preventing the salivary gland route of host-to-host transmission.
Introduction
Herpesvirus entry is a complex process accomplished by a set of envelope glycoproteins that promote attachment of virus particles to host cells, recognition of host cell entry receptors, and fusion of the viral envelope with cellular membranes. All herpesviruses use a conserved core protein machinery consisting of glycoprotein gB and the glycoprotein complex gH/gL to promote the fusion process [1–2]. Recognition and binding to entry receptors on host cells in vitro may either be accomplished by the gH/gL core complex alone, by cooperation with other glycoproteins in the viral envelope, or by forming gH/gL complexes tightly binding additional viral proteins. Such multimeric gH/gL complexes are formed during virion assembly [1].
For Epstein-Barr virus (EBV), human herpesvirus 6, and human cytomegalovirus (HCMV) alternative multimeric gH/gL complexes that promote entry into distinct host cells have been identified [3–5]. During HCMV infection, two multimeric gH/gL complexes are formed: a pentameric gH/gL/pUL(128,130,131A) complex promoting entry into epithelial, endothelial, dendritic, and monocytic cells [6–11], and a trimeric gH/gL/gO complex promoting entry predominantly into fibroblasts ([12]; reviewed in [5]). Virus particles released from gO knock-out (ko) mutants are highly impaired on all cell types tested, whereas cell-associated focal virus spread in cell culture is not affected [13–14].
For EBV and HCMV it has been shown that host cells differentially route virus infection by influencing the gH/gL complex outfit of their virus progeny. In the case of EBV infection, replication in epithelial cells leads to production of virions rich in gH/gL/gp42 complexes targeting B cells, whereas replication in B cells mainly leads to incorporation of gp42-negative complexes into virions and thus to a virus progeny that targets epithelial cells [15]. Hence, replication in either B cells or epithelial cells induces a switch in cell type tropism. HCMV-infected cells have been shown to produce virus progeny heterogeneous in the amounts of the two gH/gL complexes and consequently in their cell type tropism. HCMV-infected fibroblasts release viruses that contain high or low amounts of gH/gL/pUL(128,130,131A) and are endotheliotropic or non-endotheliotropic, respectively [16]. Endothelial cells (EC), in contrast, release only virions that contain low amounts of gH/gL/pUL(128,130,131A) and retain those with a high gH/gL/pUL(128,130,131A) content, which renders spread of the latter cell-associated. Although host cells targeted by specific gH/gL complexes have been identified in vitro, it is not at all understood how alternative gH/gL complexes contribute to the infection in vivo. Clarification of the roles gH/gL complexes play in vivo will not only provide new insights into virus spread and host cell targeting, but will help to understand the roles of specific host cells in virus infection.
Infection of mice with murine cytomegalovirus (mCMV) is an accepted animal model for a CMV infection in its natural host and has revealed many general principles of CMV-host interaction. We have previously characterized the gH/gL/gO complex of mCMV, which in vitro is functionally homologous to the gH/gL/gO complex of HCMV [17]. Specifically, when mCMV gO is knocked-out, viral infectivity present in supernatants of infected fibroblast cultures is strongly reduced, thus driving virus dissemination in the cell monolayer towards a cell-associated pattern of focal spread. More recently, we found that mCMV forms an alternative gH/gL complex with MCK-2, the gene product of the mCMV m131–129 open reading frame (ORF). This trimeric gH/gL/MCK-2 complex facilitates infection of macrophages (MΦ) [18–19], a property also attributed to the pentameric gH/gL/pUL(128,130,131A) of HCMV in vitro [10,20]. The residual infectivity of gO-ko virus released into the supernatant of infected cells is MCK-2 dependent [18]. Besides associating with gH/gL complexes, both MCK-2 of mCMV and the UL128 protein of HCMV are also able to act as C-C chemokines attracting cells and modifying their functions [21–22]. In immunocompetent mice infected with MCK-2-ko mutants, reduced virus titers in salivary glands (SG), and reduced numbers of infected peripheral blood monocytes and tissue MΦ are observed [18,23–25]. Additionally, absence of MCK-2 is associated with a reduced recruitment of immunosuppressive inflammatory monocytes [26] and an enhanced anti-viral CD8 T-cell response [25,27]. It is currently not clear which of the observed phenotypes are due to MCK-2 functioning as a chemokine and which are due to, or modulated by, MCK-2 functioning as an entry mediator as part of the gH/gL/MCK-2 complex, nor whether these functions can be separated.
Here, we studied the in vivo host cell infection and subsequent intra-tissue spread of mCMV mutants selectively expressing either the gH/gL/gO or the gH/gL/MCK-2 complex, or lacking both of these alternative gH/gL complexes. We show that an efficient initial establishment of organ infection, with the notable
exception of SG infection, is crucially dependent on the gH/gL/gO complex. gO-transcomplementation of a genetic gO-ko mutant in virus ΔgO-gOtrans reversed the cell entry deficiency phenotype and, most notably, its gO-deficient progeny was then able to spread within different tissues with viral doubling times comparable to those of wild-type (WT) virus. This spread, however, required the alternative complex gH/gL/MCK-2, as revealed by absence of spread of viral progeny of the gO-transcomplemented double-ko mutant ΔgOΔMCK-2-gOtrans.
In essence, these results revealed an example for a herpesvirus for which neither the gB and gH/gL core complexes alone, nor potentially unidentified other virion envelope glycoprotein complexes, can engender efficient infection in vivo. The alternative gH/gL/MCK-2 complex can substitute for the gH/gL/gO complex for intra-tissue spread and SG infection but not for entry into most first-hit target cell types in organs implicated in CMV disease.
Results
In vivo attenuation of mCMV mutants lacking gO is reversed by gO-transcomplementation
The gene product gO of mCMV ORF m74 forms a complex with the glycoproteins gH and gL. Deletion of m74, and thus of gO, in a recombinant virus is associated with a reduced infectivity of supernatant virus and a focal spread pattern in cell culture [17]. To investigate the role of gH/gL/gO, we used a recently described gO-ko mutant ΔgO (Δm74), which lacks 532 bp at the 5’ end of ORF m74 [18], and constructed an alternative ΔgO mutant (m74stop) containing a stop cassette that interrupts ORF m74 after 120bp. Both gO mutations were introduced into the genome of the mCMV Smith strain, cloned as a bacterial artificial chromosome (BAC), in which a preexisting m129/MCK-2 frameshift mutation was repaired [28]. Production of infectious virus in fibroblast cell cultures was reduced by a factor of ~100 when compared to WT virus (S1 Fig.). This reduction corresponded to a switch from wide-spread infection of the cell monolayer to focal spread (S2A Fig.). When spread via supernatant virus was experimentally inhibited by methylcellulose overlay, spread of WT virus was reduced to a focal spread pattern exhibiting foci comparable in size to foci formed by ΔgO mutants (S2B Fig.), whereas foci formed by ΔgO mutants were not altered. This indicated that short-distance spread between neighboring cells was not affected by the lack of gO. Neutralizing, but not non-neutralizing anti-gB antibodies, also rendered spread of WT virus focal and, furthermore, reduced the size of foci formed by WT virus as well as ΔgO mutants. (S2C–S2D Fig.). This finding suggests that focal spread involves transfer of released virions from the surface of infected cells to the surface of neighboring cells, a process accessible to antibodies. This conclusion is supported by in vivo inhibition of cell-to-cell spread of WT mCMV in liver tissue upon intravenous (i.v.) infusion of virus-neutralizing serum antibodies [29]. The residual spread in presence of neutralizing anti-gB antibodies likely reflects a component of direct cell-to-cell transmission in cell culture.
The thus characterized ΔgO mutants, in comparison to WT virus and a gO-transcomplemented virus ΔgO-gOtrans (virion pictograms in Fig. 1A), were then used to investigate the importance of the gH/gL/gO complex for virulence in vivo. gO-transcomplementation by propagation of ΔgO virus in gO-expressing cells (NIH-gO) generates phenotypically WT-like virions carrying gH/gL/gO complexes in the virion envelope available upon first entry into target cells [17–18]. In further rounds of infection, however, progeny of virus ΔgO-gOtrans are again ΔgO. This makes gO-transcomplementation an elegant approach to distinguish between gO requirement for first target cell entry and subsequent intra-tissue spread.

In a first set of experiments and first model situation (Fig. 1), we intraperitoneally (i.p.) infected newborn BALB/c mice known to be particularly susceptible to mCMV infection [30]. While mice infected with WT virus succumbed to CMV disease from day 7 onward, all those infected with either of the two ΔgO mutants survived (Fig. 1B). This indicated strong virulence attenuation of the mutants in clinical terms. Notably, virulence of ΔgO virus was restored and newborn mice died of CMV disease when gO was transcomplemented in virus ΔgO-gOtrans, although gO was available only upon first cell entry. The survival/mortality rates corresponded to titers of infectious virus in diverse organs differing in cell type composition and tissue architecture, including spleen, lungs, and liver (Fig. 1C). Specifically, and consistently in all organs tested, virus titers with either of the ΔgO mutants were significantly lower than with WT virus or virus ΔgO-gOtrans. These principles were essentially reproduced in a second model situation (S3 Fig.), the ‘immunocompromised host’ model involving i.v. infection of adult BALB/c mice after hematoablative total-body γ-irradiation (reviewed in [31]).
Reversal of the growth deficiency phenotype of the gO-knockout results from gO-transcomplementation rather than from recombination
Since transcomplementation of genetic ΔgO virions with gO can only restore virus entry into the first-hit target cells, but not into neighboring cells in subsequent rounds of infection, reversion of the growth deficiency phenotype by gO-transcomplementation was an unexpected yet highly important result, as it revealed for the first time different molecular requirements for infection of first-hit target cells and subsequent intra-tissue spread. To exclude genetic recombination within the NIH-gO cells during propagation of virus ΔgO-gOtrans, we tested the virion preparation for the absence of the deleted m74 sequence by qPCR. In addition, for excluding the possibility of an in vivo selection and expansion of trace amounts of recombined virus after infection with virus ΔgO-gOtrans, we chose a two-color in situ hybridization (2C-ISH) strategy (S4A Fig.). Genomes from WT and mutant viruses are both stained red with hybridization probe m74.1 directed against the shared, undeleted region of m74, whereas absence of black stain after hybridization with probe m74.2 − specific for the 532-bp deletion in virus ΔgO − verified the genetic deletion status of the transcomplemented mutant. S4B Fig. shows 2C-ISH images for consecutive sections of liver tissue from mice infected with either WT virus or virus ΔgO-gOtrans, or co-infected with both viruses. In none of the liver sections from mice infected only with the mutant virus ΔgO-gOtrans could the m74.2 sequence (black stain) be detected. This finding refutes the objection that recombination between the Δm74 mCMV genome and the gO-expressing vector used for transcomplementation might possibly have genetically restored ORF m74 in virus ΔgO-gOtrans.
Studies with cell lines predicted cell-type specific differences in the requirement of the gH/gL/gO complex
Since host tissues are composed of diverse cell types, we wondered if the observed in vivo attenuation phenotype of ΔgO mutants (Figs. 1 and S3) reflects a general cell entry deficit or a deficit in infecting particular cell types that account for most of the virus productivity, such as fibroblasts and epithelial cells. For a prediction from cell culture experiments, we infected cell lines representing fibroblasts (NIH3T3), epithelial cells (TCMK-1), EC (MHEC-5T), and MΦ (ANA-1) with WT mCMV, the two independent ΔgO mutants, and the gO-transcomplemented virus ΔgO-gOtrans (S5 Fig.). Compared to WT virus and normalized to infection of MEF (in which the viruses were grown and quantitated), both ΔgO mutants showed a reduced capacity to infect NIH3T3 fibroblasts and a loss of the capacity to infect TCMK-1 epithelial cells, phenotypes that were reverted by gO-transcomplementation. In sharp contrast, infection of ANA-1 MΦ was not affected and infection of MHEC-5T EC was even enhanced. Notably, enhanced infection of EC by ΔgO mutants was not reversed by gO-transcomplementation, a phenomenon that might be explained by non-physiological ratios of the alternative gH/gL complexes affecting the infection efficiency for EC [32–33]. In conclusion, the cell culture data predicted an entry deficit of ΔgO mutants for fibroblasts and epithelial cells but not for EC and MΦ.
gO is required for an efficient initial virus entry into main cell types of the liver
Reduced virus titers measured several days after host infection can result from inefficient infection of first-hit target cells as the starting point, or from inefficient subsequent spread within tissue from initially infected cells to neighboring cells, or from a combination of both. For identifying and quantitating first-hit target cells in vivo, we used an approach allowing time too short for completion of the productive viral replication cycle, thus revealing the rate of cell entry uninfluenced by spread. For quantitating entry events without hindrance by immune defense on the route to target organs, we infected γ-irradiated mice i.v. (via the vena cava inferior) so that virus reaches its target tissues with the circulation within seconds, initiating an almost synchronized infection. Such a scenario has a clinical correlate in the early infection of patients conditioned by hematoablative treatment for a subsequent hematopoietic cell transplantation (HCT) (for a clinical review, see [34]). Under such defined conditions and at 24h after infection with WT mCMV, ~90% of the infected liver cells had proceeded to the second kinetic phase of viral gene expression, the early (E) phase, as indicated in 2-color immunohistochemical (2C-IHC) staining of intranuclear viral proteins immediate-early (IE)1 [35] and E1 [36–37] (IE1+E1+cells), whereas only ~10% of the cells expressed IE1 but not yet detectable amounts of E1 (IE1+E1-cells), indicating they were still in the IE phase (Fig. 2). Importantly, infected liver cells had not proceeded to expression of the essential glycoprotein gB (M55), replication of viral DNA, and expression of the essential late (L) phase major capsid protein MCP (M86), which proves that the first cycle was not completed and thus spread excluded (Fig. 2B).
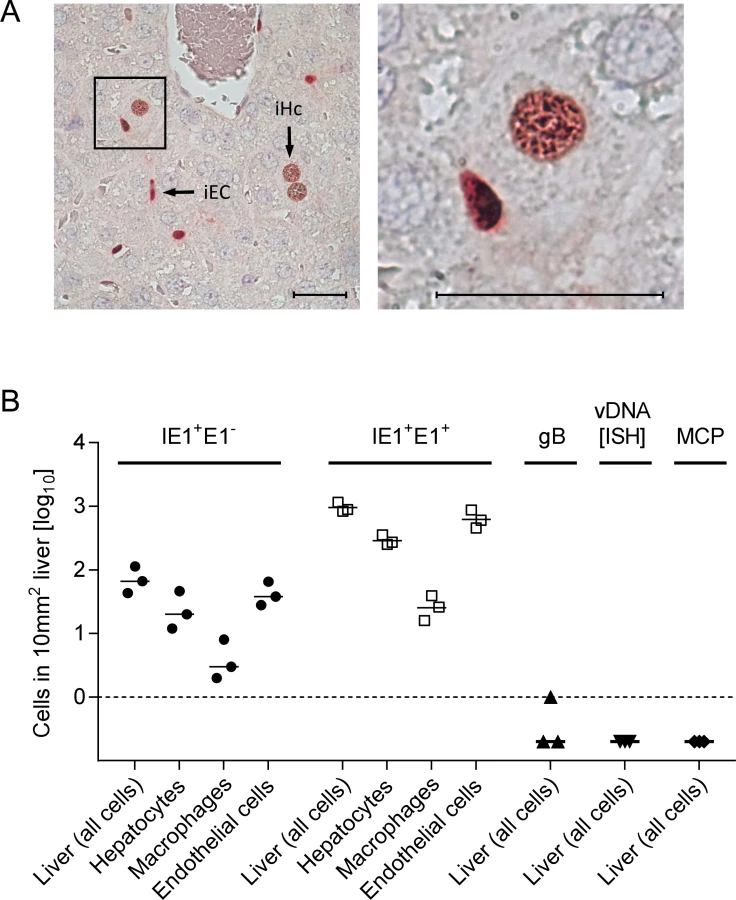
With a focus on the liver, for quantitating successful entry events differentiated by liver cell type, we combined IHC detection of the IE1 protein with cell type-specific markers (Fig. 3). In the liver sinusoids (for a sketch, see Fig. 3A; modified from [38]), virions directly meet CD31+ liver sinusoidal endothelial cells (LSEC), which form the lining of the sinusoids and are noted targets of acute [39] and latent [40] mCMV infection. Virions also directly meet liver-resident F4/80 (Ly71)+ MΦ, known as Kupffer cells, which localize to the sinusoidal lumen attached to the sinusoidal lining and are also recognized targets of mCMV infection [18]. Although hepatocytes (Hc) are separated from the sinusoidal lumen by the fenestrated endothelium and the space of Disse, they are also first-hit target cells of mCMV because virions can pass through the fenestrae. This has originally been indicated by detection of recombined rec-egfp virus in Hc within 24h after infection of Alb-cre mice with floxed reporter virus. Reciprocally, Hc were infected with unrecombined reporter virus 24h after infection of Tie2-cre mice, in which rec-egfp virus was still confined to EC [39].
A three-color IHC (3C-IHC) approach distinguished between infected Hc (IE1+ iHc, red nuclear staining), which are distinctive by cytomorphology, infected MΦ (IE1+F4/80+ iMΦ, red and turquoise-green), and infected EC (IE1+CD31+ iEC, red and black) (Fig. 3B). Most cells expressing IE1 at 24h after infection with WT virus were EC, followed by Hc and MΦ (Figs. 2B and 3C). As shown in Fig. 2B, most of the IE1+ cells co-expressed protein E1, indicating they were in the E phase. Remarkably, this applied to all three cell types, revealing for the first time synchronicity of in vivo viral gene expression in EC, MΦ, and Hc despite their different localization in the tissue. The ranking of the cell types in the absolute numbers of infected cells might reflect their quantitative representation in the liver; alternatively, it might also reflect cell-type specific differences in the cells’ propensity to become infected. To answer this question, we related the numbers of infected cells of each cell type to the corresponding numbers of all cells (S6 Fig.). Whereas the percentages of iHc and iMΦ were ~1%, the percentage of iEC was significantly higher, namely ~5%. This does not necessarily indicate a higher susceptibility of EC to infection in molecular terms; rather, this might reflect a better accessibility of EC that—by lining the sinusoids—provide a huge surface ideal for virion entry.
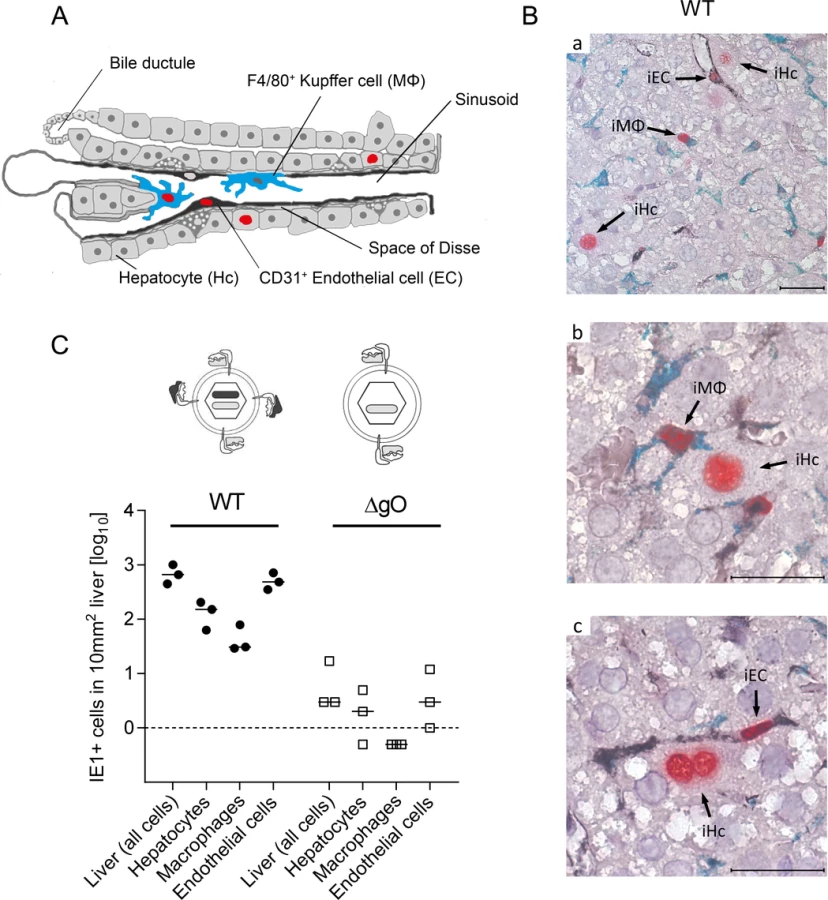
Importantly, absence of gO in virus ΔgO substantially reduced the number of infected cells, and this consistently applied to all three cell types (Fig. 3C), demonstrating that gH/gL/gO is critical for efficient virus entry into quite diverse cells. It may be of interest to note that co-infection with WT and ΔgO viruses did not inhibit WT virus infection, a finding that excludes the possibility of ΔgO defective particle interference in the entry process or enhanced innate/intrinsic defenses elicited by high numbers of ΔgO particles as alternative explanations for poor infectivity of ΔgO viruses.
Since ΔgO viruses still carry the alternative complex gH/gL/MCK-2, the data imply that gH/gL/MCK-2 is not an efficient entry mediator on its own, although our previous work has shown that gH/gL/MCK-2 improves the efficacy of entry, specifically into F4/80+ liver MΦ, in viruses co-expressing gH/gL/gO [18]. Altogether, with respect to cell entry of virus arriving from the circulation, gH/gL/MCK-2 cannot substitute for gH/gL/gO. Strikingly, the requirement of gH/gL/gO for cell entry in vivo applied to all main cell types of the liver. This finding was not predicted by the specific cell lines used for the in vitro studies (recall S5 Fig.).
Intra-tissue virus spread proceeds virtually unabated also in the absence of gO
A reduced increase in virus titers in organs over time may be due to an impaired virus spread within the respective organ or to reduced initial numbers of infected cells. To understand the problem, one must consider that virus multiplication follows an exponential function, so that lower initial numbers of infected cells develop into increasing differences in absolute titers over time, even when the viral capacity for spread within tissue, which is characterized by the virus doubling time (vDT), is actually unaffected by the mutation under study. Exponential functions are linearized by log-transformation of the measured values of the dependent variable, the Y-axis values, to make the data accessible to linear regression analysis. This allows calculation of vDT from the slope of the regression line (reviewed in [41]). In essence, in a comparison of two viruses, parallel regression lines indicate identical spread capacities, whereas diverging regression lines indicate different spread capacities.
After infection with WT virus, gH/gL/gO is available for first entry and for spread, whereas after infection with ΔgO it is unavailable throughout (Fig. 4A). Comparing these two viruses for growth in the liver resulted in fairly parallel log-linear regression lines, though with 1-log distance from each other in numbers of infected cells at any time, which reflects the known difference in initial infection (recall Fig. 3) followed by an almost equally efficient intra-tissue spread (Fig. 4B, outer left panel). Analysis of specific liver cell types (Fig. 4B, remaining panels) revealed almost identical vDT for WT and ΔgO virus in Hc, whereas there is a trend to somewhat slower spread of the mutant among EC and MΦ. The generally poor spread in MΦ likely relates to the fact that MΦ are solitary cells not establishing firm contacts with each other or with other cell types, which hampers virus transfer from cell to cell, whereas Hc form a three-dimensional parenchyma with intimate cell contacts. It has to be taken into account that virus spread occurs not only between cells of the same type but also between different cell types, as was documented previously by bidirectional spread between Hc and EC [38]. The conclusion that spread is unaffected or only minimally affected by deletion of gO is visually confirmed by IHC images directly showing a lower number but comparable size of ΔgO foci (Fig. 4C).

gO-transcomplementation reverts the entry deficit of ΔgO virus and restores virus growth and histopathology
Virus ΔgO-gOtrans carries the gH/gL/gO complex upon first cell entry, but its progeny are ΔgO again (Fig. 5A). Notably, unlike the situation seen above for ΔgO, comparing viruses WT and ΔgO-gOtrans for growth in the liver now revealed superposable regression lines with regard to all liver cells as well as to the individual liver cell types (Fig. 5B), suggesting equivalent growth properties in terms of both initial entry and subsequent spread. Alternatively, identical numbers of infected liver cells could have resulted from many small foci of infection with one of the viruses and fewer but larger foci for the other. This alternative explanation is refuted, however, by IHC images of liver tissue sections demonstrating comparable numbers and size distributions of infectious foci for the two viruses in the time course (Fig. 6).
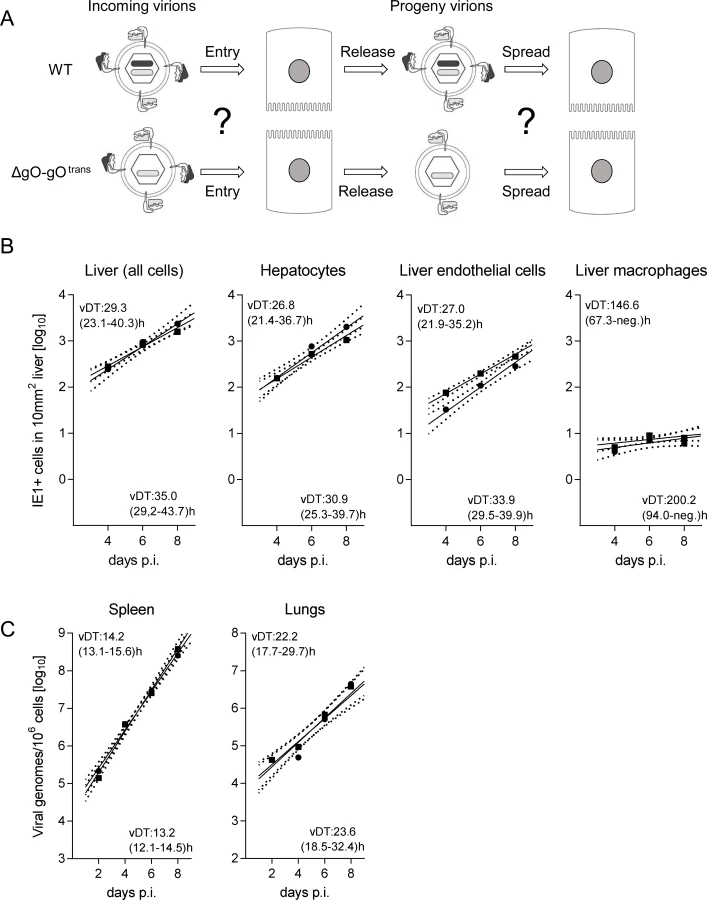

To test if the same rules apply also to other organs involved in CMV disease, we determined log-linear growth regression lines in spleen and lungs by quantitating the increase in viral genome load over time, and found identical growth of WT and ΔgO-gOtrans (Fig. 5C). In conclusion, despite marked differences to the liver in terms of cell type composition and overall tissue architecture, virus spread can proceed in the absence of gH/gL/gO also in organs other than the liver.
The alternative complex gH/gL/MCK-2 is required for intra-tissue virus spread only in the absence of gH/gL/gO
gO-independence of intra-tissue virus spread suggested involvement of an alternative viral envelope glycoprotein complex. Although infection of most organs has been shown not to depend on MCK-2 when gH/gL/gO is present ([23,24] and S7 Fig.), it remained possible that spread can be promoted in a redundant fashion by either gH/gL/gO or gH/gL/MCK-2. If that was true, co-deletion of both complexes should strongly diminish virus spread within organs. A first hint for such a function of MCK-2 was given by data on viral spread in fibroblast cell cultures (S8 Fig.). As double-ko virus ΔgOΔMCK-2 does not produce infectious progeny in cell culture [18], it was necessary to transcomplement gO. When compared to virus ΔgO-gOtrans still carrying the alternative gH/gL/MCK-2 complex (S8 Fig., left images), foci from ΔgOΔMCK-2 progeny of ΔgOΔMCK-2-gOtrans virus barely expanded (S8 Fig., center images), indicating an involvement of gH/gL/MCK-2 in viral spread in vitro. This conclusion was further corroborated by inhibition of viral spread in the presence of antibodies directed against MCK-2 (S8 Fig., right images).
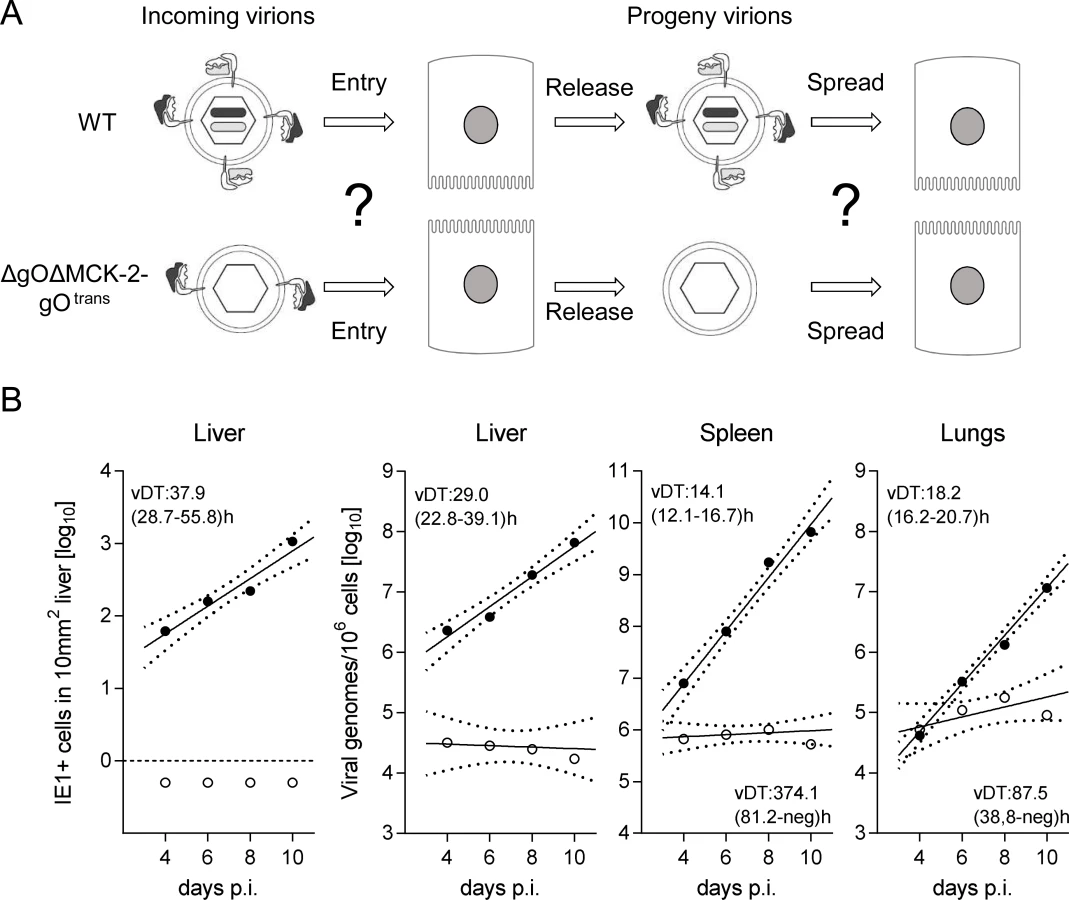
Since in vitro foci from virus ΔgOΔMCK-2-gOtrans still existed, though substantially condensed in size, we asked how simultaneous absence of both alternative gH/gL complexes would translate to virus growth in vivo in liver, spleen, and lungs (Fig. 7). In the liver, the number of cells infected by ΔgOΔMCK-2 progeny of ΔgOΔMCK-2-gOtrans virus remained below the detection limit and the number of viral genomes from the initial infection even slowly declined over time, thus indicating complete absence of intra-tissue spread. Spread was also undetectable in the spleen, whereas one might discuss some residual − yet very inefficient − spread in the lungs.
Salivary gland infection does not follow the rule
An unexpected result was obtained by the analysis of gH/gL complex requirements for the infection of SG (Fig. 8). Unlike what was seen for the other organs, even uncomplemented ΔgO virus replicated like WT virus (Fig. 8A), indicating gO-independence of entry into glandular epithelial cells, the main virus-producing cell type in SG [42]. Notably, SG infection by ΔgO virus apparently depended on the expression of MCK-2, since double deletion in virus ΔgOΔMCK-2-gOtrans strongly reduced SG infection, resulting in a 3-log difference in the viral genome number on day 10 compared to WT and ΔgO virus (Fig. 8A). From these findings it was tempting to conclude that the gH/gL/gO complex is not involved in either step of SG infection and that instead the gH/gL/MCK-2 complex plays a non-redundant, essential role.
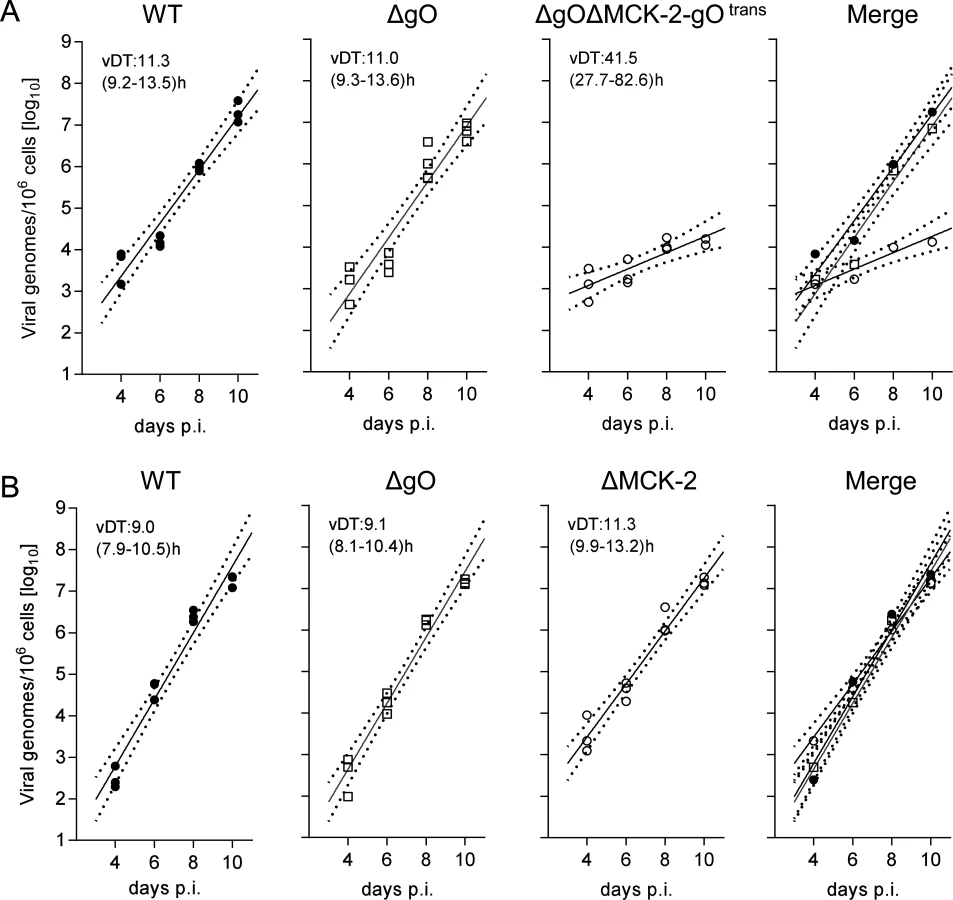
This interpretation, however, was corrected by an independent experiment comparing WT and ΔgO virus with a ΔMCK-2 virus lacking the gH/gL/MCK-2 complex but still expressing the gH/gL/gO complex (Fig. 8B). Surprisingly, ΔMCK-2 virus replicated in the SG like WT and ΔgO virus, indicating that MCK-2 is not essential but can be substituted by gO.
In conclusion, the alternative gH/gL complexes gH/gL/gO and gH/gL/MCK-2 mediate efficient viral growth and cannot be substituted in their roles by any other virion envelope glycoprotein complexes. Whereas gH/gL/gO and gH/gL/MCK-2 mediate intra-tissue spread as well as SG infection in a redundant fashion capable of replacing each other, gH/gL/gO is essential for entry into first-hit target cells in most organs, with the notable exception of the SG.
Discussion
For HCMV, two gH/gL complexes, gH/gL/gO and gH/gL/pUL(128,130,131A), were characterized and corresponding target cells identified in vitro (see the Introduction). In vivo identification of infected cell types is usually based on autopsy or biopsy material derived from immunocompromised patients with overt disease [43], so that one cannot distinguish between first-hit target cells and secondarily infected cells, and virus intra-tissue spread in a time course is difficult, if not impossible, to assess in humans, as it would require repeated biopsies in patients. Due to the host restriction of CMVs, in vivo studies with viruses mutated in their outfit with envelope glycoprotein complexes are limited to natural host animal models, of which infection of mice with mCMV is the most intensively explored. For mCMV, also two gH/gL complexes, gH/gL/gO and gH/gL/MCK-2, are known [17–18]. Depending on route of infection and immune status, local fibrocytes, MΦ in lymphoid tissues and liver, EC, Hc as an epithelial cell type, and more recently also mast cells [44] and alveolar MΦ [19] are noted first-hit target cells of mCMV [39,45].
Cell culture analyses of HCMV and mCMV ΔgO mutants in fibroblasts agreed in demonstrating a markedly reduced infectivity of virus progeny and a focal spread pattern [13,14,17]. A critical requirement for the mCMV gH/gL/gO complex was here also documented for entry into cells of the epithelial cell line TCMK-1. We expanded on these insights with the primary objective to identify the in vivo role of gO with the aim to confirm the predictions. In accordance with the cell culture studies, we found that ΔgO mutants of mCMV are indeed strongly attenuated in virulence in terms of virus replication and pathogenesis in vital organs. In contrast, infection of the EC cell line MHEC-5T and of ANA-1 MΦ revealed gO-independence in vitro, a prediction from cell culture that did not hold true in vivo for liver EC and MΦ, the infection of which proved to be gO-dependent.
By following an experimental strategy to distinguish between first virus entry into liver cell types and subsequent intra-hepatic virus spread, we found that absence of gO strongly reduced the numbers of initially infected EC, MΦ, and Hc. In contrast, the capacity for subsequent intra-hepatic spread, as reflected by virus doubling times (vDT), was unaffected by the mutation. These rules applied also to spleen and lungs. For the lungs, this finding is remarkable in that − for entering lung parenchyma from the circulation − virus has to pass EC that form a barrier of continuous lung endothelium [39], a step during which trans-complemented gO is necessarily lost. This indicates gO-independent spread from pulmonary vascular EC to interstitial cells and alveolar epithelium [44]. Thus, like in the liver, the first cell entry after arrival via the circulation proved to be the gO-dependent critical step for organ infection.
In our model of i.v. liver infection after hematoablative treatment, not only cells directly accessible from the lumen of the sinusoids, such as EC and MΦ, but also Hc, which are separated from the circulation by the fenestrated sinusoidal endothelium, became infected synchronously without a preceding viral replication cycle in any other cell type. This argues against a general involvement of an interposed hematopoietic cell type on the route to target tissues. An exception appears to be the long-distance virus dissemination to the SG. Previous work has already indicated a special situation for the SG − distinguishing infection of SG from that of other organs − in that mCMV does not appear to reach this site as free virions but needs to hijack CX3CR1hi patrolling monocytes (PM) to serve as vehicles transporting it to the SG, a mechanism that was concluded to depend on MCK-2 [21,25]. In this context, our data add the important new information that virus entry into glandular epithelial cells, the main virus-producing cell type in SG, can take place independently of gO, because MCK-2 can substitute for gO in its role. The result that, in our infection model, gO can in turn substitute for MCK-2 in SG infection, as seen with virus ΔMCK-2, was quite unexpected in view of previous work in immunocompetent mice having documented an SG growth deficiency of virus mutants not expressing MCK-2 [18,46–47]. A first hint for a model-dependent difference was given by the previous finding that the prototype of BAC-cloned mCMV [48]), in which an m129/MCK-2 frameshift mutation prevents the expression of full-length MCK-2 [49], replicated like WT virus in the SG of γ-irradiated mice even after local, intraplantar infection [30]. This argues against a critical role of the route of infection and leaves the hematoablative treatment as the differential parameter. Future studies will be aimed at identifying the hematopoietic cell that, in immunocompetent mice, restricts virus dissemination to the SG making it dependent on MCK-2. The PM discussed above [21,25] is an obvious candidate.
Interestingly, horizontal host-to-host transmission occurs through free monocapsid virions released with packed vacuoles from the glandular epithelial cells into the salivary duct [42] and thus the need for an efficient docking of free virions to first-hit target cells may have been the evolutionary driver for the acquisition of the gH/gL/gO complex.
The apparent question remains why intra-tissue spread does not require the gH/gL/gO complex. There exist examples for other viruses indicating that infection of cells by free virions arriving at first target cells can differ from the transfer of virus from an infected cell to closely neighboring cells. Mechanisms discussed for cell-to-cell spread include the formation of polarized contacts, so-called ‘virological synapses’ [49–50], transit through cell junctions [51–53], and transfer of extracellular virus involving the formation of membrane protrusions [54]. These modes of cell-to-cell transfer have in common a high local virus concentration associated with a high efficiency of infection and may differ from infection by free virions in the requirements for virion constituents [50]. Interpreting our findings, we propose that sufficient avidity for the binding of free virions to target cells depends on the gH/gL/gO complex, whereas for cell-to-cell spread either of the alternative gH/gL complexes is sufficient so that gH/gL/MCK-2 can substitute for gH/gL/gO in the spread of ΔgO viruses. If one of the alternative gH/gL complexes is preferentially used during spread of WT virus is open to question and might also depend on the relative amounts of these complexes incorporated into the virion envelope. These may vary with the cell type in which the virus has replicated [16] and may also differ between virus strains [32–33].
As alternative gH/gL complexes of HCMV strains cannot be studied in vivo with respective mutants, it must necessarily remain speculative whether our findings in the mCMV model exactly predict the roles the corresponding alternative gH/gL complexes play in human infection. In cell culture, the gH/gL/gO complexes of HCMV and mCMV were functionally comparable [13,14] and reduced capacities to infect MΦ apply to both, HCMV lacking gH/gL/pUL(128,130,131A) and mCMV lacking gH/gL/MCK-2 [10,18]. Reduced capacity of gH/gL/pUL(128,130,131A) deletion mutants of HCMV to infect EC and epithelial cells in vitro [6], was not seen with mCMV mutants lacking the gH/gL/MCK-2 complex [18]. Yet, data on cell tropism found in cell culture need not necessarily extrapolate to in vivo cell tropism. Cells in cell culture, and in particular immortalized and often clonal cell lines, likely differ in many respects from cells of the same cell types in vivo, and also cell contacts and cytokine milieu in cell monolayers do not always reflect those in the context of tissues. Our own data on EC and MΦ cell lines MHEC-5T and ANA-1, respectively, showed gO-independence of the infection, which did not apply to in vivo infection of these two cell types in the liver. Similarly, recent findings showed that mast cells in vivo, but not cultured mast cells, are susceptible to productive mCMV infection [44,55], and that PM, but not inflammatory monocytes, are infected by mCMV in vivo, although MΦ derived from both monocyte populations were found to be equally susceptible in vitro [25].
In summary, this first report on the roles alternative gH/gL complexes of a CMV play in vivo shows redundance in mediating intra-tissue virus spread as well as infection of SG, the site of virus host-to-host transmission, and revealed a critical role for gO in the initiation of infection by free virions. This makes the gH/gL/gO complex an interesting target for prevention of primary infection.
Materials and Methods
Mice and infection conditions
BALB/c mice were bred and maintained under SPF conditions at the Laboratory Mouse Breeding and Engineering Centre of the Faculty of Medicine, University of Rijeka, or in the Central Laboratory Animal Facility at the University Medical Center Mainz.
For immunosuppression, hematoablative conditioning of 8–9 week-old female BALB/c mice was achieved by total-body γ-irradiation with a single dose prior to infection. Adult mice were infected i.v. with tissue culture (NIH3T3)-derived mCMV WT or mutants in 500 μl of PBS. Neonatal mice were infected i.p. with the indicated viruses in 50 μl of PBS at 6 h post-partum. All mice were sacrificed by CO2 inhalation or cervical dislocation.
Ethics statement
Animal research protocols of the University Medical Center Mainz were approved by the ethics committee of the Landesuntersuchungsamt Rheinland-Pfalz, permission no. 23 177–07/G09–1–004, according to German Federal Law §8 Abs. 1 TierSchG (animal protection law). All experiments done at the University of Rijeka, Croatia, were in accordance with the University of Rijeka animal use and care policies in accordance to the guidelines of the animal experimentation law (SR 455.163; TVV) of the Swiss Federal Government.
Cells, viruses, and studies in cell culture
Primary mouse embryonic fibroblasts (MEF) from BALB/c mice, NIH3T3 cells (ATCC: CRL-1658), the endothelial cell line MHEC5-T [56] and the epithelial cell line TCMK-1 (ATCC: CCL-139) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum. The macrophage cell line ANA-1 [57] was maintained in RPMI medium supplemented with 10% fetal calf serum. NIH3T3 cell lines stably expressing m74/gO (NIH-gO) were used for gO-transcomplementation [17]. BAC (pSM3fr-MCK-2fl)-derived virus [28] was used as WT mCMV. The mCMV ORF m74 deletion mutant (ΔgO) and the m74/m131–129 double-knockout mutant (ΔgOΔMCK-2) have been described previously [18]. For analysis of virus spread and dissemination in cell culture, MEF were seeded in flat-bottomed 96-well plates and cell monolayers were infected with 50 PFU per well. One hour after infection, cell monolayers were washed and incubated for further 3 days with culture medium supplemented, depending on the question, with neutralizing anti-gB antibody (mAb, clone 97.3) [58], non-neutralizing anti-gB antibody (mAb, clone 5F12; kindly provided by Michael Mach, University Erlangen-Nürnberg, Germany), or rabbit anti-MCK-2 serum WU1073 [59]. Foci of infection or percentages of infected cells were visualized by indirect immunofluorescent staining of mCMV gB protein using anti-gB mAb (clone 97.3), or of mCMV intranuclear IE1 protein pp89/76 using mAb CROMA101. For counterstaining of cell nuclei, cells were incubated in PBS containing 5 μg/ml Hoechst 333258 (Invitrogen). To monitor virus infection of cells in suspension, intracellular cytofluorometric staining was performed. Briefly, cells were detached with 0.5 mM Na-EDTA, fixed with 1% paraformaldehyde for 10 min, and then stained in PBS containing 0.3% Saponin and 1% BSA using anti-IE1 antibody and Fluor488-coupled goat anti-mouse antibody (Invitrogen). Cells were washed with PBS containing 0.03% Saponin. After staining, cells were resuspended in 1% paraformaldehyde and analyzed on a FACSCalibur using CellQuest software (BD Biosciences).
BAC mutagenesis and reconstitution of recombinant virus
Markerless BAC mutagenesis was performed to introduce a stop cassette in the m74 ORF in the pSM3fr-MCK-2fl BAC as described previously [60]. For constructing the pSM3fr-m74stop mutant (virus: ΔgO; m74stop), the primers m74stop-for (5’-GGA GGT TCG GTC GCA TCG ATT GTA TCA TAA CCT CCG TCT TCA TAA TCA TCG GCT AGT TAA CTA GCC AGG ATG ACG ACG ATA AGT AGG G-3’) and m74stop-rev (5’-AAA GTG TAG CAT ACA ACC CGG CCG TTA CCG GCT ATA TCG AGA TGA GCG AAG GCT AGT TAA CTA GCC GAT GAT TAT GAA GAC GGA GGC AAC CAA TTA ACC AAT TCT GAT TAG-3’) were used. The sequences of the stop cassette are indicated by italic type. Insertion of the stop cassette was controlled by restriction enzyme pattern analysis and sequencing. Recombinant CMVs were reconstituted by transfection of purified BAC DNA into MEF using Superfect transfection reagent (Qiagen). Transfected cells were propagated until viral plaques appeared and supernatants from these cultures were used for further propagation. Virus stocks were prepared from supernatants of infected NIH3T3 cells, or from NIH-gO cells in case of gO-transcomplementation, by sucrose-gradient ultracentrifugation as described [41]. Virus titers were determined by TCID50 assay or standard plaque assay performed on MEF.
Quantitation of viral genomes and infectious virus in host tissues
The in vivo replication of WT and mutant mCMV was determined by establishing log-linear virus growth curves for various host tissues of interest. At defined times post-infection, viral genomes present in the respective organ lysates were quantitated by M55 (encoding gB)- specific qPCR normalized to cell number by pthrp specific qPCR [41]. In vivo infectivity was determined from homogenates of infected organs by plaque assay on MEF under conditions of centrifugal enhancement of infectivity.
In situ hybridization specific for viral genes
To distinguish between WT and mutant virus genomes in liver tissue sections, 2-color in situ hybridization (2C-ISH) was applied essentially as described previously [41] with hybridization probes adapted to detect or exclude m74 sequences. Probe m74.1 was synthesized using Fluorescein-12-dUTP (Roche Applied Science) in dNTP mix and primer pair m74.1_probe_rev (104.541-CAG AGA CGG TAC GTG TTG-104.558) (GenBank accession no. NC_004065) and m74.1_probe_for (105.150-CGT GTT GGT GAC CGA ATC-105.133). For probe m74.2, Digoxigenin-11-dUTP (Roche Applied Science) was incorporated by PCR using primer pair m74.2_probe_rev (105.280-CCA TGG ATC GGT GAC ACG AAA G-105.301) and m74.2_probe_for (105.774-ATC CGC CGC GAA AGT GAA C-105.746). After DNA hybridization on deparaffinized serial 1-μm sections of liver tissue, red staining was achieved by using alkaline phosphatase-conjugated anti-Fluorescein antibody (Roche Applied Science) with Fuchsin+ Substrate-Chromogen System (Dako) as the chromogenic substrate. Black staining was achieved by using peroxidase-conjugated anti-digoxigenin antibody (Roche Applied Science) with diaminobenzidine tetrahydrochloride (DAB, Sigma-Aldrich) as the substrate, followed by color enhancement with ammonium nickel sulfate hexahydrate.
To detect viral genomes (vDNA) in cell nuclei of infected liver cells for quantitating cells in the late (L) phase of the viral productive cycle, ISH specific for gene M55 was performed on 2-μm sections of liver tissue as described previously [61].
Immunohistochemical (IHC) analyses of viral protein expression
To simultaneously detect intranuclear viral proteins IE1 and E1 for distinguishing mCMV-infected cells in the immediate-early (IE) and early (E) phase of infection, two-color IHC (2C-IHC) specific for the viral proteins IE1 and E1 was performed on 2-μm liver tissue sections essentially as described [41], with modifications. IE1 was labeled with mAb CROMA 101. Black staining was achieved by using the ImmPRESS HRP anti-mouse Ig detection kit (Vector Laboratories) with DAB as substrate and ammonium nickel sulfate hexahydrate for color enhancement. E1 was labeled with mAb CROMA 103 and stained in red with alkaline phosphatase-conjugated polyclonal goat anti-mouse IgG (AbD Serotec) and Fuchsin+ Substrate-Chromogen System. A light blue counterstaining was achieved with hematoxylin. Single-color IHC specific for glycoprotein B (gB) and the late (L) phase protein MCP (major capsid protein) were performed as described [41].
For quantitating infected cells differentiated by cell type, 3C-IHC analysis was performed on 2-μm liver tissue sections combining intranuclear IE1-specific IHC labeling [41] with the detection of cell type-specific markers CD31 for EC and F4/80 (Ly71) for MΦ: (i) Rat mAb anti-CD31 (PECAM-1; clone SZ31; Dianova) followed by biotin-conjugated polyclonal anti-rat Ig antibody (BD Biosciences) and a peroxidase-coupled avidin biotin complex (Vectastain Elite ABC Kit, Vector Laboratories). (ii) DAB with color enhancement by ammonium nickel sulfate hexahydrate used to stain EC in black, followed by trypsin digestion. (iii) Rat mAb anti-F4/80 (clone BM8; acris antibodies), biotin-conjugated polyclonal anti-rat Ig antibody (BD Biosciences), and peroxidase-coupled avidin biotin complex (Vectastain Elite ABC Kit), followed by HRP-Green Solution Set (42 life sciences) for turquoise-green staining of MΦ. (iv) Red staining of intranuclear IE1 protein with Fuchsin+ Substrate-Chromogen System.
Statistical analyses and calculation of viral doubling times in host tissues
Statistical tests used are specified in the respective figure legends and were performed using GraphPad Prism version 6.04 for Windows, GraphPad Software. Differences are considered statistically significant for P values of <0.05. Viral doubling times (vDT = log2/a) and the corresponding 95% confidence intervals were calculated by linear regression analysis from the slopes a of log-linear growth curves [41].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. Connolly SA, Jackson JO, Jardetzky TS, Longnecker R (2011) Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol 9:369–381. doi: 10.1038/nrmicro2548 21478902
2. Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, et al. (2012) Herpes virus fusion and entry: a story with many characters. Viruses 4:800–832. doi: 10.3390/v4050800 22754650
3. Hutt-Fletcher LM (2007) Epstein-Barr virus entry. J Virol 81:7825–7832. 17459936
4. Pedersen SM Hollsberg P (2006) Complexities in human herpesvirus-6A and-6B binding to host cells. Virology 356:1–3. 16959284
5. Adler B, Sinzger C (2013) Cytomegalovirus inter-strain variance in cell-type tropism. In: Reddehase, editor. Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, UK. Vol. II, pp. 297–322.
6. Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, et al. (2004) Human cytomegalovirus UL131–128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol 78:10023–10033. 15331735
7. Gerna G, Percivalle E, Lilleri D, Lozza L., Fornara C., et al. (2005) Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131–128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J Gen Virol 86:275–284. 15659746
8. Wang D, Shenk T (2005) Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc Natl Acad Sci USA 102:18153–18158. 16319222
9. Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C., et al. (2006) Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J Gen Virol 87:2451–2460. 16894182
10. Sinzger C, Eberhardt K, Cavignac Y, Weinstock C, Kessler T, et al. (2006) Macrophage cultures are susceptible to lytic productive infection by endothelial-cell-propagated human cytomegalovirus strains and present viral IE1 protein to CD4+ T cells despite late downregulation of MHC class II molecules. J Gen Virol 87:1853–1862. 16760387
11. Ryckman BJ, Chase MC, Johnson DC (2008) HCMV gH/gL/UL128–131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc Natl Acad Sci USA 105:14118–14123. doi: 10.1073/pnas.0804365105 18768787
12. Vanarsdall AL, Chase MC, Johnson DC (2011) Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J Virol 85:11638–11645. doi: 10.1128/JVI.05659-11 21880752
13. Jiang XJ, Adler B, Sampaio KL, Digel M, Jahn G, et al. (2008) UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J Virol 82:2802–2812. doi: 10.1128/JVI.01550-07 18184717
14. Wille PT, Knoche AJ, Nelson JA, Jarvis MA, Johnson DC (2010) An HCMV gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts, epithelial, and endothelial cells. J Virol 84:2585–2596. doi: 10.1128/JVI.02249-09 20032184
15. Borza CM, Hutt-Fletcher LM (2002) Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8:594–599. 12042810
16. Scrivano L, Sinzger C, Nitschko H, Koszinowski UH, Adler B (2011) HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog 7:e1001256. doi: 10.1371/journal.ppat.1001256 21249233
17. Scrivano L, Esterlechner J, Muhlbach H, Ettischer N, Hagen C, et al. (2010) The m74 gene product of murine cytomegalovirus (MCMV) is a functional homolog of human CMV gO and determines the entry pathway of MCMV. J Virol 84:4469–4480. doi: 10.1128/JVI.02441-09 20181688
18. Wagner FM, Brizic I, Prager A, Trsan T, Arapovic M, et al. (2013). The viral chemokine MCK-2 of murine cytomegalovirus promotes infection as part of a gH/gL/MCK-2 complex. PLoS Pathog. 9:e1003493. doi: 10.1371/journal.ppat.1003493 23935483
19. Stahl FR, Keyser KA, Heller K, Bischoff Y, Halle S., et al. (2014) Mck2-dependent infection of alveolar macrophages promotes replication of MCMV in nodular inflammatory foci of the neonatal lung. Mucosal Immunol. doi: 10.1038/mi.2014.42 25515629
20. Nogalski MT, Chan GC, Stevenson EV, Collins-McMillen DK, Yurochko AD (2013). The HCMV gH/gL/UL128–131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog 9:e1003463. doi: 10.1371/journal.ppat.1003463 23853586
21. Noda S., Aguirre SA, Bitmansour A, Brown JM, Sparer TE, et al. (2006) Cytomegalovirus MCK-2 controls mobilization and recruitment of myeloid progenitor cells to facilitate dissemination. Blood 107:30–38. 16046529
22. Straschewski S, Patrone M, Walther P, Gallina A., Mertens T, et al. (2011) Protein pUL128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. J Virol 85:5150–5158. doi: 10.1128/JVI.02100-10 21367908
23. Fleming P, Davis-Poynter N, Degli-Esposti M, Densley E, Papadimitriou J, et al. (1999) The murine cytomegalovirus chemokine homolog, m131/129, is a determinant of viral pathogenicity. J Virol 73:6800–6809. 10400778
24. Saederup N, Aguirre SA, Sparer TE, Bouley DM, Mocarski ES (2001) Murine cytomegalovirus CC chemokine homolog MCK-2 (m131–129) is a determinant of dissemination that increases inflammation at initial sites of infection. J Virol 75:9966–9976. 11559829
25. Daley-Bauer LP, Roback LJ, Wynn GM, Mocarski ES (2014) Cytomegalovirus hijacks CX3CR1(hi) patrolling monocytes as immune-privileged vehicles for dissemination in mice. Cell Host Microbe 15:351–362. doi: 10.1016/j.chom.2014.02.002 24629341
26. Daley-Bauer LP, Wynn GM, and Mocarski ES (2012) Cytomegalovirus impairs antiviral CD8+ T cell immunity by recruiting inflammatory monocytes. Immunity 37:122–133. doi: 10.1016/j.immuni.2012.04.014 22840843
27. Wikstrom ME, Fleming P, Comerford I, McColl SR, Andoniou CE, et al. (2013) A chemokine-like viral protein enhances alpha interferon production by plasmacytoid dendritic cells but delays CD8+ T cell activation and impairs viral clearance. J Virol 87:7911–7920. doi: 10.1128/JVI.00187-13 23658453
28. Jordan S, Krause J, Prager A, Mitrovic M, Jonjic S, et al. (2011) Virus progeny of murine cytomegalovirus bacterial artificial chromosome pSM3fr show reduced growth in salivary glands due to a fixed mutation of MCK-2. J Virol 85:10346–10353. doi: 10.1128/JVI.00545-11 21813614
29. Wirtz N, Schader SI, Holtappels R, Simon CO, Lemmermann NA, et al. (2008) Polyclonal cytomegalovirus-specific antibodies not only prevent virus dissemination from the portal of entry but also inhibit focal virus spread within target tissues. Med Microbiol Immunol 197:151–158. doi: 10.1007/s00430-008-0095-0 18365251
30. Cekinovic D, Lisnic VJ, Jonjic S (2014) Rodent models of congenital cytomegalovirus infection. Methods Mol Biol 1119:289–310. doi: 10.1007/978-1-62703-788-4_16 24639229
31. Holtappels R, Ebert S, Podlech J, Fink A, Böhm V, et al. (2013) Murine model for cytoimmunotherapy of CMV disease after haematopoietic cell transplantation. In: MJ Reddehase, editor. Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, UK. Vol. II, pp.354–381.
32. Murrell I, Tomasec P, Wilkie GS, Dargan DJ, Davison AJ, et al. (2013) Impact of sequence variation in the UL128 locus on production of human cytomegalovirus in fibroblast and epithelial cells. J Virol 87:10489–10500. doi: 10.1128/JVI.01546-13 23885075
33. Zhou M, Yu Q, Wechsler A, Ryckman BJ (2013) Comparative analysis of gO isoforms reveals that strains of human cytomegalovirus differ in the ratio of gH/gL/gO and gH/gL/UL128–131 in the virion envelope. J Virol 87:9680–9690. doi: 10.1128/JVI.01167-13 23804643
34. Seo S, Boeckh MClinical Cytomegalovirus Research: Haematopoietic Cell Transplantation In: MJ Reddehase, editor. Cytomegaloviruses: from molecular pathogenesis to intervention. Caister Academic Press, Norfolk, UK. Vol. II, pp.337–353.
35. Keil GM, Fibi MR, Koszinowski UH (1985) Characterization of the major immediate-early polypeptides encoded by murine cytomegalovirus. J Virol 54:422–428. 2985805
36. Bühler B, Keil GM, Weiland F, Koszinowski UH (1990) Characterization of the murine cytomegalovirus early transcription unit e1 that is induced by immediate-early proteins. J Virol 64:1907–1919. 2157860
37. Ciocco-Schmitt GM, Karabekian Z, Godfrey EW, Stenberg RM, Campbell AE, et al. (2002) Identification and characterization of novel murine cytomegalovirus M112–113 (e1) gene products. Virology 294:199–208. 11886278
38. Frevert U, Engelmann S, Zougbédé S, Stange J, Ng B, et al. (2005) Intravital observation of Plasmodium berghei sporozoite infection of the liver. PLoS Biol 3:e192. 15901208
39. Sacher T, Podlech J, Mohr CA, Jordan S, Ruzsics Z, et al. (2008) The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3:263–272. doi: 10.1016/j.chom.2008.02.014 18407069
40. Seckert CK, Renzaho A, Tervo HM, Krause C., Deegen P.,et al. (2009) Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J Virol 83:8869–8884. doi: 10.1128/JVI.00870-09 19535440
41. Lemmermann NA, Podlech J, Seckert CK, Kropp KA, Grzimek NK, et al. (2010) CD8 T-cell immunotherapy of cytomegalovirus disease in the murine model. In D Kabelitz, SHE Kaufmann, editors. Methods in Microbiology: Immunology of Infection, Academic Press, London UK, pp. 369–420.
42. Jonjić S, Mutter W, Weiland F, Reddehase MJ, Koszinowski UH (1989) Site-restricted persistent cytomegalovirus infection after selective long-term depletion of CD4+ T lymphocytes. J Exp Med 169:1199–1212. 2564415
43. Plachter B, Sinzger C, Jahn G (1996) Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res 46:195–261. 8824701
44. Ebert S, Becker M, Lemmermann NA, Büttner JK, Michel A, et al. (2014). Mast cells expedite control of pulmonary murine cytomegalovirus infection by enhancing the recruitment of protective CD8 T cells to the lungs. PLoS Pathog 10:e1004100. doi: 10.1371/journal.ppat.1004100 24763809
45. Hsu KM, Pratt JR, Akers WJ, Achilefu SI, Yokoyama WM (2009) Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J Gen Virol 90:33–43. doi: 10.1099/vir.0.006668-0 19088270
46. Manning WC, Stoddart CA, Lagenaur LA, Abenes GB, Mocarski ES (1992) Cytomegalovirus determinant of replication in salivary glands. J Virol 66:3794–3802. 1316482
47. Lagenaur LA, Manning WC, Vieira J, Martens CL, Mocarski ES (1994) Structure and function of the murine cytomegalovirus sgg1 gene: a determinant of viral growth in salivary gland acinar cells. J Virol. 68:7717–7727. 7966561
48. Wagner M, Jonjic S, Koszinowski UH, Messerle M (1999) Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol 73:7056–7060. 10400809
49. Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, et al. (2003) Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716. 12589003
50. Sattentau Q. (2008) Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol 6:815–826. doi: 10.1038/nrmicro1972 18923409
51. Dingwell KS, Brunetti CR, Hendricks RL, Tang Q, Tang M, Rainbow AJ, Johnson DC et al. (1994) Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J Virol 68:834–845. 8289387
52. Mulder W, Pol J, Kimman T, Kok G, Priem J, et al. (1996) Glycoprotein D-negative pseudorabies virus can spread transneuronally via direct neuron-to-neuron transmission in its natural host, the pig, but not after additional inactivation of gE or gI. J Virol 70:2191–2200. 8642642
53. Johnson DC, Webb M, Wisner TW, Brunetti C (2001) Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J Virol 75:821–833. 11134295
54. Gill MB, Edgar R, May JS, Stevenson PG (2008) A gamma-herpesvirus glycoprotein complex manipulates actin to promote viral spread. PLoS ONE 3:e1808. doi: 10.1371/journal.pone.0001808 18350146
55. Becker M, Lemmermann NA, Ebert S, Baars P, Renzaho A, et al. (2014) Mast cells as rapid innate sensors of cytomegalovirus by TLR3/TRIF signaling-dependent and-independent mechanisms. Cell Mol Immunol doi: 10.1038/cmi.2014.73 25544504
56. Plendl J, Sinowatz F, Auerbach R (1995) A transformed murine myocardial vascular endothelial cell clone: characterization of cells in vitro and of tumours derived from clone in situ. Virchows Arch 426:619–628. 7655744
57. Cox GW, Mathieson BJ, Gandino L, Blasi E, Radzioch D, et al. (1989) Heterogeneity of hematopoietic cells immortalized by v-myc/v-raf recombinant retrovirus infection of bone marrow or fetal liver. J Natl Cancer Inst 81:1492–1496. 2778838
58. Cekinovic D, Golemac M, Pugel EP, Tomac J, Cicin-Sain L, et al. (2008) Passive immunization reduces murine cytomegalovirus-induced brain pathology in newborn mice. J Virol 82:12172–12180. doi: 10.1128/JVI.01214-08 18842707
59. MacDonald MR, Burney MW, Resnick SB, Virgin HW IV (1999) Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a beta chemokine of novel structure. J Virol 73:3682–3691. 10196260
60. Tischer BK, von Einem J, Kaufer B, Osterrieder N (2006) Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. 16526409
61. Podlech J, Holtappels R, Wirtz N, Steffens HP, Reddehase MJ (1998) Reconstitution of CD8 T cells is essential for the prevention of multiple-organ cytomegalovirus histopathology after bone marrow transplantation. J Gen Virol 79:2099–2104. 9747717
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 2
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- Control of Murine Cytomegalovirus Infection by γδ T Cells
- ATPaseTb2, a Unique Membrane-bound FoF1-ATPase Component, Is Essential in Bloodstream and Dyskinetoplastic Trypanosomes
- Rational Development of an Attenuated Recombinant Cyprinid Herpesvirus 3 Vaccine Using Prokaryotic Mutagenesis and In Vivo Bioluminescent Imaging
- Telomeric ORFS in : Does Mediator Tail Wag the Yeast?