
The Early Stage of Bacterial Genome-Reductive Evolution in the Host
The equine-associated obligate pathogen Burkholderia mallei was developed by reductive evolution involving a substantial portion of the genome from Burkholderia pseudomallei, a free-living opportunistic pathogen. With its short history of divergence (∼3.5 myr), B. mallei provides an excellent resource to study the early steps in bacterial genome reductive evolution in the host. By examining 20 genomes of B. mallei and B. pseudomallei, we found that stepwise massive expansion of IS (insertion sequence) elements ISBma1, ISBma2, and IS407A occurred during the evolution of B. mallei. Each element proliferated through the sites where its target selection preference was met. Then, ISBma1 and ISBma2 contributed to the further spread of IS407A by providing secondary insertion sites. This spread increased genomic deletions and rearrangements, which were predominantly mediated by IS407A. There were also nucleotide-level disruptions in a large number of genes. However, no significant signs of erosion were yet noted in these genes. Intriguingly, all these genomic modifications did not seriously alter the gene expression patterns inherited from B. pseudomallei. This efficient and elaborate genomic transition was enabled largely through the formation of the highly flexible IS-blended genome and the guidance by selective forces in the host. The detailed IS intervention, unveiled for the first time in this study, may represent the key component of a general mechanism for early bacterial evolution in the host.
Published in the journal:
. PLoS Pathog 6(5): e32767. doi:10.1371/journal.ppat.1000922
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000922
Summary
The equine-associated obligate pathogen Burkholderia mallei was developed by reductive evolution involving a substantial portion of the genome from Burkholderia pseudomallei, a free-living opportunistic pathogen. With its short history of divergence (∼3.5 myr), B. mallei provides an excellent resource to study the early steps in bacterial genome reductive evolution in the host. By examining 20 genomes of B. mallei and B. pseudomallei, we found that stepwise massive expansion of IS (insertion sequence) elements ISBma1, ISBma2, and IS407A occurred during the evolution of B. mallei. Each element proliferated through the sites where its target selection preference was met. Then, ISBma1 and ISBma2 contributed to the further spread of IS407A by providing secondary insertion sites. This spread increased genomic deletions and rearrangements, which were predominantly mediated by IS407A. There were also nucleotide-level disruptions in a large number of genes. However, no significant signs of erosion were yet noted in these genes. Intriguingly, all these genomic modifications did not seriously alter the gene expression patterns inherited from B. pseudomallei. This efficient and elaborate genomic transition was enabled largely through the formation of the highly flexible IS-blended genome and the guidance by selective forces in the host. The detailed IS intervention, unveiled for the first time in this study, may represent the key component of a general mechanism for early bacterial evolution in the host.
Introduction
The genomes of host-adapted bacteria, including endosymbionts and obligatory intracellular pathogens, go through reductive evolution [1], [2], [3]. Such changes are partly due to a reduced pressure to maintain genes that are not essential for survival in the host. Similarly, decreased efficiency of purifying selection, resulting from the reduced population size from a restricted life, results in inactivated genes, including beneficial genes, through genetic drift [3]. During the early stage of the genome reduction process, the majority of genes are lost as large chromosomal fragments spanning multiple genes. Such genome reduction has been documented in diverse bacterial groups, including Firmicutes, Chlamydiae, Spirochetes, and γ-Proteobacteria [1], [3], [4], [5], [6], [7]. Most of these bacteria have large expansion of IS elements (insertion sequences), and thus it has been suggested that the IS elements may play an essential role during the genome reduction process [1], [3], [8], [9], [10].
Burkholderia pseudomallei and Burkholderia mallei belong to the ß-Proteobacteria family, and are the causative agents of melioidosis and glanders, respectively [11], [12], [13], [14], [15], [16], [17]. B. mallei has very recently (∼3.5 myr) evolved from a clone of B. pseudomallei through extensive genome reduction [18], [19], accounting for as much as 1.41 Mb or 20% of the genome, as estimated by the size difference between the genomes of B. mallei ATCC 23344 and B. pseudomallei K96243 [18], [20], [21]. Concomitant with this process, B. mallei became constantly associated with mammalian hosts, specifically equines [22], [23], while B. pseudomallei maintains an opportunistic pathogenic lifestyle [17]. Preliminary analyses of the two type strains, B. mallei ATCC 23344 and B. pseudomallei K96243, have suggested that genome reduction and rearrangement in B. mallei were mediated by IS elements that are widely spread throughout the genome [20], [21]. Genes that have been deleted from the B. mallei genome but are maintained in B. pseudomallei include genes that are required for environmental survival. Many of these genes encode metabolic functions for the synthesis of metabolites or the utilization of various sugars and amino acids, without which bacterial propagation in the environment could be significantly hindered [20].
While the genomic reduction during bacterial restriction to their hosts has been well documented [1], [8], [10], most of the stepwise processes have not yet been elucidated. The B. mallei genome has unique significance, as it is much younger than the other genomes in which the genome-reductive evolutionary processes have been most studied to date, including Buchnera (>150 million years) and other much older groups [1], [3], [4], [5], [6], [7]. The studies with these older genomes have been challenging due to the subsequent genomic- and nucleotide-level mutations that accumulated over a long evolutionary history. In this study, we dissected 10 genomes each of B. pseudomallei and B. mallei to understand the early-stage processes that drive genome-reductive evolution in host-associated bacteria.
Results/Discussion
Multiple IS elements with massive proliferation
It is well known that bacteria specialized to a (host) niche, often have a large number of IS elements compared to their free-living relatives [1], [3], [8], [9], [10]. Likewise, by comparing genome sequences, we found that three types of IS elements, ISBma1, ISBma2, and IS407A, were significantly increased in B. mallei compared to B. pseudomallei (Fig. 1A). By contrast, other types of IS, including IS1356, ISBma3, ISBma4, and ISBma5 were found in low copy number in both species of bacteria. These elements appeared to be mostly degenerate evolutionary remnants (i.e., part of the IS disrupted or deleted) of the Burkholderia lineage. ISBma1, ISBma2, and IS407A also had degenerate elements in each species; the ISBma1 elements had the highest levels of degeneration (44%), followed by ISBma2 (20%), and by IS407A (5%) (Fig. 1A).
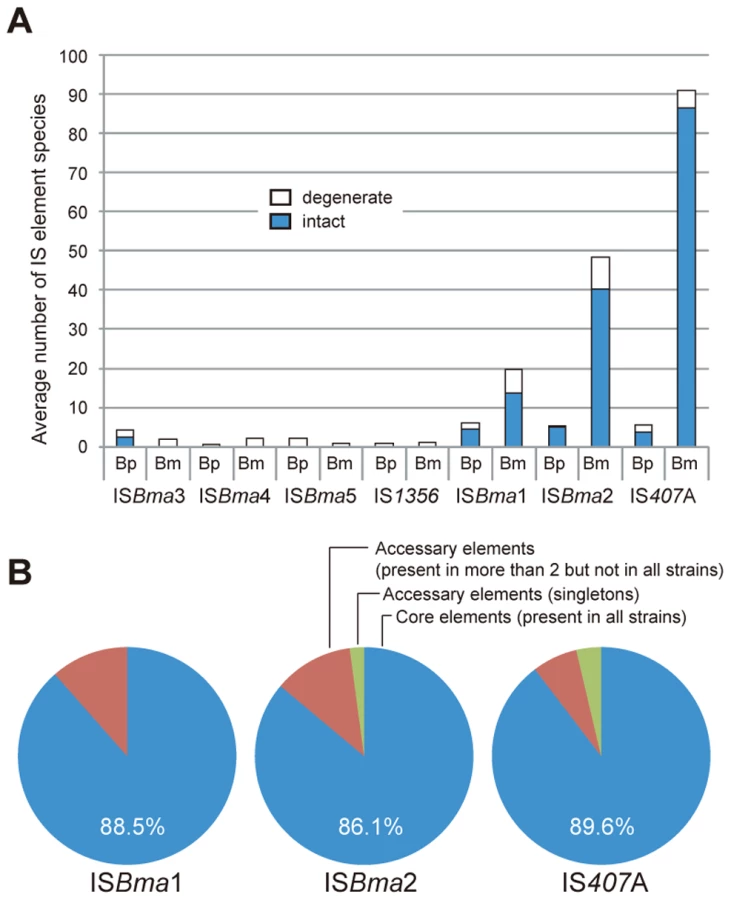
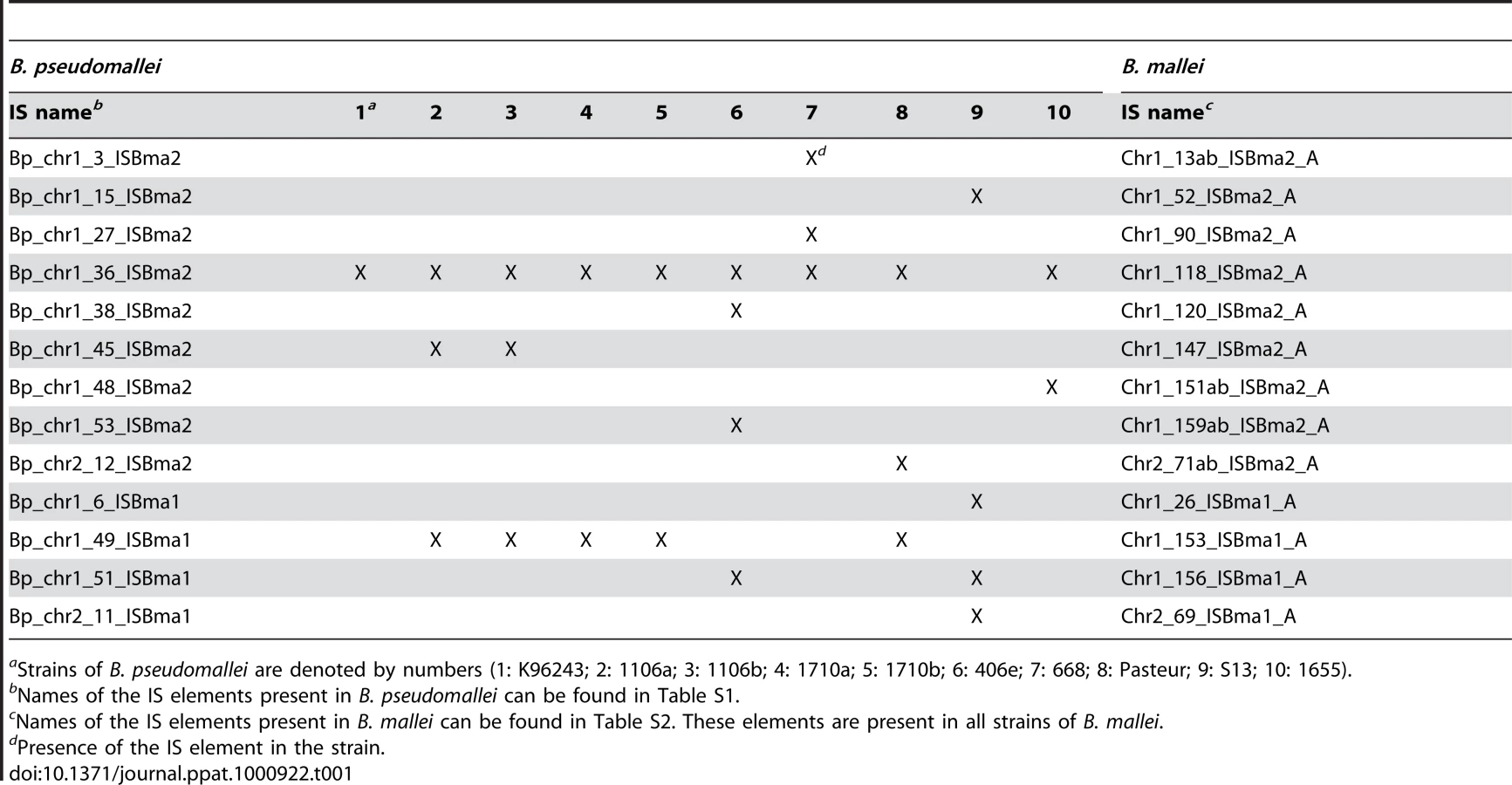
Intriguingly, up to almost 90% of ISBma1, ISBma2, and IS407A (88.5%, 86.1%, and 89.6%, respectively) were found to be present at the corresponding loci in all 10 B. mallei strains, when examined after the rearranged genomic fragments in each strain were aligned against a reference genome of B. pseudomallei K96243 (Fig. 1B; for a scaled map with the IS insertion sites in all B. mallei and B. pseudomallei strains, see Fig. S1; for the patterns of genomic rearrangements in the strains of each species, see Fig. S2; for the actual comparative blast data, see Tables S1 and S2). In contrast to these “core” elements, those elements that were not present in all (singletons and those found in a few strains), collectively referred to as “accessory” elements, were much less common. That the core elements, expected to be associated with the speciation of B. mallei from B. pseudomallei, accounted for most of the elements clearly reflects the common origin of B. mallei strains from a clone of B. pseudomallei [18], [20]. More importantly, it also suggests that further transpositions were significantly slowed after subsequent geographical segregation of the bacteria. There are 13 core elements in B. mallei that have matching IS elements located at the same sites in B. pseudomallei strains (Table 1). These elements were found to be composed of elements of ISBma1 and ISBma2 but not of IS407A. This finding suggests that ISBma1 and ISBma2 have a longer history of association with B. pseudomallei than IS407A does.
IS-mediated large genomic deletions and rearrangements
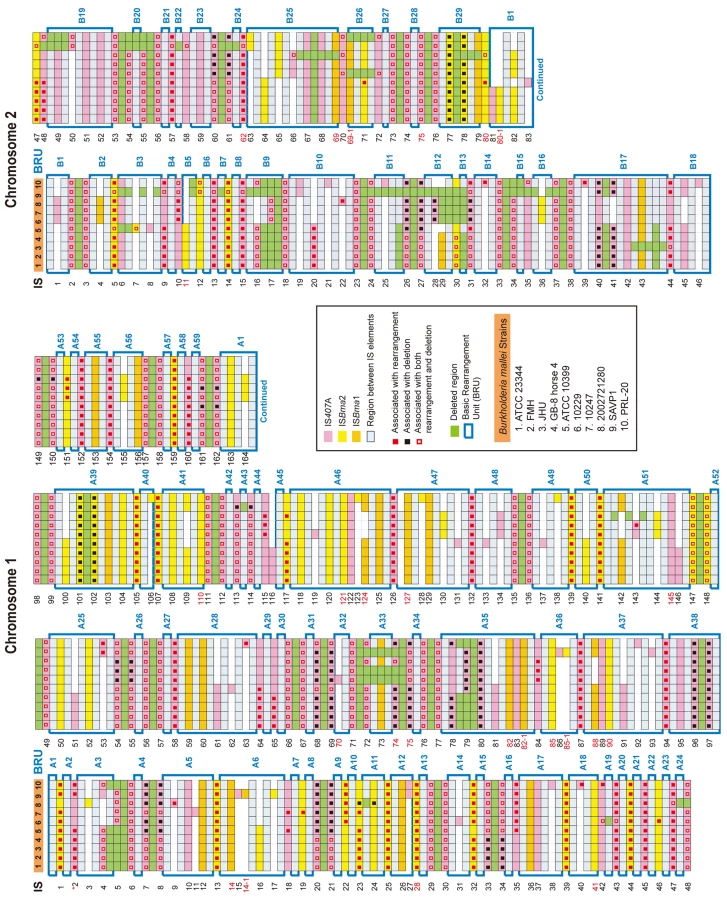
Among the three largely expanded elements, we found that IS407A and ISBma2 were associated with almost all of the large genomic deletions and rearrangements in the B. mallei strains (Fig. 2; Figs. S1 and S2; Tables S1 and S2). The only exception to this was a large deletion found in the strain ATCC 23344 and its direct derivatives, FMH, JHU, and GB8 horse 4 [24], between the 43rd and the 44th elements in chromosome 2 (Fig. 2; Table S1). No genomic rearrangement was mediated by features other than the two IS elements. ISBma1, which was significantly increased in B. mallei, was not directly involved in any of the genomic deletions or rearrangements, however as many as 35% of it served as secondary entry points for IS407A. The majority of the core elements of IS407A, 71.8% and 63.3% in chromosomes 1 and 2, respectively, mediated rearrangements, deletions, or both (Fig. 3A). By contrast, accessory elements of IS407A contributed less, but were more active in chromosome 2 than in chromosome 1. By contrast, 50.4% and 53.2% of the core elements of ISBma2 in chromosomes 1 and 2, respectively, contributed to rearrangements and/or deletions, and the accessory elements in both chromosomes were very rarely involved (Figs. 2 and 3). We identified 59 and 28 genomic fragments in chromosomes 1 and 2, respectively, which were encompassed by core elements of IS407A or ISBma2; these core elements mediated genomic rearrangements in at least one strain (Figs. 2; Table S1). We referred to these genomic fragments as BRUs (basic rearrangement units), a set of basic units for genomic reduction and rearrangement in B. mallei. The BRUs formed various rearrangement patterns in the B. mallei strains (Fig. S2A). By contrast, B. pseudomallei strains had little variation in genome arrangement among one another due to low levels of IS elements- a few rearrangements were found but were around non-IS repeat sequences (Fig. S2B).
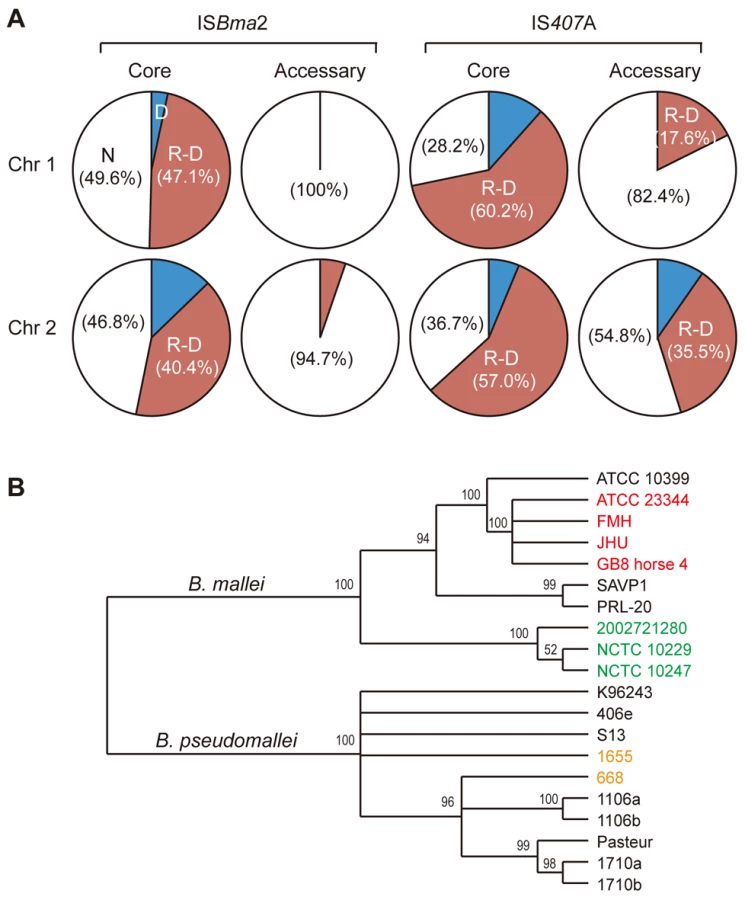
When the pattern of the IS insertions and their involvement in genome-reductive and rearrangement processes in strains were used to construct a phylogenetic tree, strains sharing a recent common ancestry (e.g., ATCC 23344 and its immediate derivative isolates, FMH, JHU, and GB8 horse 4) or common recent geographical origins (e.g., strains NCTC 10257, NCTC 10229, and 2002721280 from European countries) were grouped together (Fig. 3B). This phylogenic relationship supports the hypothesis that the accessory IS elements, which provided the major determinants for the tree rather than the common core elements, occurred following the speciation and geographical segregation of the B. mallei strains. By contrast, such patterns were not obvious among the B. pseudomallei strains which did not go through IS element expansions; Australian strains 1655 and 668 did not branch separately from the South Asian strains.
The deletions and rearrangements that were mediated by accessory elements were most frequently noted in strains SAVP1 and 2002721280, which lost virulence after successive passages in laboratory cultures [25] (Figs. 2 and S1). Most of the extra deletions in these strains were more prominent in chromosome 2 than in chromosome 1. In SAVP1, an IS407A-mediated deletion removed a major group of virulence genes encoding the animal-type type III secretion system in the BRU B22 (Figs. 2 and S1); this deletion may be a major cause of the avirulence of that strain. By contrast, there is no obvious deletion that may be responsible for the loss of virulence in strain 2002721280. That the strains SAVP1 and 2002721280 obtained deleterious mutations from in vitro culturing suggests that maintenance of the genomic contents in B. mallei requires selective pressure for survival in the host environment. By contrast, the fully virulent strain PRL-20 showed more frequent deletions and rearrangements mediated by accessory elements than other virulent strains. This strain may represent one of the more evolved (more genome-reduced) strains of B. mallei.
Although extra deletions and rearrangements were noted, the actual number of the accessory IS elements was not significantly increased in PRL-20, SAVP1 or 2002721280. Furthermore, none of the direct derivatives of the strain ATCC 23344 (i.e. FMH, JHU, and GB8 horse 4) had new IS insertions (Fig. 2 and Table S1). These ATCC 23344 derivatives also did not have genomic rearrangements; the only change found was a single IS407A-mediated deletion located within the BRU B17 in the strain JHU (Fig. 2 and Table S1). These lines of evidence suggest that B. mallei genomes are structurally flexible with regard to deletions, however perhaps not as much anymore for additional IS transpositions or genomic rearrangements.
Different insertion target preferences
IS407A elements are known to generate 4-bp target region duplications as direct repeats around them when they transpose [26]. We found that ISBma1 generates 8-bp target region duplications, and that ISBma2 generates longer repeats of various lengths (18–26 bp) (Table 2; for the entire data, see Table S3). In addition to the various lengths of duplications, these target regions of the three types of IS had different nucleotide compositions and patterns. Most notably, the sequences of ISBma1 contained homopolymers of A and/or T in up to 8-bp stretches of nucleotides (Fig. 4A). The target sequences of ISBma2 had a loose pattern in which the GC-rich central region was encompassed by strands of As and Ts on either side. Target sequences of IS407A had the least characteristic composition. It is intriguing to note that each IS element showed different levels of copy number expansion, ISBma1 with the lowest (3.3×), ISBma2 with an increased level (9.5×), and IS407A with the highest (16.7×) (Fig. 1A). Perhaps this difference, at least in part, resulted from the availability of genomic sites suited for insertion targets.
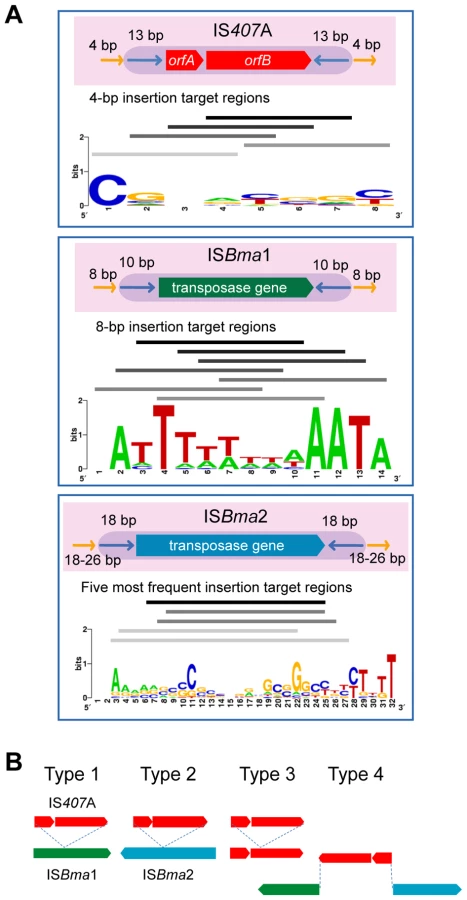
There were concordant patterns of disruption of the core elements of one type by another, in that ISBma1 and ISBma2 were intersected by transposed IS407A (Fig. 4B), while the reverse (IS407A disrupted by ISBma1 or ISBma2) was not found. A possible explanation for these insertion patterns may be that ISBma1 and ISBma2 could not transpose into IS407A due to the lack of sites suited for their rather uncommon target preferences, while IS407A did not have this problem. Consistent with this hypothesis, ISBma1 and ISBma2 also did not have self-disrupted elements, while there were several self-disrupted IS407A elements. The involvement of the three IS elements with different target sites increased the total number of IS insertions in the genome. Furthermore, this increase led to further spread of IS407A, because ISBma1 and ISBma2 provided neutral insertion points for the element. This in turn directly improved the efficiency of IS407A-mediated recombination in the genome, resulting in more sophisticated deletions and rearrangements.
We estimated that 83.7% of IS407A and 65.6% of ISBma2 elements in the B. mallei genomes lost their matching target duplicates, while all of the elements from intact ISBma1 elements were maintained (Table S3). Almost all of the IS407A (see Table S3 for details) and all of the ISBma2 elements that contained matching repeats were not involved in genomic rearrangements in B. mallei. This indicates that recombination among the elements were the major cause of the loss of the matching target duplicates.
Nucleotide-level mutations in B. mallei
B. mallei still has a high nucleotide-level identity (99%) to B. pseudomallei. Consistent with this, there was no AT-biased genome deviation in B. mallei, unlike that seen in many old symbionts or obligatory host-associated pathogens [1], [3]. Although the overall identity is still very high, significant nucleotide-level divergence exists, especially at the SSRs (simple sequence repeats), where there are intrinsically high mutation rates [27]. These SSRs were abundant in both B. mallei and B. pseudomallei at corresponding sites in the genomes. However, there were more genes that were disrupted by frameshift mutations in B. mallei compared to B. pseudomallei (Table S4). Most of these disrupted genes were commonly present in all B. mallei strains, reflecting the clonal origin of the strains. Some of these gene disruptions may have contributed to better adaptation of the bacteria (increased persistence) in the host environment or simply became obsolete [28]. One of the most characteristic loss of function or of surface structure in B. mallei is the loss of flagella [20]. A gene essential for flagellum biogenesis, fliP, [29] in the strain ATCC23344 was disrupted by a 65-kb fragment flanked by IS407A elements, and this mutation completely turned B. mallei flagella-less. This disruption in fliP is present in all B. mallei strains (Table S1; Fig. 2, between BRUs A2 and A3), implying the significance of losing flagella in the evolution of host-restricted B. mallei. The loss of flagella has been noted in other bacteria, including Bordetella pertussis and Bordetella parapertussis during their host specialization, derived from the strains of Bordetella bronchiseptica, [9] and Yersinia pestis during its conversion from a gut to a systemic pathogen [30]. Additional disrupted genes not present in all strains were found at approximately the same levels as in B. pseudomallei, suggesting that there were no significant increases in mutation rates in B. mallei after geographical segregation. There also was no significant level of erosion of these, so called, pseudogenes by purifying selection at levels high enough to contribute to the actual genome size reduction (data not shown).
Genomic potential for gene expression divergence
The extensiveness of the genome-wide reduction and rearrangements as well as additional nucleotide-level mutations may suggest that there is a potential for altered gene expression patterns in B. mallei. A total of 341 potential regulatory genes survived the general IS-mediated genomic reduction in B. mallei (not taking into account the diverse strain-specific deletions that occurred after speciation). Among these genes, only a small fraction (about 10) in each strain had deleterious (e.g. frameshift, null, or IS-insertion) mutations (for the list of the genes, see Table S5). In addition, none of the predicted operons in B. mallei, which correspond to the putative operons previously found in B. pseudomallei K96243 [31], were disrupted by IS elements (data not shown). We also estimated the potential for changes in promoters. There were 2,473 upstream sequences of genes, many of which may overlap or contain promoters, in the reference genome of B. pseudomallei K96243 that have homologous sequences (with at least 95% identity over at least 95% of their lengths) in all other strains of B. pseudomallei. We found that up to 99% of these sequences also matched the corresponding regions in B. mallei ATTC23344 at the same homology levels (see Table S6 for the list of the 2,473 upstream sequences, associated gene information, and the blast data). Together, all these data from the analyses of the conserved genomic regions suggest that there is only a low potential for the genes in B. mallei to have significantly divergent gene expression patterns from B. pseudomallei.
By contrast, there were 56 genes with putative regulatory functions that were lost along with the commonly deleted genomic fragments of the B. mallei genome. These genes include potential global regulatory genes, such as those encoding a quorum-sensing system (genes BPSS1176 and BPSS1180 in the reference genome of B. pseudomallei K96243), a two-component regulatory system (the pair BPSS1994 and BpSS1995 in B. pseudomallei K96243), and a number of regulators of various families (Table S7). Whether the loss of any of these 56 regulatory genes affects the expression of the remaining genes in the B. mallei genome was yet to be examined.
Similar gene expression profiles in B. mallei and B. pseudomallei
To experimentally estimate the possible transcriptomic divergence between B. pseudomallei and B. mallei, we infected female BALB/c mice with B. mallei ATCC 23344 or B. pseudomallei K96243, employing the previously established aerosol models of acute glanders and melioidosis [32]. Gene expression was compared in the bacteria that colonized the lungs and the spleens of the mice. Both B. mallei- and B. pseudomallei-challenged animals showed increases in the bacterial loads within these organs over time, with B. pseudomallei having slightly faster growth rates (Fig. 5). In our experience, B. pseudomallei also grew faster than B. mallei in vitro (data not shown). Unlike the mice infected by B. mallei, sampling the B. pseudomallei-challenged animals after 72 hr was not possible due to animal mortality from the more rapid disease progression. When gene expression profiles in the spleens and lungs were compared between B. mallei and B. pseudomallei at middle- (i.e., 24 hr for both bacteria) and late stages (i.e., 48 hr for B. pseudomallei and 72 hr for B. mallei) of infection (a total of four comparison pairs), conserved B. mallei and B. pseudomallei orthologs showed nearly identical patterns with high Pearson correlation coefficient (R) values ranging from 0.94 to 0.97, regardless of the host tissue type (Fig. 5). Therefore, there was no indication of significant modifications of the expression schemes in the genes required by B. mallei to thrive in BALB/c mice compared to those in B. pseudomallei. This is consistent with the findings of our previous gene expression studies in culture and in vivo, which also showed similar gene expression patterns in B. mallei and B. pseudomallei [20], [33], [34], [35]. These data suggest that, during the early stage, genomic reduction proceeds conservatively, not seriously affecting the indigenous gene expression patterns. In contrast to B. mallei, most of the transcription units in the insect symbiont Buchnera were altered, most likely due to complex genomic alterations accumulated over a long period of time [2].
Conclusions
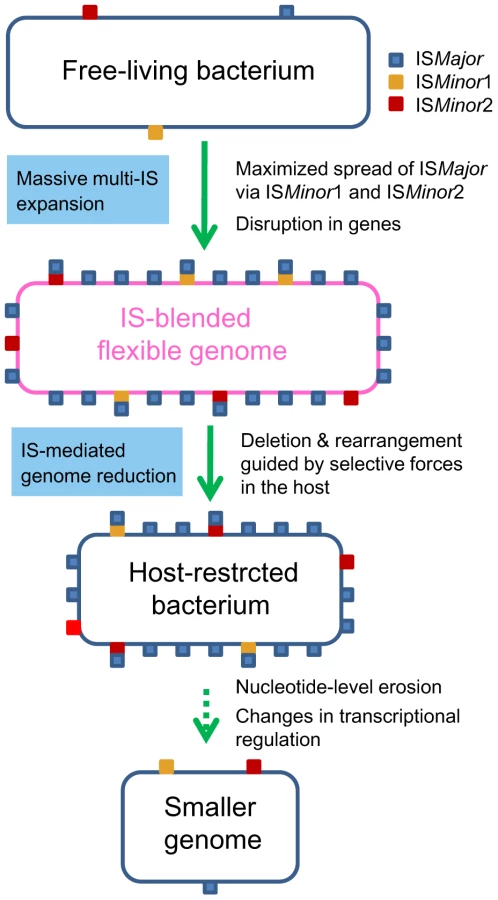
In this study, we unveiled the mechanics of genomic deletions and rearrangements that occur in the early stage of bacterial specialization in the host, by conducting comparative analyses of B. mallei and its parental species, B. pseudomallei. It became clear that stepwise IS intervention was the main driving force mediating a large genomic reduction in B. mallei. Expansion of ISBma1 and ISBma2 in a clone of B. pseudomallei set the stage for the wide spread of IS407A, allowing its proliferation to sites, to which the element itself may rarely target. Actual genomic deletions and rearrangements occurred through recombination reactions mainly among IS407A and also among ISBma2 (Fig. 2). These processes achieved highly efficient deletions of dispensable genomic regions, causing only small disruptions to the portions of the genome that were maintained. This was possible due to the guidance by selective forces in the host and via the intrinsic flexibility of the compactly IS-blended genome. The B. mallei genome currently appears to still be structurally flexible with regard to deletions but is now less flexible with regard to genomic rearrangements and additional transpositions. This may indicate that the genomic evolution in B. mallei has been moving into a second stage, in which large-scale genomic alterations are reduced and nucleotide-level erosion has become more important. On the other hand, a large number of genes disrupted by frameshift mutations in SSRs were found in the B. mallei genome. The loss of function encoded by these genes and of flagella via disruption in fliP by IS407A (Table S1), could be part of the adaptive evolution for survival in the host environment, which will eventually lead to genome size reduction by erosion over time. Widespread relics of IS elements found in diverse symbionts and obligate pathogens [1], [3], [8] clearly suggest that a similar sequential IS intervention, modeled in Figure 6, may illustrate a general mechanism, by which elaborate genome transition occurs during early bacterial evolution after establishing constant association with the host.
Materials and Methods
Ethics statement
All research involving live animals was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996. All mouse experiments conducted in the USAMRIID (US Army Medical Research Institute of Infectious Diseases) were approved by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Sequencing and annotation
The type strains for B. mallei (ATCC23344) [20] and B. pseudomallei (K96243) [21] were previously sequenced. Strains FMH, JHU, and GB8 horse 4 were direct derivatives of strain ATCC 23344 after passages in the human or horse, and these strains were also sequenced previously [24]. B. mallei strains NCTC10229, NCTC10247, and SAVP1 were sequenced with full closure and manually annotated as previously described [20]. The remaining three strains (2002721280, ATCC10399, and PRL-20) were sequenced to 8× Sanger sequence coverage by the whole genome shotgun method [36] without closure, and assembled using the Celera Assembler [37], and contigs were oriented by alignment to the reference strain ATCC23344 using PROMER [38]. ORFs were predicted and annotated automatically using GLIMMER [39], [40]. Pseudo-chromosomes were constructed from the ordered scaffolds, using manual examination where necessary. Similarly, B. pseudomallei strains 1106a, 1710b, and 668 were sequenced with full closure and manual annotation, while 1655, 406e, S13, and Pasteur 6068 were sequenced without closure and annotated automatically.
Comparative genomic analyses with B. mallei and B. pseudomallei
For the analyses of genomic deletions and rearrangements in B. mallei and B. pseudomallei, 5,799 predicted protein sequences from the B. pseudomallei type strain K96243 were compared with the nucleotide sequences of the genomes of B. mallei (ATCC 23344, 2002721280, ATCC 10399, FMH, JHU, GB8 horse 4, PRL-20, NCTC 10229, NCTC 10247, SAVP1) and the other strains of B. pseudomallei (1106a, 1106b, 1655, 1710a, 1710b, 406e, 668, Pasteur, S13) using tblastn (http://blast.wustl.edu). For the mapping of the insertions of ISBma1, ISBma2, and IS407A in the genomes of B. mallei and B. pseudomallei, the entire sequences of the IS elements were searched against the 20 genomes using blastn (http://blast.wustl.edu). For the analysis of association of the IS elements with genomic deletions and rearrangements in B. mallei and of the target sequences in the genomes, strain ATCC 23344 represented all of its immediate derivatives, FMH, JHU, and GB8 horse 4, to avoid redundancy in the data, because the three strains showed identical patterns. To compare the patterns of genome rearrangements in the B. mallei strains, the positions of the BRUs in each strain of B. mallei relative to B. pseudomallei K96243 were visualized using a genome-comparative software tool ACT ([41]; http://www.sanger.ac.uk/Software/ACT), and the displays were compared in parallel among the strains.
We also examined B. mallei and B. pseudomallei for intergenic regions that potentially containing promoters, putative regulatory genes, and disruptions of putative operons to estimate the possibility of causing gene expression divergence. For intergenic region comparisons, up to 100 bp upstream of the start codon, or up to as much as available if the neighboring gene was closer, of the genes that contain at least 50 bp of an untranslated upstream region were retrieved from the genome of B. pseudomallei K96243. Then, these sequences (2,268 and 1,566 from chromosomes 1 and 2, respectively) were searched against the genomes of B. mallei and B. pseudomallei using blastn (http://blast.wustl.edu), and the length-match as well as the identity values of the orthologous regions were calculated. Putative operons reported by Rodrigues et al. from the genome of B. pseudomallei K6243 [31] were used to match the orthologous gene clusters in the genome of B. mallei ATCC 23344, and these gene clusters were examined for any disruptions caused by IS elements. All the genome sequences of B. mallei and B. pseudomallei used in this study are available through the Pathema web site (http://pathema.jcvi.org/cgi-bin/Burkholderia/PathemaHomePage.cgi) at the J. Craig Venter Institute (http://www.jcvi.org/).
Construction of the phylogenetic tree
A phylogenetic tree was constructed with the strains of B. mallei and B. pseudomallei based on the insertion patterns of and the role played in the genomic deletions and rearrangements by the three major IS elements, ISBma1, ISBma2, and IS407A. All the data used are shown in Tables S1 and S2 and Figure 2. Bootstrapped maximum parsimony trees were calculated using the PAUP package with default parameters, and a consensus tree was produced from the bootstrap replicates. Branches with bootstrap scores of less than 50 were collapsed in the tree.
Determination of the target sequence patterns
Among the duplicated target regions encompassing the IS elements ISBma1, ISBma2, and IS407A, those regions that had perfectly matching sequences were first collected. Then, among the sequences from unmatched pairs, those that occurred in more than two strains were assumed to be un-mutated valid sequences and, therefore, were added to the data pool for the analysis. Strain ATCC 23344 represented all its direct derivatives (FMH, JHU, and GB8 horse 4) in this analysis to avoid redundancy in the data. The collected sequences were aligned with Clustal X, and the alignments were graphically visualized using Sequence logos [42].
Mouse infection, bacterial load estimation, and RNA preparation
Exposure of mice to bacterial aerosol was performed as described by Roy et al. [43]. Fresh overnight cultures of B. pseudomallei DD503 [44] and B. mallei ATCC 23344 were prepared in LB or in LBG (LB supplemented with 4% glycerol), respectively, at 37°C with aeration (250 rpm). Thirty female BALB/c mice six to eight weeks old (National Cancer Institute, Frederick, MD, USA) were infected with these bacteria: nine mice each with B. pseudomallei and B. mallei for the gene expression studies, and six mice each for the bacterial load assays. The mice infected with B. mallei received an inhaled dose of 7.2×103 cfu (7.2×LD50), and those mice infected with B. pseudomallei received 1.8×104 cfu (18×LD50), as estimated by colony counting on agar plates. The infected mice were provided with rodent feed and water ad libitum and maintained on a 12-hr light cycle. After 24 and 48 hr (for both B. mallei and B. pseudomallei) or 72 hr (for B. mallei) of infection, five mice from each point in time were euthanized in a CO2 chamber, and their spleens and lungs were removed. Due to animal mortality, a 72 hr point in time was not possible for B. pseudomallei. The organs from two randomly picked mice were saved for bacterial load estimations, and the rest were homogenized in 1 ml of Trizol (Invitrogen Corp., Carlsbad, CA, USA) using a Tissue-Tearor (BioSpec Products, Bartlesville, OK, USA). Total RNA was purified according to the manufacturer's recommendations (Invitrogen Corp., Carlsbad, CA, USA). The bacterial load in the mouse organs was estimated as described by Ulrich and DeShazer [32].
RNA labeling and microarray analysis
Total RNA, both bacterial and mouse, from the same organ types from three mice was pooled to compensate for potential individual variation. These pooled RNA samples were used for the experiments without further purification of the bacterial RNA because RNA from mice does not cross-hybridize to the B. mallei microarray at a level affecting the legitimate interactions between the B. mallei array and the Burkholderia transcriptome [35]. The B. mallei whole genome array used in this study for both B. mallei and the closely related B. pseudomallei (average gene identity at the nucleotide level of 99%) was described in detail previously [33]. The B. mallei- and B. pseudomallei-infected organ samples were paired for the hybridization reactions based on early and late pathological states. A total of eight hybridization reactions or four different comparisons were performed, each of which was replicated in flip-dye pairs and the final ratios were calculated as log2 (B. pseudomallei gene expression intensity/B. mallei gene expression intensity). Labeling of the probes, slide hybridization, and slide scanning were carried out as previously described [35]. The independent TIFF slide images from each channel were analyzed using TIGR Spotfinder to assess the relative expression levels, and the data were normalized using a local regression technique LOWESS (LOcally WEighted Scatterplot Smoothing) with the MIDAS software (<http://www.jcvi.org/cms/research/software>, The J. Craig Venter Institute, Rockville, MD, USA). The resulting data were averaged from triplicate genes on each microarray and from duplicate flip-dye arrays for each experiment.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. MoranNA
2003 Tracing the evolution of gene loss in obligate bacterial symbionts. Current Opinion in Microbiology 6 512 518
2. MoranNA
MiraA
2001 The process of genome shrinkage in the obligate symbiont Buchnera aphidicola. Genome Biology 2 research0054.0051 0054.0012
3. MoranNA
PlagueGR
2004 Genomic changes following host restriction in bacteria. Current Opinion in Genetics & Development 14 627 633
4. NilssonAI
KoskiniemiS
ErikssonS
KugelbergE
HintonJCD
2005 Bacterial genome size reduction by experimental evolution. Proc Natl Acad Sci U S A 102 12112 12116
5. SallstromB
AnderssonSGE
2005 Genome reduction in the [alpha]-Proteobacteria. Current Opinion in Microbiology 8 579 585
6. BatutJ
AnderssonSGE
O'CallaghanD
2004 The evolution of chronic infection strategies in the [alpha]-proteobacteria. Nat Rev Micro 2 933 945
7. MoranNA
McLaughlinHJ
SorekR
2009 The Dynamics and Time Scale of Ongoing Genomic Erosion in Symbiotic Bacteria. Science 323 379 382
8. MiraA
PushkerR
Rodríguez-ValeraF
2006 The Neolithic revolution of bacterial genomes. Trends in Microbiology 14 200 206
9. ParkhillJ
SebaihiaM
PrestonA
MurphyLD
ThomsonN
2003 Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35 32 40
10. TreangenTJ
AbrahamA-L
TouchonM
RochaEPC
2009 Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiology Reviews 33 539 571
11. DanceD
2000 Ecology of Burkholderia pseudomallei and the interactions between environmental Burkholderia spp. and human-animal hosts. Acta Trop 74 159 168
12. DharakulT
SongsivilaiS
1999 The many facets of melioidosis. Trends Microbiol 7 138 140
13. McGilvrayC
1944 The transmission of glanders from horse to man. Can J Public Health 35 268 275
14. BenensonA
1995 Control of Communicable Diseases Manual Washington, DC American Public Health Association
15. DeShazerD
WaagD
2004 Glanders: New Insights into an Old Disease.
LindlerL
LebedaF
KorchGW
Biological Weapons Defense: Infectious Diseases and Counterbioterrorism The Humana Press Inc 209 237
16. ChengAC
CurrieBJ
2005 Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev 18 383 416
17. InglisTJJ
SagripantiJL
2006 Environmental factors that affect the survival and persistence of Burkholderia pseudomallei. Appl Environ Microbiol 72 6865 6875
18. GodoyD
RandleG
SimpsonA
AanensenD
PittT
2003 Multilocus sequence typing and evolutionary relationships among the causative agents of Melioidosis and Glanders, Burkholderia pseudomallei and Burkholderia mallei. J Clin Microbiol 41 2068 2079
19. LinCH
BourqueG
TanP
2008 A Comparative Synteny Map of Burkholderia Species Links Large-Scale Genome Rearrangements to Fine-Scale Nucleotide Variation in Prokaryotes. Mol Biol Evol 25 549 558
20. NiermanWC
DeShazerD
KimHS
TettelinH
NelsonKE
2004 Structural flexibility in the Burkholderia genome. Proc Natl Acad Sci USA 101 14246 14251
21. HoldenMTG
TitballRW
PeacockSJ
Cerdeño-TárragaAM
AtkinsT
2004 Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci USA 101 14240 14245
22. WilkinsonL
1981 Glanders: medicine and veterinary medicine in common pursuit of a contagious disease. Med Hist 25 363 384
23. WhitlockGC
EstesDM
TorresAG
2007 Glanders: off to the races with Burkholderia mallei. FEMS Microbiol Lett 277 115 122
24. RomeroC
DeShazerD
FeldblyumT
RavelJ
WoodsD
2006 Genome sequence alterations detected upon passage of Burkholderia mallei ATCC 23344 in culture and in mammalian hosts. BMC Genomics 7 228
25. SchutzerSE
SchlaterLR
RonningCM
DeShazerD
LuftBJ
2008 Characterization of clinically-attenuated Burkholderia mallei by whole genome sequencing: candidate strain for exclusion from Select Agent lists. PLoS ONE 3 e2058
26. DeShazerD
WaagDM
FritzDL
WoodsDE
2001 Identification of a Burkholderia mallei polysaccharide gene cluster by subtractive hybridization and demonstration that the encoded capsule is an essential virulence determinant. Microbial Pathogenesis 30 253 269
27. LevinsonG
GutmanGA
1987 Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol 4 203 221
28. DeitschKW
LukehartSA
StringerJR
2009 Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat Rev Micro 7 493 503
29. MalakootiJ
ElyB
MatsumuraP
1994 Molecular characterization, nucleotide sequence, and expression of the fliO, fliP, fliQ, and fliR genes of Esherichia coli. J Bacteriol 176 189 197
30. ParkhillJ
WrenBW
ThomsonNR
TitballRW
HoldenMTG
2001 Genome sequence of Yersinia pestis, the causative agent of plague. Nature 413 523 527
31. RodriguesF
Sarkar-TysonM
HardingSV
SimS-H
ChuaH-H
2006 Global map of growth-regulated gene expression in Burkholderia pseudomallei, the causative agent of melioidosis. J Bacteriol 188 8178 8188
32. UlrichRL
DeShazerD
2004 Type III secretion: a virulence factor delivery system essential for the pathogenicity of Burkholderia mallei. Infect Immun 72 1150 1154
33. TuanyokA
KimHS
NiermanWC
YuY
DunbarJ
2005 Genome-wide expression analysis of iron regulation in Burkholderia pseudomallei and Burkholderia mallei using DNA microarrays. FEMS Microbiol Lett 252 327 335
34. MooreRA
Reckseidler-ZentenoS
KimH
NiermanB
YuY
2004 Contribution of gene loss to the pathogenic evolution of Burkholderia pseudomallei and Burkholderia mallei. Infect Immun 72 4172 4187
35. KimH
SchellMA
YuY
UlrichRL
SarriaSH
2005 Bacterial genome adaptation to niches: Divergence of the potential virulence genes in three Burkholderia species of different survival strategies. BMC Genomics 6 174
36. FleischmannRD
AdamsMD
WhiteO
ClaytonRA
KirknessEF
1995 Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269 496 512
37. MyersEW
SuttonGG
DelcherAL
DewIM
FasuloDP
2000 A whole-genome assembly of Drosophila. Science 287 2196 2204
38. DelcherAL
PhillippyA
CarltonJ
SalzbergSL
2002 Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res 30 2478 2483
39. DelcherAL
HarmonD
KasifS
WhiteO
SalzbergSL
1999 Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27 4636 4641
40. SalzbergSL
DelcherAL
KasifS
WhiteO
1998 Microbial gene identification using interpolated Markov models. Nucleic Acids Res 26 544 548
41. CarverTJ
RutherfordKM
BerrimanM
RajandreamM-A
BarrellBG
2005 ACT: the Artemis comparison tool. Bioinformatics 21 3422 3423
42. SchneiderTD
StephensRM
1990 Sequence logos: a new way to display consensus sequences. Nucl Acids Res 18 6097 6100
43. RoyCJ
HaleM
HartingsJM
PittL
DunihoS
2003 Impact of inhalation exposure modality and particle size on the respiratory deposition of ricin in BALB/c mice. Inhal Toxicol 15 619 638
44. MooreRA
DeShazerD
ReckseidlerS
WeissmanA
WoodsDE
1999 Efflux-mediated aminoglycoside and macrolide resistance in Burkholderia pseudomallei. Antimicrob Agents Chemother 43 465 470
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 5
- Stillova choroba: vzácné a závažné systémové onemocnění
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Diagnostika virových hepatitid v kostce – zorientujte se (nejen) v sérologii
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
Nejčtenější v tomto čísle
- Quorum Sensing Inhibition Selects for Virulence and Cooperation in
- The Role of Intestinal Microbiota in the Development and Severity of Chemotherapy-Induced Mucositis
- Susceptibility to Anthrax Lethal Toxin-Induced Rat Death Is Controlled by a Single Chromosome 10 Locus That Includes
- Demonstration of Cross-Protective Vaccine Immunity against an Emerging Pathogenic Ebolavirus Species