
Insights into the Genomic Landscape: Comparative Genomics Reveals Variations in Ploidy and Nutrient Utilisation Potential amongst Wine Isolates
The yeast Dekkera bruxellensis is a major contaminant of industrial fermentations, such as those used for the production of biofuel and wine, where it outlasts and, under some conditions, outcompetes the major industrial yeast Saccharomyces cerevisiae. In order to investigate the level of inter-strain variation that is present within this economically important species, the genomes of four diverse D. bruxellensis isolates were compared. While each of the four strains was shown to contain a core diploid genome, which is clearly sufficient for survival, two of the four isolates have a third haploid complement of chromosomes. The sequences of these additional haploid genomes were both highly divergent from those comprising the diploid core and divergent between the two triploid strains. Similar to examples in the Saccharomyces spp. clade, where some allotriploids have arisen on the basis of enhanced ability to survive a range of environmental conditions, it is likely these strains are products of two independent hybridisation events that may have involved multiple species or distinct sub-species of Dekkera. Interestingly these triploid strains represent the vast majority (92%) of isolates from across the Australian wine industry, suggesting that the additional set of chromosomes may confer a selective advantage in winery environments that has resulted in these hybrid strains all-but replacing their diploid counterparts in Australian winery settings. In addition to the apparent inter-specific hybridisation events, chromosomal aberrations such as strain-specific insertions and deletions and loss-of-heterozygosity by gene conversion were also commonplace. While these events are likely to have affected many phenotypes across these strains, we have been able to link a specific deletion to the inability to utilise nitrate by some strains of D. bruxellensis, a phenotype that may have direct impacts in the ability for these strains to compete with S. cerevisiae.
Published in the journal:
. PLoS Genet 10(2): e32767. doi:10.1371/journal.pgen.1004161
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004161
Summary
The yeast Dekkera bruxellensis is a major contaminant of industrial fermentations, such as those used for the production of biofuel and wine, where it outlasts and, under some conditions, outcompetes the major industrial yeast Saccharomyces cerevisiae. In order to investigate the level of inter-strain variation that is present within this economically important species, the genomes of four diverse D. bruxellensis isolates were compared. While each of the four strains was shown to contain a core diploid genome, which is clearly sufficient for survival, two of the four isolates have a third haploid complement of chromosomes. The sequences of these additional haploid genomes were both highly divergent from those comprising the diploid core and divergent between the two triploid strains. Similar to examples in the Saccharomyces spp. clade, where some allotriploids have arisen on the basis of enhanced ability to survive a range of environmental conditions, it is likely these strains are products of two independent hybridisation events that may have involved multiple species or distinct sub-species of Dekkera. Interestingly these triploid strains represent the vast majority (92%) of isolates from across the Australian wine industry, suggesting that the additional set of chromosomes may confer a selective advantage in winery environments that has resulted in these hybrid strains all-but replacing their diploid counterparts in Australian winery settings. In addition to the apparent inter-specific hybridisation events, chromosomal aberrations such as strain-specific insertions and deletions and loss-of-heterozygosity by gene conversion were also commonplace. While these events are likely to have affected many phenotypes across these strains, we have been able to link a specific deletion to the inability to utilise nitrate by some strains of D. bruxellensis, a phenotype that may have direct impacts in the ability for these strains to compete with S. cerevisiae.
Introduction
Dekkera (Brettanomyces) bruxellensis has been described in the population ecology of various fermented beverages, such as wine, beer and cider [1]–[3], and is of increasing relevance to the biofuel industry [4]. Recent genomic sequencing of this species is beginning to reveal the mechanisms by which it is able to survive the harsh environment of alcoholic fermentation, primarily through gene-family expansions in membrane transporters and oxidoreductase enzyme classes that are predicted to facilitate nutrient scavenging and maintain redox homeostasis respectively [5]. However, our understanding of how other industrially important traits have evolved in D. bruxellensis lags well behind what is known for S. cerevisiae [6].
In general, D. bruxellensis utilises a make-accumulate-consume strategy similar to that found in S. cerevisiae [7], however traits, including carbon and nitrogen source utilisation [8], vary considerably between D. bruxellensis strains. For example, it was recently shown that nitrate utilisation enables D. bruxellensis to out-compete S. cerevisiae in continuous industrial fermentations [9], and key genes involved in nitrate assimilation were found in a cluster in the partial genome sequence of D. bruxellensis strain CBS2499 [10]. Nonetheless, nitrate utilisation is not a defining feature of this species. Nearly one third of D. bruxellensis isolates from a range of sources do not grow on nitrate as a sole nitrogen source [8]; presumably nitrate assimilation is less important for D. bruxellensis in some fermentation ecosystems. In another recent study, variation in sulphite tolerance in D. bruxellensis was linked to amplified fragment length polymorphism (AFLP) and 26S rDNA genetic markers [11], inferring a genetic basis for previously reported regional variation and groupings of this yeast across Australian wineries [1].
To date, de-novo assemblies exist for genomes of two D. bruxellensis wine isolates, AWRI1499 [5] and CBS2499 [12], from Australia and France, respectively. Unlike AWRI1499, CBS2499 has the same 26S rDNA sequence as the D. bruxellensis type strain CBS 74 (unpublished data), a lambic-beer isolate. This data, in combination with AFLP genotyping [1], infers the two sequenced strains are likely to be highly divergent. AWRI1499, a representative isolate of a sulphite-tolerant genotype group [11], is an allotriploid comprising a moderately heterozygous diploid, and divergent haploid complements [5]. Thus far it remains unclear to what degree intra-specific differences observed using methods such as AFLP may simply reflect presence or absence of all or part of the divergent haploid genome found in AWRI1499. CBS2499 was assembled as a pseudo-haploid [12], preventing such comparison with other de-novo assemblies.
To improve our understanding of genome diversity amongst D. bruxellensis wine isolates and gain insights into the evolution of industrially relevant traits in this important microorganism, we have performed mapping-assemblies of CBS2499 and two newly sequenced Australian D. bruxellensis wine strains against the reference genome sequence of AWRI1499. Comparative genomics of the four strains reveals that presence of a divergent haploid genome is not a feature restricted to AWRI1499, but has arisen through at least two independent ‘hybridisation’ events. In addition to large-scale ploidy variation, gene conversion and allelic expansion appear to be key molecular mechanisms driving strain divergence. Some phenotypes, such as nitrate/nitrite utilisation, on the other hand, are determined by genomic insertions and deletions (InDels).
Results
Analysis of Dekkera genomes
In order to compare the genomic complements of D. bruxellensis strains, a re-sequencing strategy was used to align genome data from short-read sequencing (2×100 bp) for three strains against the published draft genome assembly of D. bruxellensis strain AWRI1499 [5]. Two of the assemblies were for strains sequenced specifically for this work; AWRI1608 and AWRI1613. For the third, CBS2499, comparable data (2×100 bp format genome data) used as part of the D. bruxellensis CBS2499 draft genome assembly [12] was obtained from the NCBI short read archive. The two newly sequenced strains were chosen because they have divergent AFLP genotypes and, with AWRI1499 represent 98% of D. bruxellensis isolates associated with Australian wineries [1]. In all, the divergence between all four strains, as determined from AFLP analysis, is considerable and therefore should provide insights into the genomic landscape of wine isolates of this important yeast.
Given the unusual nature of the D. bruxellensis AWRI1499 genome (triploid hybrid comprised of a closely related diploid set of alleles and a third distantly related genomic complement), it was of interest to determine whether this genomic organisation is a defining characteristic of this species. Sequence alignments were therefore interrogated globally to determine genomic ploidy and the levels of both inter - and intra-allelic genetic diversity (Fig. 1, Datasets S1 and S2).
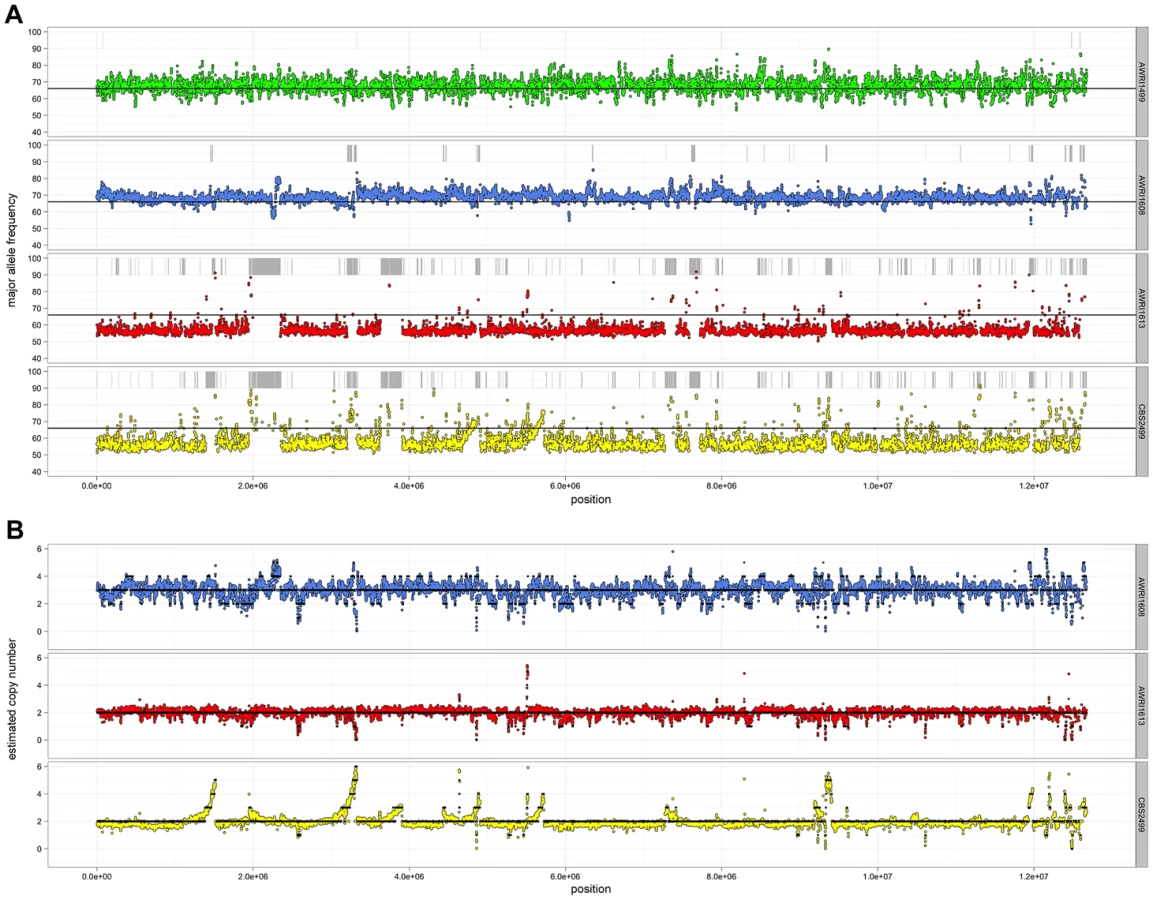
Ploidy levels across the genomes were estimated by taking advantage of allele proportions. In a triploid genome, it is expected that the maximum average frequency of a particular allele at a heterozygous site will be approximately 0.66 (due to a base difference in a single allele), while this number will be closer to 0.5 for heterozygous sites in a diploid. The observed average major allele frequency was therefore calculated across the entire genome of each isolate using a sliding window approach (Fig. 1A). As a triploid control, RNA-seq data for AWRI1499 was mapped to the AWRI1499 genome and this showed a maximum average allele proportion consistent with its triploid state (0.68±0.04, data not shown). AWRI1608 also displayed an average allele proportion (0.69±0.03), consistent with this isolate being triploid. However, both CBS2499 (0.58±0.05) and AWRI1613 (0.57±0.03) displayed maximum allele frequencies consistent with these isolates being diploid.
For all strains, while the average maximum allele frequency approximated to either 0.66 or 0.5 there were many localised regions that differed from these values, including significant portions of the genomes that displayed loss of heterozygosity (LOH; 17.9% of AWRI1613, 16.3% of CBS2499 and 3% of AWRI1608) (Fig. 1A, Fig. 2). As these differences in local allele proportions may be due to copy number variation (CNV), such as heterozygous deletions or genomic duplications, CNV was also determined globally for each of the genomes (Fig. 1B). While copy number was relatively stable, there were many instances of localised copy number variation in each strain, and of opposing copy number changes in the same genomic region between strains (Fig. 2A). Copy number amplification was especially prominent in CBS2499 with several genomic regions displaying effective copy numbers of 4 n or greater (Fig. 1B, Fig. 2B). Regions of increased copy number in CBS2499 appear to coincide with the ends of genomic scaffolds in both the AWRI1499 and CBS2499 [12] assemblies (Fig. S1), a feature not described in the CBS2499 de-novo assembly. This may be indicative of sub-telomeric amplification of sequences, which is common to other yeasts including S. cerevisiae and Cryptococcus neoformans [13], [14]. Examination of the functional annotation for the genes in these expanded regions revealed a statistically significant enrichment for those encoding proteins involved in carbohydrate metabolic processes (p = 6.5×10-7) and may therefore indicate adaptation to utilisation of specific carbon sources by this strain.
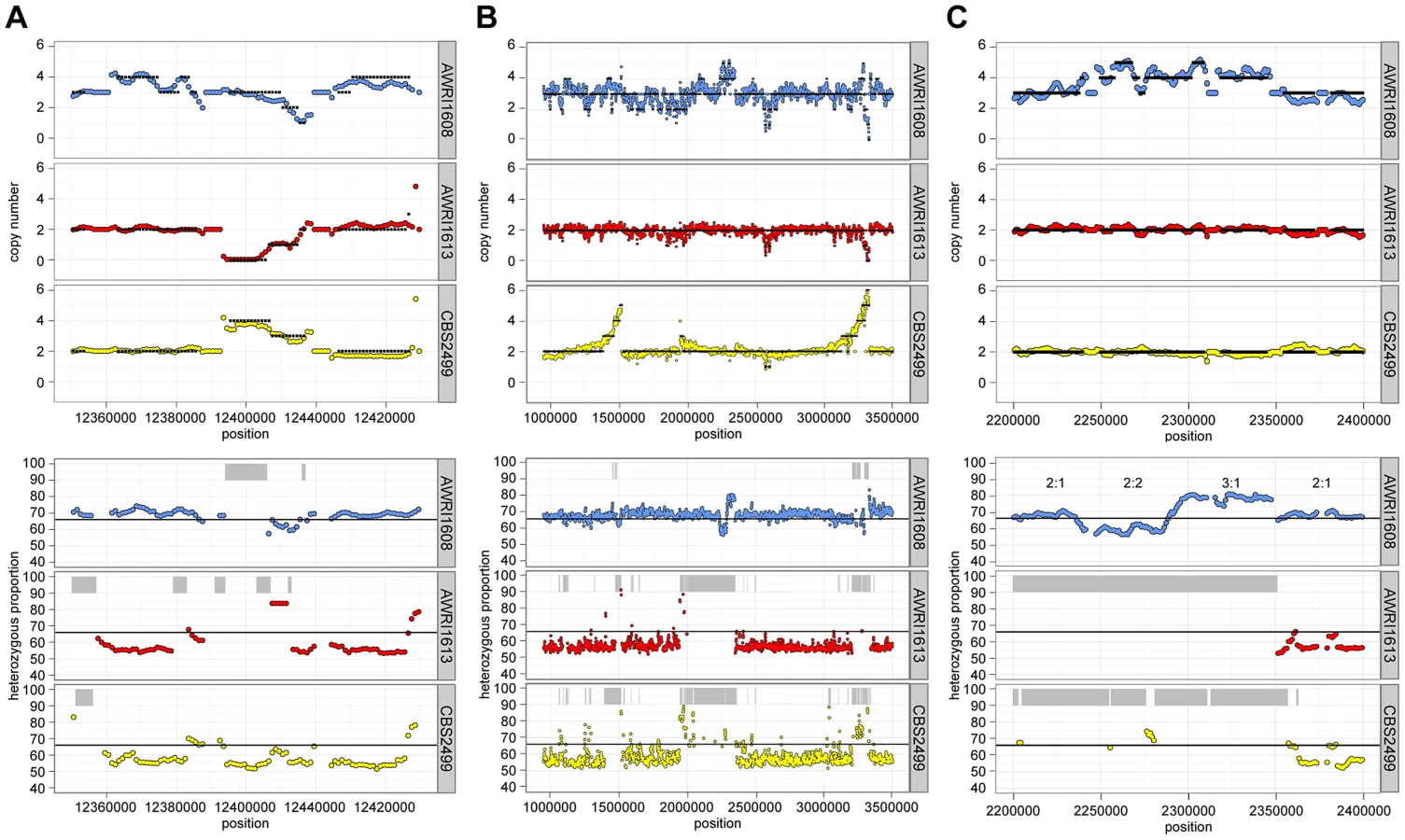
It was also apparent that there was co-localisation of copy number variation and alterations in allele frequencies (including the majority of LOH events). This is consistent with gene conversion, rather than heterozygous deletion, being responsible for the majority (95%) of genomic regions displaying LOH across the strains. For example, Fig. 2C describes a 100 kb genomic locus in which loss of heterozygosity has occurred in AWRI1613 and CBS2499 without altering the normal diploid genomic complement of these strains, while in AWRI1608, the otherwise triploid state is predicted to have been amplified to a tetraploid complement. Interestingly, this amplification in AWRI1608 is complicated by the fact that the allelic ratios change from 2∶2 to 3∶1 within the amplified region. This change in allelic ratios is indicative of either gene conversion of one allele following amplification of the region, or two amplification events that each amplified adjacent parts of the region carrying different homologs.
Allelic relationship across Dekkera isolates
It has been shown previously that the allotriploid genome of AWRI1499 consists of two highly related sets of chromosomes in addition to a third, more distantly related, set [5]. As the genomic analyses of AWRI1608, AWRI1613 and CBS2499 predicted only one of these strains to also be a triploid, it was of interest to determine the relationship between each of the haplotypes across all of the strains in order to ascertain whether either of the diploid strains contained the “divergent” haplotype of AWRI1499. Seven loci that displayed three clearly defined haplotypes in AWRI1499 [5] were selected with individual haplotypes derived for each locus in each strain by taking advantage of co-occurring SNPs within individual reads. This resulted in a total of ten possible haploid sequences (3+3+2+2) for each locus for which maximum-likelihood phylogenies [15], [16] were constructed (Fig. 3, Dataset S3). Consistent with whole genome alignments (Fig. S2), AWRI1613 and CBS2499 alleles were identical for five of seven loci, and exhibited only minor differences for the remaining two (AWRI1499_1134 and AWRI1499_1822). Furthermore, in the majority of cases, the phylogeny was resolved into a relationship whereby two of the alleles from AWRI1499 and AWRI1608 and both alleles from AWRI1613 and CBS2499 formed a highly related clade, while the third alleles from AWRI1499 and AWRI1608 were both divergent from this conserved clade and also distinct from one another. For the remaining two loci, it appears likely that gene conversion has resulted in either one or both of these divergent alleles being replaced by an allele that is consistent with the conserved clade.
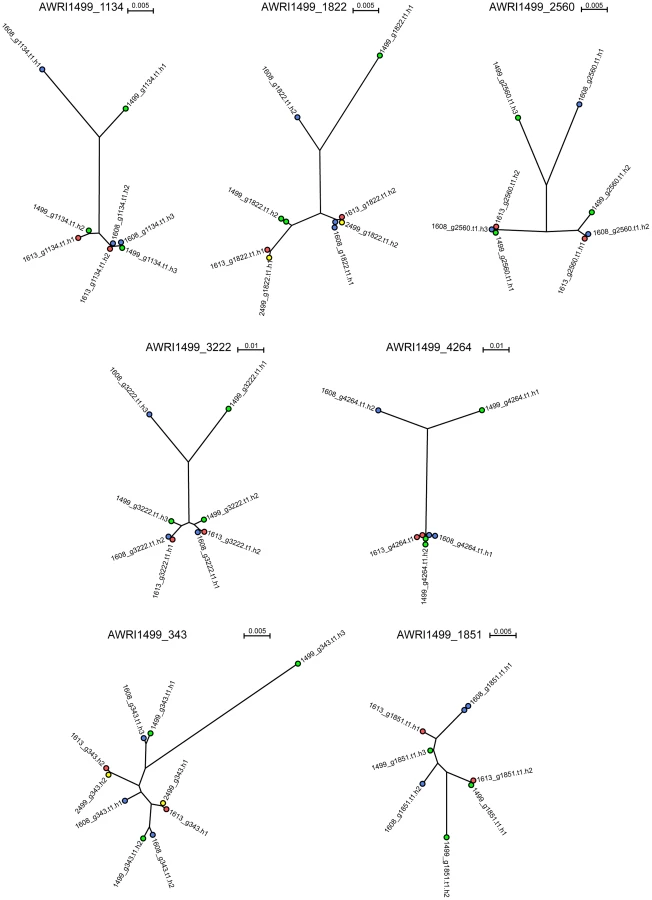
Maximum likelihood phylogenies for individual haplotypes derived at five additional genomic loci (Fig. S3, Dataset S3), previously sequenced for a collection of international D. bruxellensis strains [17], revealed similar topologies. Together with a 26S rDNA phylogeny (Fig. S4), these data provide evidence that predominant Australian D. bruxellensis strain AWRI1499 [1] is similar to South African wine-related D. bruxellensis strains CBS4481 (Y900) and CBS5206 (Y908). Of note, the DbHAD phylogeny (Fig. S3E) suggested that the horizontal transfer of an adenyl deaminase from an unknown Proteobacterial species [10] occurred prior to divergence of the AWRI1499 and AWRI1608 ‘third haplotype’ donors. A protein-based phylogeny (Fig. S5) suggests that DbHAD1 may have descended vertically from the common progenitor of D. bruxellensis and Ogataea parapolymorpha.
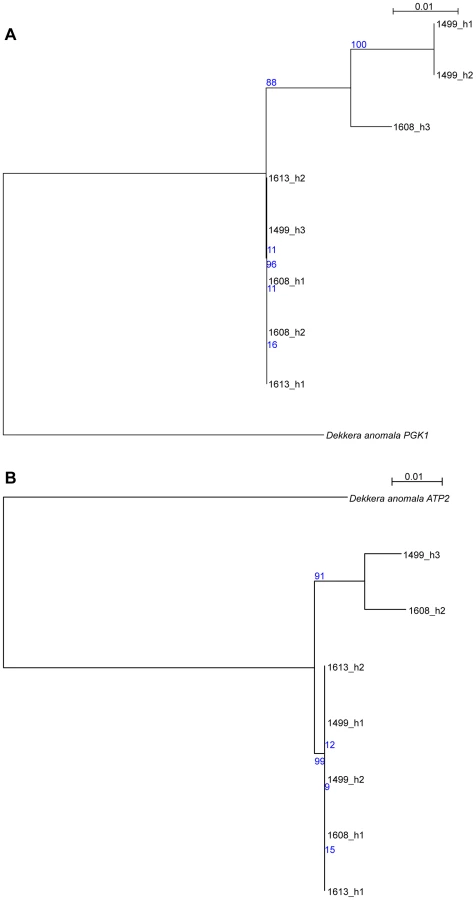
While the initial analysis of the AWRI1499 genome failed to identify a potential ‘donor’ species for the divergent alleles [5], we sought to determine if there was sequence data now available that would shed new light on this. Protein-based maximum-likelihood phylogenies were therefore produced for each of the haplotype groups from D. bruxellensis for three of the open reading frames (ORFs) presented in Fig. 3, in addition to homologs identified in the Genbank non-redundant protein database (Fig. S6). This analysis clearly shows all of the D. bruxellensis alleles to be far more closely related to each other than to any other available protein sequences. A small number of gene sequences available for D. anomala, the closest known relative of D. bruxellensis according to 26S rDNA based phylogenies [18], were then used as nucleotide queries against the AWRI1499 blast database. Two accessions, annotated as ATP2 and PGK1, were strong positive matches to AWRI1499 open reading frames, with 92% and 93% nucleotide identity. Nucleotide-based maximum-likelihood phylogenies for these ORFs, with haplotypes extracted from AWRI1499, 1608 and 1613, were performed (Fig. 4). The D. anomala sequences were not closely related to any of the D. bruxellensis haplotypes. As such, the potential source of the divergent alleles in the triploid strains remains to be determined.
Variation in nitrate assimilation potential
While there was significant variation in SNP diversity across strains, strain-specific genomic deletions were found to be far less common, with an average of only 0.15% of the genome lost, across the three strains relative to AWRI1499. The majority (97%) of these deletions were found in AWRI1613.
Of the genomic loci that did display strain-specific deletions, one region that was lost specifically in AWRI1613 was of particular interest as it involved the D. bruxellensis nitrate assimilation cluster (Fig. 5). While nitrate utilisation is common throughout Ascomycota, genes associated with the nitrate assimilation cluster are generally confined to Pezizomycotina; Dekkera, Ogataea, Wickerhamomyces and Blastobotrys are the only genera within the Saccharomycotina where this cluster has been identified. In these species the nitrate cluster appears to have been retained from the last common ancestor with the Pezizomycotina (Fig. S7). However, despite this ancient evolutionary conservation, it is apparent that both the nitrate and nitrite reductase genes have been lost in AWRI1613, along with an adjacent β-galactosidase gene. Furthermore, while this cluster is present in AWRI1608 it is predicted to have undergone LOH via gene conversion, resulting in three identical alleles. In contrast, the nitrate cluster of CBS2499 is predicted to have undergone a duplication event resulting in four copies of this genomic region being present in a 1∶1 ratio of two alleles (Fig 2A, Fig 5A).
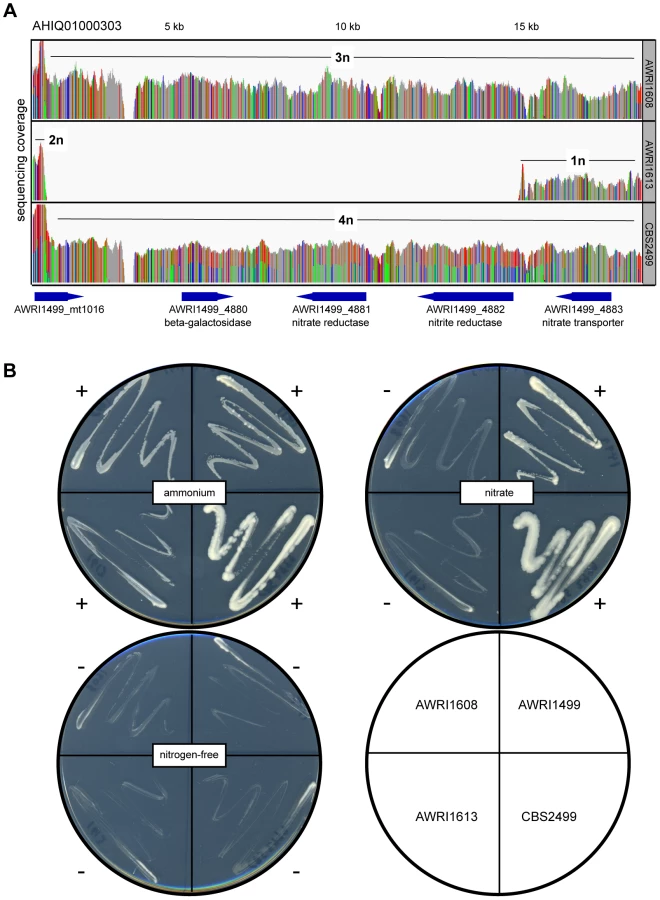
Consistent with presence or absence of ORFs encoding these key assimilatory enzymes, AWRI1499 and CBS2499 displayed robust growth on nitrate as a sole nitrogen source whereas AWRI1613 was unable to grow on this medium (Fig. 5B). Interestingly, despite the presence of the nitrate assimilation locus, AWRI1608 was also unable to utilise nitrate. In order to determine if this loss of nitrate utilisation was due to frameshift or nonsense mutations in the homozygous coding sequences of the AWRI1608 cluster, the ORFs of the nitrate and nitrite reductases were compared to haplotyped sequences from CBS2499 (Dataset S4). In each case, there were no obvious truncated coding regions or non-synonymous mutations present in the AWRI1608 ORFs, compared to either of the CBS2499 haplotypes that would be expected to cause the drastic changes to enzyme function that could account for the loss of nitrate utilisation in AWRI1608.
To further investigate the cause of the non-nitrate utilisation phenotype of AWRI1608, a high-affinity nitrate transporter gene that lies adjacent to the two reductases and the genomic region surrounding the two putative nitrate assimilation transcription factors of D. bruxellensis [10] was also investigated in these strains. The gene encoding the nitrate transporter was shown to be homozygous in both strains that could not grow on nitrate (AWRI1608 and AWRI1613) but was heterozygous in AWRI1499 and CBS2499 (data not shown). However, even in the two homozygous strains the ORF is predicted to encode a full-length, functional, nitrate transporter. Similarly, the genomic region encompassing both putative regulatory proteins was predicted to be present in all four strains; as for the nitrate and nitrite reductase genes in AWRI1608, both ORFs displayed LOH in AWRI1608, AWRI1613 and CBS2499 (data not shown). In contrast, this region was shown to be heterozygous in AWRI1499.
Discussion
The advent of next generation sequencing has enabled a significant increase in knowledge regarding the genomic makeup of important, but often genetically intractable, industrial yeasts. This manuscript describes the first analysis of inter-strain variation in the genomic landscape of D. bruxellensis. The most prominent finding of these genome comparisons was the common occurrence of triploid hybrids in the strains examined. AWRI1613 and CBS2499 were determined to be diploid in this study, with each strain possessing a pair of closely related chromosomes with moderate levels of heterozygosity. However, AWRI1499 and AWRI1608, the most common strains found in Australian wineries, were shown to be triploid. While triploid, both AWRI1499 and AWRI1608 contain pairs of chromosomes that are closely related to those found in the diploid strains suggesting that a diploid complement comprises the basis of the D. bruxellensis genome. The third, complete, set of more distantly related chromosomes present in AWRI1499 and AWRI1608 are therefore likely to have been introduced via hybridisation with a distantly related strain of D. bruxellensis or possibly another closely related but as yet undescribed species (Fig. 6). Furthermore, the divergent third sets of chromosomes present in each triploid are, in-turn, distantly related to each other, indicating that the two triploid strains likely arose from independent hybridisation events.
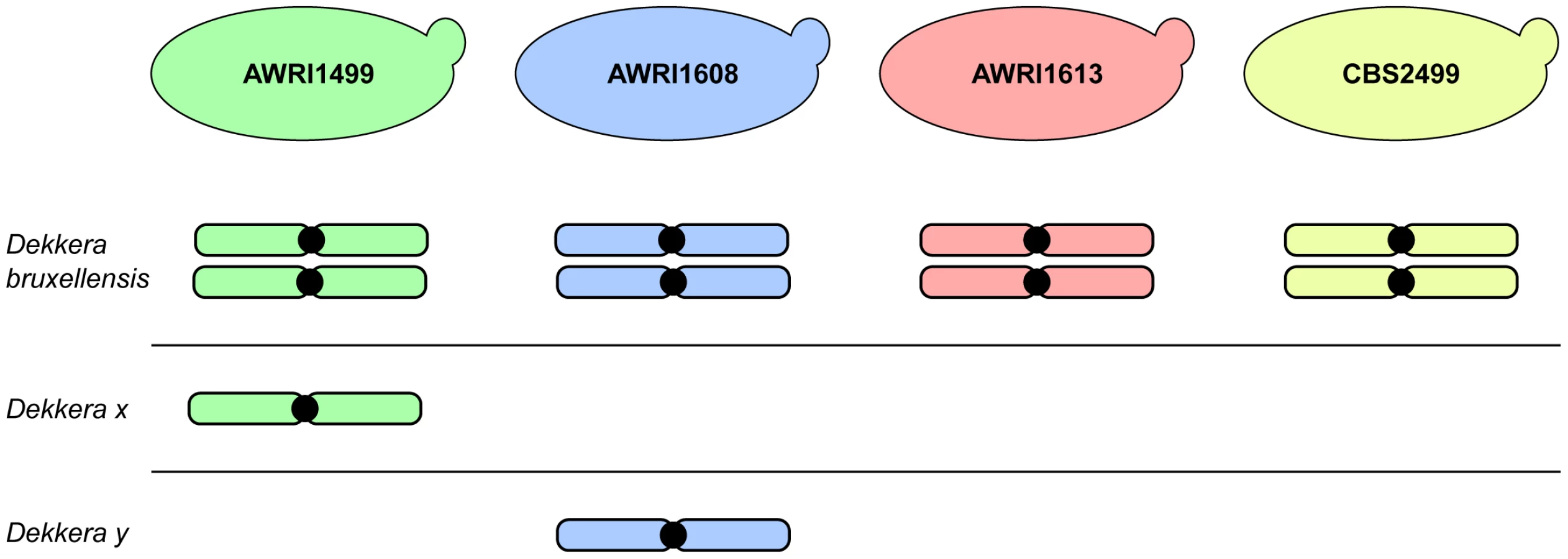
The results of this genomic study therefore suggest that the D. bruxellensis genomic landscape is similar to counterparts in the Saccharomyces sensu stricto clade, where inter-specific hybrids between S. cerevisiae, S. kudriavzevii, S. uvarum and S. eubayanus can be found in natural environments and in industrial fermentations [19]–[22]. Furthermore, Saccharomyces spp. interspecific hybrids are often allotriploid, with a ‘diploid’ complement coming from S. cerevisiae and a ‘haploid’ input from a non-cerevisiae parent. These Saccharomyces sensu stricto hybrids have been isolated from cold winemaking and brewing environments, where it is suggested the hybrid has a selective advantage over its parents. In these situations, the S. cerevisiae genomic component provides the means to efficiently ferment sugar to ethanol while genomic contributions from cold tolerant Saccharomyces spp. allow the hybrid strains to ferment at temperatures that are normally too low for S. cerevisiae [6], [19], [23]. In fact, strains of S. pastorianus, the yeast species responsible for the vast majority of lager beer fermentations, are hybrids generated from matings between S. cerevisiae and Saccharomyces eubayanus. At least one line of these hybrids has allotriploid origins, as was observed for the two D. bruxellensis hybrids analysed in this work [19], [20], [24].
At this time it is not possible to determine whether the ‘additional haploid’ inputs in the karyotypes of the two triploid D. bruxellensis strains described in this paper originate from one or more non-D. bruxellensis species or distantly related D. bruxellensis strains. However, as for lager brewing, it appears that the formation of these triploid hybrid strains may have resulted in a population replacement event, with the hybrid strains representing 92% of isolates from across the Australian wine industry. Based upon a limited multi-locus analysis, some previously analysed international strains [17] bear resemblance to AWRI1499 and AWRI1608, therefore it is possible that the current population structure in Australian wineries reflects historical gene flows and bottlenecks. It remains to be determined whether the additional sets of chromosomes in AWRI1499 and AWRI1608 confer a selective advantage in the winery environment, although increased levels of resistance to sulphite, the primary means of D. bruxellensis control in a winery setting, may be at least partially responsible [11].
As for presumptive selective pressures underpinning hybrid prevalence, the driver for loss of nitrate assimilation ability in specific strains of D. bruxellensis remains unclear. Ammonium is fully utilised by S. cerevisiae and other wine yeast species during alcoholic fermentation [25]. However D. bruxellensis appears to be the only wine yeast species that can assimilate nitrate, which is reported to be at levels of between 0.9 and 53.7 mg/l in Californian wine [26]. One might predict therefore that, in ecological settings where nitrate is available and other nitrogen sources are limited, nitrate assimilation would provide D. bruxellensis with a selective advantage. Yet up to a third of D. bruxellensis wine isolates fail to grow on nitrate [8]. Interestingly, it was recently shown that during anaerobic fermentation nitrate assimilation in D. bruxellensis favours the production of acetic acid over ethanol while partially abolishing the Custers effect [27]. This impact of nitrate assimilation may be detrimental in some environmental settings, thereby providing a selective pressure for its loss.
This phenomenon of loss of nutrient utilisation is not unknown in nature. For example, there is a concerted loss of the galactose (GAL) catabolism cluster in Japanese isolates of Saccharomyces kudriavzevii when compared to European relatives. In this example, the Japanese strains show degeneration of the genes involved in the utilization of galactose to pseudogene equivalents, while this cluster is completely active in European strains [28]. Evidence was also presented for a selective pressure driving loss of function for all members of the GAL pathway thereby producing a GAL− phenotype, as the presence of partial function in the pathways were suggested to have fitness costs.
At face value, this does not appear to be the case for loss of nitrate assimilation in AWRI1613, which still retains the coding region for the nitrate transporter. However, it is currently not known whether the presence of this transporter may be due to pleiotropy; it may, for example, be required for secondary transport functions. It is also possible that it may be non-functional, although given that the nucleotide sequence of the nitrate transporter ORF in AWRI1613 represents the opposite haplotype group to the homozygous transporter sequence from AWRI1608 (Fig. S8), it would be expected that at least one of these strains would have a functional transporter as evidenced by the nitrate assimilation phenotype of CBS2499, which has one copy of each haplotype.
Further study of nitrate assimilation in D. bruxellensis will reveal the molecular mechanisms driving the phenotype towards, or away from, utilisation of this nitrogen source, augmenting knowledge gained through detailed studies of the preferred yeast model system for nitrate assimilation, Ogataea parapolymorpha [29].
Materials and Methods
Yeast strain, nucleic acid preparation, and sequencing
D. bruxellensis strains AWRI1608 and AWRI1613 were obtained from The Australian Wine Research Institute Microorganisms Culture Collection. For nitrate assimilation tests, strains were grown on solid YPD for 2 days at 30°C, then plated onto either YNB+ nitrate, YNB+ ammonium as a positive control, or YNB with no nitrogen source as a negative control. Plates were then incubated for 7 days at 30°C.
Genomic DNA was prepared using a standard zymolyase and phenol-chloroform extraction from cultures grown under standard conditions. DNA sequencing was performed using 2×100 bp paired-end chemistry on the Illumina HiSeq2000 (Ramaciotti Centre, Sydney Australia).
AWRI1499 genome sequences were obtained from Genbank (Accession number AHIQ0100000). CBS2499 short-read sequences were obtained from the NCBI short-read archive (Accession number SRR065689). Sequence data for AWRI1608 and AWRI1613 have been deposited in the NCBI short-read archive under the Bioproject accession PRJNA213658. 26S rDNA sequences for AWRI1499, 1608 and 1613 have been deposited with GenBank (accessions KF781196, KF781197 and KF781198, respectively).
Mapping assemblies and analysis
Short read sequences were mapped to the AWRI1499 genome using Novoalign v2.08.01 (www.novocraft.com). The .sam files produced by Novoalign (default parameters; -F ILM1.8 –ILQ_SKIP -i PE 100-1000 -o SAM) were converted to sorted .bam files using samtools view v0.1.18 [30]. SNPs, and regions of LOH and gain-of-heterozygosity (GOH) were identified from the alignments using the pileup2snp functionality of Varscan v2.3 (default parameters; -min-coverage 10) [31] combined with custom python scripts and presented relative to a concatenated AWRI1499 genome sequence. The position of individual AWRI1499 Genbank contigs and ORF annotations within the concatenated genome sequence are provided in Datasets S5 and S6 respectively. Sequence alignments were visualized using the Integrated Genome Browser v2.0 [32]. Any region displaying a maximum allele frequency of >95% was classed as being homozygous for that allele.
Sequencing coverage was extracted from alignments in .bam format using mpileup from the samtools v0.1.18 package [30] with actual coverage values converted to changed ploidy levels in sliding windows using custom python scripts. Segmental smoothing of copy number alterations calculated final copy number based on rounding the average value across 21 adjacent genomic windows. To provide additional robustness against single outliers producing small false-positive intervals of altered ploidy levels, a difference of ±0.75 was required between the average ploidy of the current 21 window genomic segment and the predicted ploidy level of the previous genomic segment in order to trigger a change in the final predicted ploidy level. If this threshold was not met, the average value obtained for the 21 segment window was rounded to the ploidy level of the previous genomic segment.
Phylogenies were constructed using PhyML v3.0 (GTR model; default parameters) and visualized using Seaview v4.0 [15], [16].
Supporting Information
Zdroje
1. CurtinC, BellonJR, HenschkePA, GoddenPW, de Barros LopesMA (2007) Genetic diversity of Dekkera bruxellensis yeasts isolated from Australian wineries. FEMS Yeast Res 7 : 471–481.
2. BokulichNA, BamforthCW, MillsDA (2012) Brewhouse-resident microbiota are responsible for multi-stage fermentation of american coolship ale. PLoS ONE 7: e35507.
3. CotonE, CotonM, LevertD, CasaregolaS, SohierD (2006) Yeast ecology in French cider and black olive natural fermentations. Int J Food Microbiol 108 : 130–135.
4. PassothV, BlomqvistJ, SchnürerJ (2007) Dekkera bruxellensis and Lactobacillus vini form a stable ethanol-producing consortium in a commercial alcohol production process. Appl Environ Microbiol 73 : 4354–4356.
5. CurtinC, BornemanAR, ChambersPJ, PretoriusIS (2012) De-Novo Assembly and analysis of the heterozygous triploid genome of the wine spoilage yeast Dekkera bruxellensis AWRI1499. PLoS ONE 7: e33840.
6. BornemanAR, PretoriusIS, ChambersPJ (2012) Comparative genomics: a revolutionary tool for wine yeast strain development. Curr Opin Biotech 24 : 192–199.
7. Rozpe¸dowskaE, HellborgL, IshchukOP, OrhanF, GalafassiS, et al. (2011) Parallel evolution of the make-accumulate-consume strategy in Saccharomyces and Dekkera yeasts. Nat Commun 2 : 302.
8. ConternoL, JosephCL, ArvikT, Henick-KlingT, BissonLF (2006) Genetic and physiological characterization of Brettanomyces bruxellensis strains isolated from wines. Am J Enol Viticult 57 : 139–147.
9. Barros PitaW, LeiteFCB, Souza LiberalAT, SimõesDA, de MoraisMA (2011) The ability to use nitrate confers advantage to Dekkera bruxellensis over S. cerevisiae and can explain its adaptation to industrial fermentation processes. A van Leeuw J Microb 100 : 99–107.
10. WoolfitM, Rozpe¸dowskaE, PiškurJ, WolfeKH (2007) Genome survey sequencing of the wine spoilage yeast Dekkera (Brettanomyces) bruxellensis. Eukaryotic Cell 6 : 721–733.
11. CurtinC, KennedyE, HenschkePA (2012) Genotype dependent sulfite tolerance of Australian Dekkera (Brettanomyces) bruxellensis wine isolates. Lett Appl Microbiol 55 : 56–61.
12. PiškurJ, LingZ, Marcet-HoubenM, IshchukOP, AertsA, et al. (2012) The genome of wine yeast Dekkera bruxellensis provides a tool to explore its food-related properties. Int J Food Microbiol 157 : 202–209.
13. BrownCA, MurrayAW, VerstrepenKJ (2010) Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr Biol 20 : 895–903.
14. ChowEWL, MorrowCA, DjordjevicJT, WoodIA, FraserJA (2012) Microevolution of Cryptococcus neoformans driven by massive tandem gene amplification. Molecular Biology and Evolution 29 : 1987–2000 doi:10.1093/molbev/mss066
15. GouyM, GuindonS, GascuelO (2010) SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27 : 221–224.
16. GuindonS, DufayardJF, LefortV, AnisimovaM, HordijkW, et al. (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol 59 : 307–321.
17. HellborgL, PiškurJ (2009) Complex Nature of the Genome in a Wine Spoilage Yeast, Dekkera bruxellensis. Eukaryotic Cell 8 : 1739–1749.
18. BoekhoutT, KurtzmanCP, O'DonnellK, SmithMT (1994) Phylogeny of the yeast genera Hanseniaspora (anamorph Kloeckera), Dekkera (anamorph Brettanomyces), and Eeniella as inferred from partial 26S ribosomal DNA nucleotide sequences. Int J Syst Bacteriol 44 : 781–786.
19. LibkindD, HittingerCT, ValérioE, GonçalvesC, DoverJ, et al. (2011) Microbe domestication and the identification of the wild genetic stock of lager-brewing yeast. Proc Natl Acad Sci USA 108 : 14539–14544.
20. NguyenH-V, LegrasJ-L, NeuvégliseC, GaillardinC (2011) Deciphering the hybridisation history leading to the lager lineage based on the mosaic genomes of Saccharomyces bayanus Strains NBRC1948 and CBS380T. PLoS ONE 6: e25821.
21. LopandicK, GanglH, WallnerE, TscheikG, LeitnerG, et al. (2007) Genetically different wine yeasts isolated from Austrian vine-growing regions influence wine aroma differently and contain putative hybrids between Saccharomyces cerevisiae and Saccharomyces kudriavzevii. FEMS Yeast Res 7 : 953–965.
22. BornemanAR, DesanyBA, RichesD, AffourtitJP, ForganAH, et al. (2011) The genome sequence of the wine yeast VIN7 reveals an allotriploid hybrid genome with Saccharomyces cerevisiae and Saccharomyces kudriavzevii origins. FEMS Yeast Res 12 : 88–96.
23. PerisD, LopesCA, BellochC, QuerolA, BarrioE (2012) Comparative genomics among Saccharomyces cerevisiae x Saccharomyces kudriavzevii natural hybrid strains isolated from wine and beer reveals different origins. BMC Genomics 13 : 407.
24. DunnBL, SherlockG (2008) Reconstruction of the genome origins and evolution of the hybrid lager yeast Saccharomyces pastorianus. Genome Res 18 : 1610–1623.
25. JiranekV, LangridgeP, HenschkePA (1995) Amino acid and ammonium utilization by Saccharomyces cerevisiae wine yeasts from a chemically defined medium. Am J Enol Viticult 46 : 75–83.
26. OughC, CrowellEA (1980) Nitrate determination in California musts and wines. Am J Enol Viticult 31 : 344–346.
27. GalafassiS, CapusoniC, MoktaduzzamanM, CompagnoC (2013) Utilization of nitrate abolishes the “Custers effect” in Dekkera bruxellensis and determines a different pattern of fermentation products. J Ind Microbiol Biotechnol 40 : 297–303.
28. HittingerCT, GonçalvesP, SampaioJP, DoverJ, JohnstonM, et al. (2010) Remarkably ancient balanced polymorphisms in a multi-locus gene network. Nature 464 : 54–58.
29. Rossi B, Berardi E (2009) Assimilatory nitrate reduction in Hansenula polymorpha. In: Satyanarayana T, Kunze G, editors. Yeast Biotechnology: Diversity and Applications. Springer. pp. 307–320.
30. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25 : 2078–2079.
31. KoboldtDC, ZhangQ, LarsonDE, ShenD, McLellanMD, et al. (2012) VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22 : 568–576.
32. RobinsonJT, ThorvaldsdóttirH, WincklerW, GuttmanM, LanderES, et al. (2011) Integrative genomics viewer. Nat Biotechnol 29 : 24–26.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 2
- Souvislost haplotypu M2 genu pro annexin A5 s opakovanými reprodukčními ztrátami
- Liův-Fraumeniho syndrom – indikace k testování a doporučená surveillance
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Akutní intermitentní porfyrie
- Vitamín B6 jako prevence kolorektálního karcinomu
Nejčtenější v tomto čísle
- Genome-Wide Association Study of Metabolic Traits Reveals Novel Gene-Metabolite-Disease Links
- A Cohesin-Independent Role for NIPBL at Promoters Provides Insights in CdLS
- Classic Selective Sweeps Revealed by Massive Sequencing in Cattle
- Arf4 Is Required for Mammalian Development but Dispensable for Ciliary Assembly

{kind=link}