
The Cytosine Methyltransferase DRM2 Requires Intact UBA Domains and a Catalytically Mutated Paralog DRM3 during RNA–Directed DNA Methylation in
Eukaryotic DNA cytosine methylation can be used to transcriptionally silence repetitive sequences, including transposons and retroviruses. This silencing is stable between cell generations as cytosine methylation is maintained epigenetically through DNA replication. The Arabidopsis thaliana Dnmt3 cytosine methyltransferase ortholog DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) is required for establishment of small interfering RNA (siRNA) directed DNA methylation. In mammals PIWI proteins and piRNA act in a convergently evolved RNA–directed DNA methylation system that is required to repress transposon expression in the germ line. De novo methylation may also be independent of RNA interference and small RNAs, as in Neurospora crassa. Here we identify a clade of catalytically mutated DRM2 paralogs in flowering plant genomes, which in A.thaliana we term DOMAINS REARRANGED METHYLTRANSFERASE3 (DRM3). Despite being catalytically mutated, DRM3 is required for normal maintenance of non-CG DNA methylation, establishment of RNA–directed DNA methylation triggered by repeat sequences and accumulation of repeat-associated small RNAs. Although the mammalian catalytically inactive Dnmt3L paralogs act in an analogous manner, phylogenetic analysis indicates that the DRM and Dnmt3 protein families diverged independently in plants and animals. We also show by site-directed mutagenesis that both the DRM2 N-terminal UBA domains and C-terminal methyltransferase domain are required for normal RNA–directed DNA methylation, supporting an essential targeting function for the UBA domains. These results suggest that plant and mammalian RNA–directed DNA methylation systems consist of a combination of ancestral and convergent features.
Published in the journal:
. PLoS Genet 6(10): e32767. doi:10.1371/journal.pgen.1001182
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001182
Summary
Eukaryotic DNA cytosine methylation can be used to transcriptionally silence repetitive sequences, including transposons and retroviruses. This silencing is stable between cell generations as cytosine methylation is maintained epigenetically through DNA replication. The Arabidopsis thaliana Dnmt3 cytosine methyltransferase ortholog DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) is required for establishment of small interfering RNA (siRNA) directed DNA methylation. In mammals PIWI proteins and piRNA act in a convergently evolved RNA–directed DNA methylation system that is required to repress transposon expression in the germ line. De novo methylation may also be independent of RNA interference and small RNAs, as in Neurospora crassa. Here we identify a clade of catalytically mutated DRM2 paralogs in flowering plant genomes, which in A.thaliana we term DOMAINS REARRANGED METHYLTRANSFERASE3 (DRM3). Despite being catalytically mutated, DRM3 is required for normal maintenance of non-CG DNA methylation, establishment of RNA–directed DNA methylation triggered by repeat sequences and accumulation of repeat-associated small RNAs. Although the mammalian catalytically inactive Dnmt3L paralogs act in an analogous manner, phylogenetic analysis indicates that the DRM and Dnmt3 protein families diverged independently in plants and animals. We also show by site-directed mutagenesis that both the DRM2 N-terminal UBA domains and C-terminal methyltransferase domain are required for normal RNA–directed DNA methylation, supporting an essential targeting function for the UBA domains. These results suggest that plant and mammalian RNA–directed DNA methylation systems consist of a combination of ancestral and convergent features.
Introduction
Repetitive DNA in eukaryotic genomes is often transcriptionally silent, due to genome defense mechanisms directed against transposons and other mobile DNA [1], [2]. In plant genomes repetitive sequences are associated with DNA cytosine methylation [3]–[8], which is required for transcriptional silencing and suppression of transposon activity [9]–[12]. As DNA methylation can be maintained epigenetically through DNA replication, this mark serves to stably repress expression of repeated sequences following cell division [2], [13]. Gene promoters in A.thaliana are generally not DNA methylated, though gene bodies (open reading frames) may contain DNA methylation in the CG sequence context [3]–[8]. Eukaryotic DNA methylation is catalyzed by cytosine methyltransferases that share ancestry with prokaryotic restriction modification enzymes and are characterized by 10 conserved catalytic motifs [13], [14]. During catalysis a methyl group is transferred from the donor molecule S-adenosyl methionine to the carbon-5 position of the cytosine base, using an essential cysteine residue in catalytic motif IV [13], [14].
Three functional classes of DNA methyltransferase exist in A.thaliana; METHYLTRANSFERASE1 (MET1) (orthologous to mammalian Dnmt1) which maintains CG methylation, CHROMOMETHYLASE3 (CMT3) (plant specific) which maintains methylation in non-CG sequence contexts and DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2) (orthologous to Dnmt3a/Dnmt3b) which both de novo methylates DNA and maintains non-CG methylation redundantly with CMT3 [2]. The drm2 mutation blocks all de novo DNA methylation driven by repeat containing transgenes [15]–[17]. This phenotype is shared with mutations in a diverse set of RNA interference and chromatin proteins including NUCLEAR RNA POLYMERASE D1 (NRPD1), NUCLEAR RNA POLYMERASE E1 (NRPE1), RNA DEPENDENT RNA POLYMERASE2 (RDR2), DICER-LIKE3 (DCL3), ARGONAUTE4 (AGO4), DEFECTIVE IN RNA DIRECTED DNA METHYLATION 1 (DRD1), DEFECTIVE IN MERISTEM SILENCING1 (DMS1), INVOLVED IN DE NOVO 2 (IND2), SUPPRESSOR OF VARIEGATION 3-9 HOMOLOGUE 2 (SUVH2) and SUPPRESSOR OF VARIEGATION 3-9 HOMOLOGUE 9 (SUVH9) [16]–[34]. These genes act in a pathway that generates short interfering RNAs (siRNAs) which can guide DRM2 DNA methyltransferase activity to homologous genomic sequences [2]. This is reflected in the strong correlation between genomic repeats, DNA methylation and siRNAs in A.thaliana [3]–[5], [7], [8]. Small RNAs are also required for DRM2 to maintain non-CG DNA methylation and silencing at a subset of endogenous loci, though this function is partially redundant with a second pathway consisting of CMT3 and the histone H3K9 methyltransferase KRYPTONITE/SUPPRESSOR OF VARIEGATION HOMOLOGUE4 [17], [35]–[37].
The mammalian DRM2 orthologs, Dnmt3a and Dnmt3b, are required to de novo methylate integrated retroviral sequences and imprinted genes [38], [39]. Dnmt3a forms a complex with a non-catalytic paralog Dnmt3L, which is also required for normal patterns of DNA methylation in vivo [40]–[46]. The C-terminal domains of Dnmt3a and Dnmt3L have been co-crystallized and shown to form a hetero-tetrameric complex, which promotes Dnmt3a methyltransferase activity [42]–[46]. The N-terminal PHD domain of Dnmt3L interacts with unmethylated H3K4 histone tails and recruits Dnmt3a to specific loci, including imprinted genes [45], [46]. Thus de novo methylation mediated by Dnmt3a requires interaction with non-catalytic Dnmt3L, which both stimulates catalysis and recruits the complex to chromatin. Animal PIWI proteins bind germ cell specific 25–30 nucleotide piRNAs, many of which are homologous to repeated sequences [47]. The mouse PIWI mutants miwi, mili and miwi2 are sterile and have defects in transposon de novo methylation in male germ cells, with a phenotype very similar to dnmt3l [40], [48]–[52]. Hence, small RNAs are required to target a subset of de novo DNA methylation in mammals, though as plants lack PIWI domains and piRNA, these systems appear to have evolved independently. De novo DNA methylation can also be independent of small RNAs and RNA interference, as in Neurospora crassa [53].
Here we describe a novel protein required for RNA–directed DNA methylation in A.thaliana, DOMAINS REARRANGED METHYLTRANSFERASE3 (DRM3). Based on sequence analysis and genetic observations DRM3 appears to be catalytically mutated DNA methyltransferase paralog. A conserved clade of DRM3 genes is found throughout angiosperm genomes. Although DRM3 cannot compensate for loss of DRM2 function in vivo, it is required for normal establishment and maintenance of RNA–directed DNA methylation and accumulation of specific repeat-associated siRNA. This is analogous to the function of Dnmt3L in mammals, though phylogenetic analysis indicates that DRM3 and Dnmt3L proteins diverged independently in plant and animal lineages. We also demonstrate using site-directed mutagenesis that both the catalytic methyltransferase domain and UBA domains of DRM2 are required for RNA–directed DNA methylation. We speculate that the UBA domains are involved in targeting DRM2 activity during de novo DNA methylation, potentially by recognizing a specific chromatin state. These findings extend parallels between plant and mammalian RNA–directed DNA methylation systems, despite their independent evolution.
Results
A conserved clade of catalytically mutated DRM3 paralogs within angiosperms
The A.thaliana genome encodes a previously uncharacterized protein with high amino acid identity to DRM2, which we term DRM3 (At3g17310) (Figure 1A and 1B). Like DRM2, DRM3 contains N-terminal ubiquitin associated (UBA) domains and a C-terminal region which shares identity with the Dnmt3 cytosine methyltransferase domain, but whose catalytic motifs are rearranged such that motifs VI–X precede I–V (Figure 1A and 1B) [54]. During catalysis cytosine methyltransferases form a covalent bond between a conserved cysteine in motif IV and carbon-6 of the cytosine base [14]. The catalytic cysteine is preceded by an invariant proline, which hydrogen bonds to exo-cyclic NH2 groups of cytosine to promote specific recognition and stabilize interaction between the base and catalytic site [14]. Close inspection of the DRM3 methyltransferase domain reveals the absence of highly conserved residues and notably the invariant proline-cysteine sequence is absent from motif IV (Figure 1B). Additionally, DRM3 lacks a conserved glutamic acid within motif IX and glycine within motif X (Figure 1B). It is likely that the absence of these residues inactivate DRM3 cytosine methyltransferase activity, because a drm2 mutation alone is sufficient to block de novo DNA methylation [15]–[17].
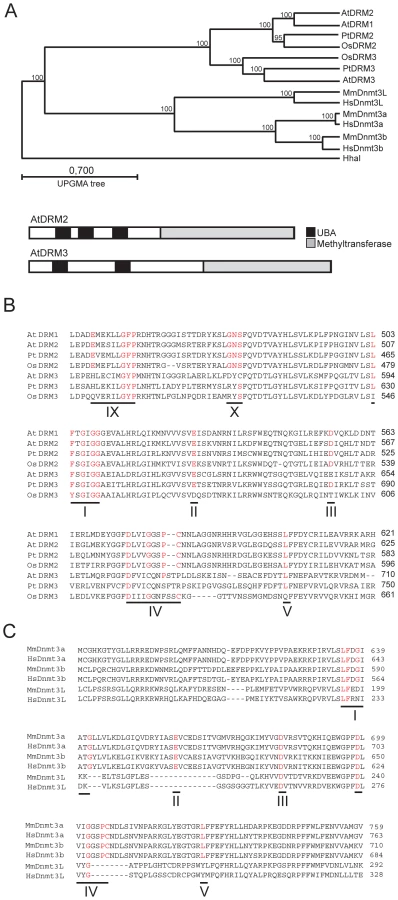
BLAST searches of the angiosperm Populus trichocarpa and Oryzae sativa genomes reveals a clade of DRM3 proteins shared between these species, indicating that distinct DRM2 and DRM3 proteins are conserved and were present prior to the divergence of the monocots and dicots (Figure 1A). All members of the DRM3 clade lack conserved motif IV proline-cysteine, motif IX glutamic acid and motif X glycine residues (Figure 1B). Although OsDRM3 possesses a cysteine within motif IV, it lacks a preceding proline and therefore is predicted to be catalytically inactive (Figure 1B) [14]. The mammalian Dnmt3L proteins also carry mutations in key catalytic motifs, including the catalytic proline-cysteine sequence in motif IV [36]–[39] (Figure 1C). However, phylogenetic analysis indicates that the Dnmt3 and DRM protein families diverged independently in plants and mammals (Figure 1A). This scenario is supported by the fact that all DRM proteins share an identical rearrangement of their catalytic motifs (Figure 1B) [54]. As Dnmt3L proteins are important for mammalian de novo DNA methylation we sought to test whether DRM3 is analogously required for RNA–directed DNA methylation in A.thaliana.
DRM3 is required for maintenance of non-CG DNA methylation
To test whether DRM3 is required for normal DRM2 function we obtained two independent transfer-DNA (T-DNA) insertions in this gene (drm3-1 and drm3-2) (Figure S1). We tested whether the drm3 mutations have a defect in maintenance of non-CG methylation at the MEA-ISR endogenous tandem repeat [36]. Non-CG methylation at MEA-ISR is lost in drm2 and RNAi mutants such as rdr2, which can be assayed by Southern blotting and hybridization after genomic DNA digestion with the methyl-sensitive restriction endonuclease MspI [36]. The higher molecular weight 4.3 kb band hybridizing to MEA-ISR represents inhibition of MspI digestion by cytosine methylation at a single CHG site [36]. The lower 2 kb band represents digested DNA and in wild type is present at levels comparable to the upper band (Figure 2A) [36]. In drm1 drm2 mutants DNA methylation at this site is eliminated and the 4.3 kb band on the Southern blot is absent (Figure 2A) [36]. Using this assay we observed that both drm3 alleles show a reduction of methylation at MEA-ISR to a level intermediate between wild type and drm1 drm2 (Figure 2A). This methylation phenotype could be complemented by transformation of drm3-1 with a genomic fragment containing the DRM3 gene (Figure 2A). As the Southern blot assay measures methylation at a single CHG site we also sequenced the MEA-ISR locus after bisulfite conversion. This independently corroborated that drm3-1 shows an intermediate level of non-CG methylation at MEA-ISR between that of wild type and drm1 drm2 (Figure 2B, Table S1 and Figure S3). Although significant differences in CG methylation are observed between genotypes, individual CG sites remain highly methylated in all samples (>70%) and reductions in the percentage of methylated sites are less than for non-CG methylation (drm3-1 shows a 16% reduction in CG versus 76% reduction in both types of non-CG methylation, while drm1 drm2 shows an 11% reduction in CG versus reduction to absence of non-CG methylation) (Figure 2B, Table S1 and Figure S3). Furthermore, drm1 drm2 and drm3 do not share developmental phenotypes characteristic of mutants that fail to maintain CG methylation such as met1, ddm1 and vim1 vim2 vim3 [55]–[57].
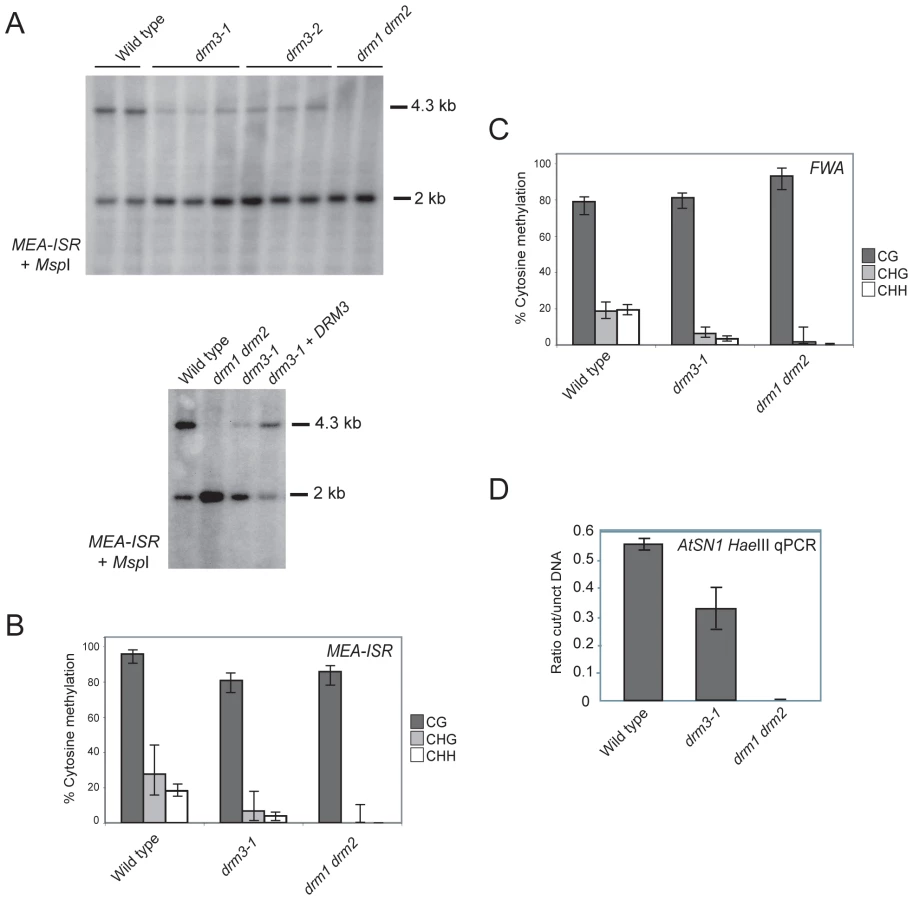
To further characterize the drm3 phenotype we performed sodium bisulfite sequencing across the promoter tandem repeats of the FWA endogene. These repeats are cytosine methylated in wild type and show an elimination of non-CG methylation in drm1 drm2 [36]. As for MEA-ISR, we observed that drm3-1 shows a strong reduction of non-CG methylation at FWA to a level intermediate between wild type and drm1 drm2 (Figure 2C, Table S2 and Figure S3). We also analyzed maintenance of DNA methylation at the SINE retroelement AtSN1 using HaeIII restriction endonuclease digestion of genomic DNA followed by quantitative PCR [18], [27]. HaeIII digestion is DNA methylation sensitive, meaning that methylated sequences are protected from digestion. In wild type AtSN1 is densely methylated and resistant to HaeIII cleavage, whereas in drm1 drm2 methylation is greatly reduced and AtSN1 DNA is digested and as a consequence not amplified (Figure 2D). We repeated this assay in drm3-1 and again observed an intermediate phenotype between wild type and drm1 drm2, indicating a defect in maintenance of DNA methylation at AtSN1 (Figure 2D). We conclude from these observations that DRM3 is required for normal maintenance of non-CG DNA methylation in vivo. However, as non-CG methylation is not completely eliminated in drm3, it is not absolutely required for DRM2 activity.
DRM2 mRNA and protein expression are independent of DRM3
One explanation for drm3 phenotypes could be an influence on DRM2 expression or protein stability. To test this idea we first measured DRM2 mRNA levels using quantitative reverse-transcriptase PCR. This demonstrated that DRM2 mRNA accumulated normally in drm3 mutants relative to wild type (Figure S1). To test the accumulation of DRM2 protein we crossed a complementing DRM2-Myc transgene into drm3-1 and performed western blotting using α-Myc antibodies [58]. DRM2-Myc protein accumulated normally in drm3-1 relative to wild type (Figure S1). We conclude from these experiments that DRM2 expression is independent of DRM3.
DRM3 is required for normal establishment of RNA–directed DNA methylation
DRM2 is required for both maintenance of non-CG methylation and de novo establishment of methylation in all sequence contexts [15], [35], [36]. To test whether DRM3 is also required for normal establishment of RNA–directed DNA methylation we used Agrobacterium-mediated transformation with a tandem repeat containing FWA transgene [15], [16], [59]. In wild type endogenous FWA is transcriptionally silenced by DNA methylation at a set of promoter tandem repeats [59]. Loss of methylation at these repeats causes a dominant late-flowering phenotype, which can be quantified by counting leaf number before flowering [59]. When FWA is transformed into wild type A.thaliana the promoter repeats on the incoming transgene are efficiently methylated, causing transcriptional silencing [15], [16], [59]. However, FWA transformation into drm2 or RdDM pathway mutants (e.g. nrpd1, ago4, rdr2, dcl3) leads to a failure in de novo methylation, meaning incoming FWA transgenes are expressed causing a dominant late-flowering phenotype in transformed individuals [15], [16]. Therefore, we transformed FWA into wild type (Col), drm3-1 and drm1 drm2 backgrounds and analyzed the flowering time of first generation transformed individuals (T1) as a measure of establishment of RNA–directed DNA methylation. Untransformed plants were grown alongside +FWA transformants as controls and analyzed for flowering time. As previously reported FWA transformants in wild type flowered slightly but significantly later than untransformed controls, reflecting incomplete establishment of silencing in the T1 generation (Figure 3A and Table 1) [15], [16]. In contrast, drm1 drm2 FWA transformants flowered far later due to a failure to silence the incoming transgenes (Figure 3A and Table 1) [15], [16]. We observed that drm3-1 FWA transformants showed a flowering time distribution intermediate between the wild type and drm1 drm2 T1 populations (Figure 3A and Table 1). This parallels our observations for maintenance of non-CG methylation with drm3-1 showing a defect in DRM2-mediated DNA methylation, though with a phenotype weaker than drm1 drm2.
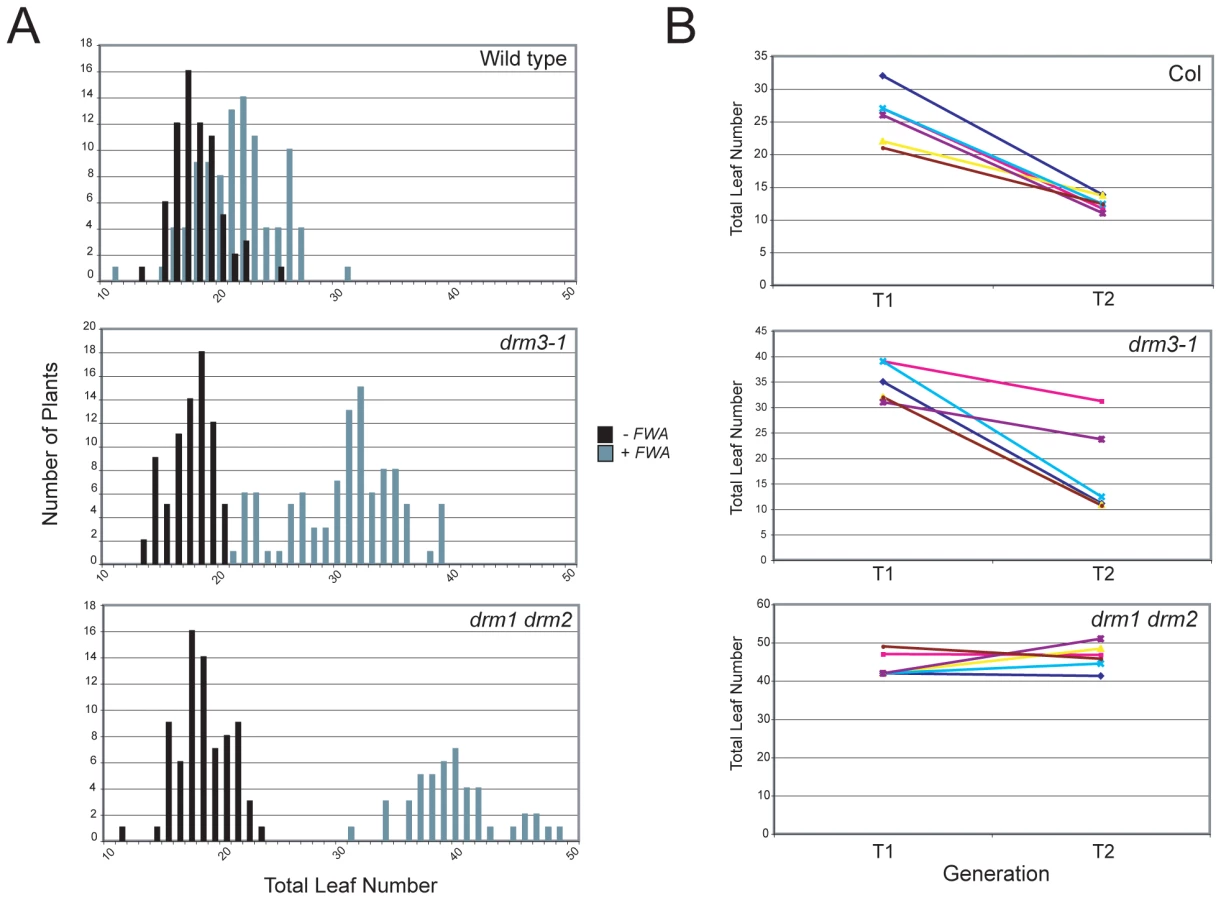

FWA silencing is frequently incomplete in the T1 generation and self-fertilization of transformed individuals results in an increase in FWA repeat DNA methylation and transcriptional silencing in the T2 progeny, which as a consequence flower earlier [59]. As drm3 appears to reduce but not abolish DRM2 activity, we predicted that wild type and drm3-1 T2 populations would become earlier flowering relative to the T1, whereas drm1 drm2 would not. Analysis of the flowering-time distribution of multiple T2 populations relative to their T1 parents showed this to be true (Figure 3B and Table S3). Consistent with a complete block to de novo DNA methylation in drm1 drm2 T2 individuals did not flower significantly earlier, which they did in the case of wild type and drm3-1 (Figure 3B and Table S3). Together, we interpret this as meaning that although DRM2 is absolutely required for de novo DNA methylation in vivo, normal levels of methyltransferase activity also require non-catalytic DRM3.
Intermediate RNA–directed DNA methylation phenotypes between wild type and drm1 drm2 have previously been observed in dcl3 mutants [16], [60], [61]. DCL3 generates 24 nucleotide siRNAs with a specialized function in RNA–directed DNA methylation and transcriptional silencing, though it acts redundantly with the other DCLs in a locus-specific manner [16], [60]–[62]. We generated drm3-1 dcl3-1 double mutants and tested for enhancement of defects during establishment of FWA RNA–directed DNA methylation, relative to the single mutants. We observed that drm3-1 dcl3-1 double mutants showed a silencing defect comparable to drm1 drm2, which was greater than either drm3-1 or dcl3-1 alone (Table 1). We interpret this enhancement as meaning that DRM3 and DCL3 act genetically in parallel to promote DRM2 activity in vivo.
Mutagenesis of DRM2 reveals a functional requirement for cytosine methyltransferase and UBA domains
DRM2 and DRM3 show a similar domain organization of N-terminal UBA domains and a C-terminal cytosine methyltransferase domain (Figure 1A). To further understand DRM2 function we performed site-directed mutagenesis of these domains and tested the consequence on RNA–directed DNA methylation. Cytosine methyltransferases require a set of 10 conserved motifs, including motif IV which contains the catalytic cysteine involved in transfer of the methyl group to cytosine (Figure 1B and 1C) [13]. We substituted the DRM2 motif IV catalytic cysteine C587 for an alanine, within an otherwise complementing DRM2 transgene containing a Myc-epitope translational fusion (hereafter termed DRM2cat-Myc) (Figure 4A) [58]. DRM proteins also possess N-terminal UBA domains, which consist of three helices connected by two conserved loop regions forming an ubiquitin interaction surface [63], [64]. The first loop contains a highly conserved MGF/MGY motif, which is required for correct folding and maintenance of UBA domain structure [64]. To test the functional importance of the DRM2 UBA domains we substituting conserved phenylalanine residues (F73, F123, F206) in each loop-I region to alanines (hereafter termed DRM2uba-Myc) (Figure 4A). The wild type DRM2-Myc and mutant DRM2cat-Myc and DRM2uba-Myc transgenes were transformed into drm1 drm2 and tested for their ability to complement mutant RNA–directed DNA methylation phenotypes.
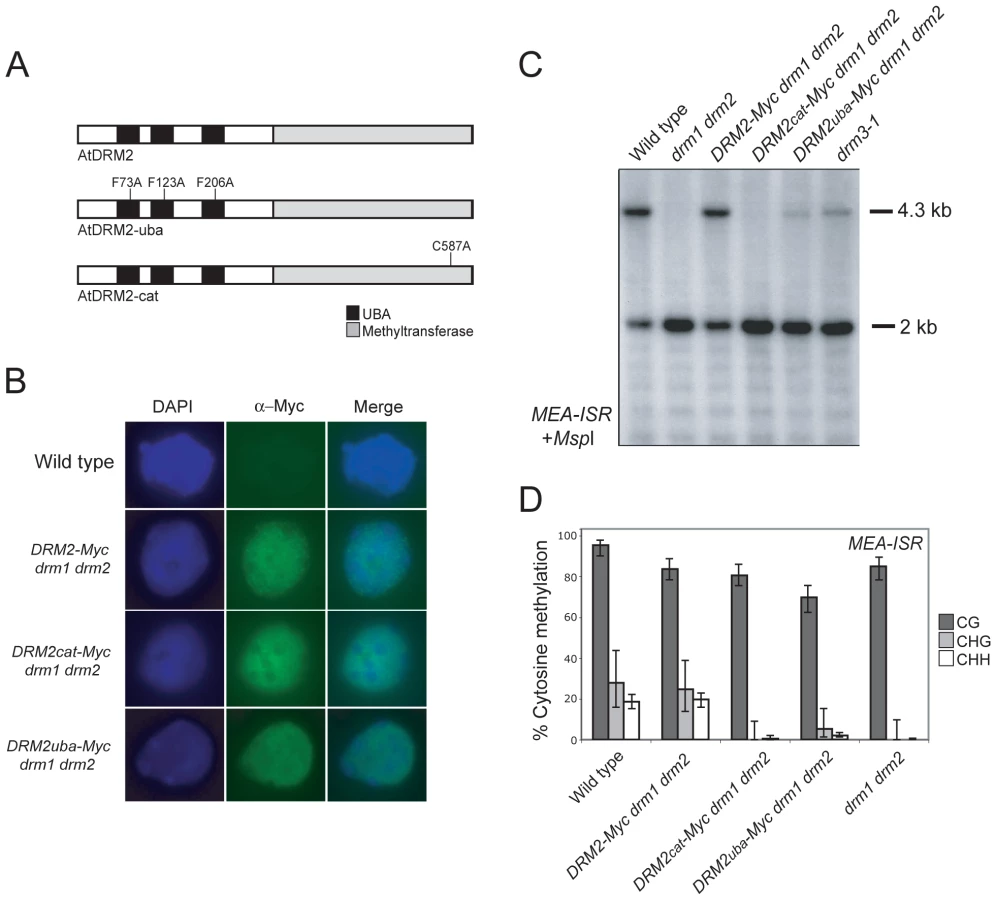
We first tested whether the catalytic and UBA mutations introduced into DRM2 had any consequence on protein accumulation and localization. Intact DRM2-Myc drm1 drm2 nuclei immunostained using α-Myc antibodies show signal throughout the nuclei with numerous bright foci and exclusion from the strongly DAPI-staining chromocentres (Figure 4B) [53]. The DRM2cat-Myc and DRM2uba-Myc mutant proteins showed comparable staining patterns to those observed for the unmutated DRM2-Myc protein (Figure 4B). This indicates that the UBA and methyltransferase mutations introduced do not cause a defect in DRM2 protein stability or localization to the nucleus.
We next tested the ability of the DRM2 transgenes to complement drm1 drm2 by assaying maintenance of non-CG methylation at MEA-ISR using methyl-sensitive MspI digestion followed by Southern blotting and hybridization as described earlier. The wild type DRM2-Myc transgene complemented drm1 drm2, whereas the DRM2cat-Myc and DRM2uba-Myc lines showed a complete and partial failure to remethylate MEA-ISR respectively (Figure 4C). We confirmed these observations by sequencing MEA-ISR following sodium bisulfite conversion; non-CG methylation was restored by DRM2-Myc but not by DRM2cat-Myc, whereas the DRM2uba-Myc transformants showed a low level of non-CG methylation, comparable to the level observed in drm3-1 (Figure 2B, Figure 4D, Table S1 and Figure S3). Therefore, intact UBA and methyltransferase domains are required for DRM2 function during maintenance of non-CG DNA methylation.
To test whether the DRM2 mutations also blocked establishment of DNA methylation we transformed DRM2-Myc drm1 drm2, DRM2cat-Myc drm1 drm2 and DRM2uba-Myc drm1 drm2 with FWA and used the flowering-time distribution within T1 populations as a measure of silencing. The FWA T1 populations generated in DRM2cat-Myc drm1 drm2, DRM2uba-Myc drm1 drm2 backgrounds flowered significantly later than the wild type DRM2-Myc drm1 drm2 control population (Figure 5A and Table 1). We interpret this as meaning that intact UBA and methyltransferase domains are also required to establish RNA–directed DNA methylation and silencing at FWA.
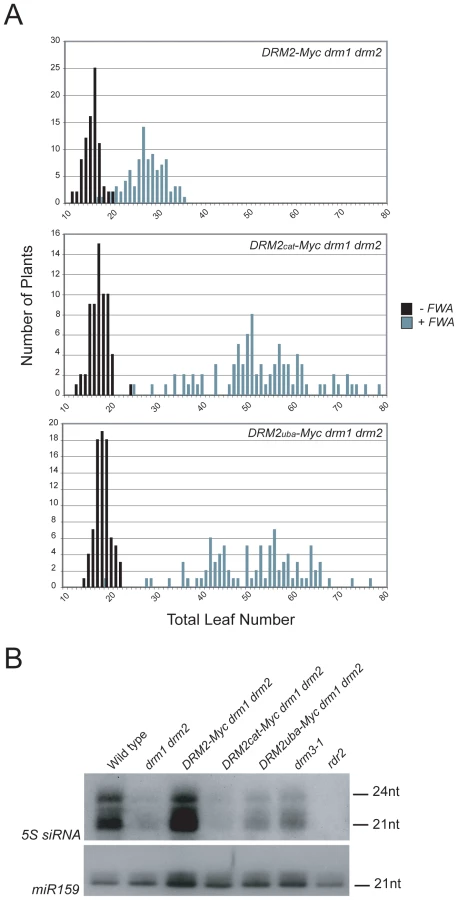
Although DRM2 is targeted by siRNAs, it is also required for accumulation of specific siRNA populations, for example siRNAs homologous to the 5S ribosomal DNA genes [29], which may reflect a feedback loop between transcriptional silencing and siRNA biogenesis. 5S siRNAs detected by northern blotting and hybridization are absent in rdr2 and greatly reduced in drm1 drm2, relative to wild type (Figure 5B) [29]. The DRM2-Myc transgene is able to restore 5S siRNA accumulation in the drm1 drm2 mutant, but this complementation is not evident with either DRM2cat-Myc or DRM2uba-Myc (Figure 5B). Consistent with DRM3 functioning to promote DRM2 activity, we also observed a strong reduction in the accumulation of 5S siRNAs in drm3-1 mutants (Figure 5B). Together these data demonstrate that both the DRM2 catalytic methyltransferase and UBA domains are required for establishment and maintenance of RNA–directed DNA methylation in vivo.
Discussion
A complex pathway of genes has been defined that generates siRNAs homologous to repeated sequences, which can guide DRM2 cytosine methyltransferase activity to target DNA sequences [16]–[34]. Here we define a new member of this pathway, the catalytically mutated DNA cytosine methyltransferase paralog DRM3, which is required for normal maintenance of non-CG DNA methylation, establishment of RNA–directed DNA methylation triggered by tandem repeats and accumulation of repeat associated siRNAs. As drm3 phenotypes are weaker than drm2 we believe that DRM3 acts to promote activity of DRM2, but is not an absolute requirement. The strength of drm3 phenotypes resembles those observed for dcl3, which is defective in accumulation of 24 nt siRNA known to promote RNA–directed DNA methylation. As the drm3-1 dcl3-1 double mutant shows an enhanced phenotype to the level of drm2, this suggests that the combined loss of 24 nt siRNA and DRM3 severely hinders DRM2 activity. The drm3 mutation does not influence DRM2 expression or protein accumulation, leading us to believe that DRM3 acts together with DRM2 at the step of catalysis. Indeed, several examples exist of catalytically inactive paralogs acting to stimulate the activity of catalytic partners [65], [66]. In this respect the stimulatory interaction between Dnmt3a and Dnmt3L is particularly notable [42]–[46]. Despite extensive testing we have been unable to find in vitro conditions where A.thaliana DRM2 is active as a cytosine methyltransferase, which has precluded us testing a direct stimulatory effect for DRM3 on catalysis. Development of these assays will be an important next step to dissect the function of DRM2 and DRM3 during RNA–directed DNA methylation.
Although an intriguing parallel exists between the functional relationships of DRM2/DRM3 and Dnmt3a/Dnmt3L, phylogenetic analysis strongly suggests that these protein families diverged independently in plants and animals (Figure 1A). There are also important differences between DRM and Dnmt3 in the mechanism of recruitment to genomic target sites. The Dnmt3L protein plays a key role in targeting Dnmt3 via an N-terminal PHD domain, which specifically recognizes histone 3 unmethylated at lysine 4 [41]–[46]. By analogy the N-terminal UBA domains of DRM2 may be involved in targeting DNA methyltransferase activity. Supporting this idea we have shown that the DRM2 UBA domains are critical for RNA directed DNA methylation. We do not currently know whether DRM3 UBA domains are also functionally important. Sequence analysis shows that all DRM3 proteins possess two UBA domains, the second of which contains conserved residues known to be important for proper domain folding, including the MGF/MGY motif in loop-I (Figure S2) [64]. Interestingly, the first UBA domain of DRM3 proteins show substitution of the MGF/MGY glycine for either lysine or asparagine, which would be predicted to abolish proper folding [64]. However, as UBA domains are able to bind ubiquitin independently, DRM3 UBA domains may still play a functional role [63], [64]. The precise functions of the DRM UBA domains are currently unknown, though it is interesting to speculate that they may recognize specific chromatin modifications during RdDM.
In mammals PIWI proteins together with interacting piRNA act upstream of Dnmt3a/Dnmt3L to DNA methylate homologous repeats during germ line development [40], [42]–[48]. Plants lack orthologs of the PIWI proteins and have not so far been observed to produce piRNA [67]. Thus, small RNA–directed DNA methylation pathways appear to have evolved independently in plants and animals. Equally, the specific rearrangement of methyltransferase catalytic motifs shared between all DRM proteins indicates that DRM and Dnmt3 families have diverged independently in plants and animals (Figure 1). Here we demonstrate a further example of convergence in the parallel evolution of catalytically mutated cytosine methyltransferase paralogs (DRM3 and Dnmt3L) in both lineages. It is also important to note that information other than small RNAs must be involved in targeting DNA methylation, as this mark can be established and maintained in Neurospora crassa RNAi mutants [53], and no evidence exists of small RNAs acting upstream of Dnmt3a in the mammalian female germ line [48]. In conclusion, comparison of DRM3 and Dnmt3L function provides an example of mechanistic convergence between plant and mammalian silencing systems.
Materials and Methods
Plant materials
The drm3-1 allele is T-DNA insertion Salk_136439 and drm3-2 allele is T-DNA insertion Sail_1215_B08. The presence of the drm3-1 insertion can be genotyped by PCR amplification using JP3192 together with a primer to the T-DNA left border JP2410 (all oligonucleotide sequences are listed in Table S4). The presence of the drm3-2 insertion can be genotyped using primers JP3510 and JP2027. The drm1 drm2 and dcl3-1 T-DNA insertions have been described previously [7], [16]. DRM3 complementation was performed by PCR amplification of an 8kb DRM3 genomic region from Columbia DNA using oligonucleotides JP3477 and JP3479 and cloned into pCR2.1 (Invitrogen). The DRM3 genomic region then was cloned into the pCAMBIA1300 binary vector as a SalI restriction fragment and used to transform drm3-1 plants by floral dipping using Agrobacterium strain ASE. The DRM2-Myc transgene has been described previously [58]. This construct was mutagenized using Quikchange (Stratagene) using oligonucleotides JP603/JP604 (catalytic mutation C587A), JP2631/JP2632 (UBA domain 1 F73A), JP2633/JP2634 (UBA domain 2 F123A) and JP2635/JP2636 (UBA domain 3 F206A).
Phylogenetic tree construction
Multiple sequence alignment was performed using the European Bioinformatics Institute CLUSTALW server (http://www.ebi.ac.uk/clustalw/). NCBI accession numbers or gene numbers for the proteins analyzed are (Arabidopsis thaliana) AtDRM1 At5g15380, AtDRM2 At5g14620, AtDRM3 At3g17310, (Populus trichocarpa) PtDRM2 DMT905, PtDRM3 DMT907, (Oryzae sativa) OsDRM2 Os3g02010, OsDRM3 Os5g04330, (Homo sapiens) HsDnmt3a NP_783328, HsDnmt3b CAB53071, HsDnm3L BAA95556, (Mus musculus) MmDnmt3a O88508, MmDnmt3b CAM27225, MmDnmt3L AAH83147 and (Haemophilus parahaemolyticus) HhaI P05102. The phylogenetic tree was constructed using the MEGA2 program using the neighbor-joining method. Bootstrap values were calculated with 1,000 replicates.
Analysis of DNA methylation
Southern blotting and hybridization were performed using 1µg of genomic DNA digested overnight using MspI and separated by gel electrophoresis using 0.8% agarose and blotted onto N+Hybond (Amersham) nitrocellulose. The MEA-ISR probe was amplified using primers JP980 and JP981. 1µg of genomic DNA was converted using a Methyleasy kit (Human Genetic Signatures) and MEA-ISR amplified using primers JP1026 and JP1027, corresponding to positions 53456–53602 of BAC clone T14P4 [15], [36]. PCR products were cloned using the TOPO TA cloning kit (Invitrogen) and sequenced using the T7 primer. The FWA promoter region bisulfite sequenced corresponds to co-ordinates 46062 to 46551 of BAC clone M7J2. This region was amplified using primers JP2004 and JP2005 followed by nested amplification with primers JP2004 and JP4423. During analysis of sodium bisulfite sequencing data the percentage of methylated sites across all clones were calculated for each sequence context. The Wilson score interval was used to find the 95% confidence intervals for these percentages. Percentages were compared with the appropriate wild type percentage using the Pearson chi square test (Table S1 and Table S2). To compensate for multiple testing using the Bonferroni adjustment, a p-value of 0.002 was used as the threshold for statistical significance. To analyze cytosine methylation at AtSN1 genomic DNA was digested with HaeIII in 40 µl alongside controls without restriction endonuclease. Samples were then analyzed by quantitative real-time PCR amplifying with primers JP6699 and JP6700 combined with Biorad SYBR Green Supermix using a MX3000 Stratagene cycler. A ratio was then calculated between the amount of DNA amplified for each sample with and without HaeIII digestion.
FWA transformation and flowering-time analysis
Plants were transformed via floral dipping using Agrobacterium tumefaciens strain ASE carrying a genomic FWA clone in the binary vector pCAMBIA1300. T1 transformant seed was selected by germination and growth on MS plates containing 50µg/ml hygromycin followed by transfer to soil under continuous light. Vegetative and cauline leaves were counted until production of the first flowers as a measure of flowering-time. Untransformed plants were grown alongside as controls.
RNA analysis
1 gram of floral buds were used to extract and purify small RNAs as described previously [60]. Small RNAs were separated by gel electrophoresis, blotted onto nitrocellulose membrane and hybridized with end-labeled oligonucleotide probes miR159 (5′-TAGAGCTCCCTTCAATCCAAA-3′) and 5S siRNA (5′-ATGCCAAGTTTGGCCTCACGGTCT-3′) [61]. End-labeling was performed using T4 polynucleotide kinase (New England Biolabs) and γ-32P-ATP (Amersham). To analyze mRNA accumulation total RNA was extracted using Trizol (Invitrogen) and converted to cDNA as described previously [53]. DRM2 expression was analyzed using qRT-PCR with primers JP6069 and JP6070 and DRM3 analyzed by RT-PCR using primers JP3192 and JP3193.
Immunohistochemistry
Immunostaining of fixed nuclei was performed as previously reported [68].
Supporting Information
Zdroje
1. AravinAA
Bourc'hisD
2008 Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev 22 970 975
2. LawJA
JacobsenSE
2010 Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11 204 220
3. CokusSJ
FengS
ZhangX
ChenZ
MerrimanB
2008 Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452 215 219
4. LippmanZ
GendrelAV
BlackM
VaughnMW
DedhiaN
2004 Role of transposable elements in heterochromatin and epigenetic control. Nature 430 471 476
5. ListerR
O'MalleyRC
Tonti-FilippiniJ
GregoryBD
BerryCC
2008 Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133 523 536
6. TranRK
HenikoffJG
ZilbermanD
DittRF
JacobsenSE
2005 DNA methylation profiling identifies CG methylation clusters in Arabidopsis genes. Curr Biol 15 154 159
7. ZhangX
YazakiJ
SundaresanA
CokusS
ChanSW
2006 Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 126 1189 1201
8. ZilbermanD
GehringM
TranRK
BallingerT
HenikoffS
2007 Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39 61 69
9. KatoM
MiuraA
BenderJ
JacobsenSE
KakutaniT
2003 Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr Biol 13 421 426
10. MirouzeM
ReindersJ
BucherE
NishimuraT
SchneebergerK
2009 Selective epigenetic control of retrotransposition in Arabidopsis. Nature 461 427 430
11. MiuraA
YonebayashiS
WatanabeK
ToyamaT
ShimadaH
2001 Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature 411 212 214
12. TsukaharaS
KobayashiA
KawabeA
MathieuO
MiuraA
2009 Bursts of retrotransposition reproduced in Arabidopsis. Nature 461 423 426
13. GollMG
BestorTH
2005 Eukaryotic cytosine methyltransferases. Annu Rev Biochem 74 481 514
14. BestorTH
VerdineGL
1994 DNA methyltransferases. Curr Opin Cell Biol 6 380 389
15. CaoX
JacobsenSE
2002 Role of the arabidopsis DRM methyltransferases in de novo DNA methylation and gene silencing. Curr Biol 12 1138 1144
16. ChanSW
ZilbermanD
XieZ
JohansenLK
CarringtonJC
2004 RNA silencing genes control de novo DNA methylation. Science 303 1336
17. HendersonIR
JacobsenSE
2008 Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev 22 1597 1606
18. AusinI
MocklerTC
ChoryJ
JacobsenSE
2009 IDN1 and IDN2 are required for de novo DNA methylation in Arabidopsis thaliana. Nat Struct Mol Biol 16 1325 1327
19. El-ShamiM
PontierD
LahmyS
BraunL
PicartC
2007 Reiterated WG/GW motifs form functionally and evolutionarily conserved ARGONAUTE-binding platforms in RNAi-related components. Genes Dev 21 2539 2544
20. HeXJ
HsuYF
PontesO
ZhuJ
LuJ
2009 NRPD4, a protein related to the RPB4 subunit of RNA polymerase II, is a component of RNA polymerases IV and V and is required for RNA–directed DNA methylation. Genes Dev 23 318 330
21. HuangL
JonesAM
SearleI
PatelK
VoglerH
2009 An atypical RNA polymerase involved in RNA silencing shares small subunits with RNA polymerase II. Nat Struct Mol Biol 16 91 93
22. JohnsonLM
LawJA
KhattarA
HendersonIR
JacobsenSE
2008 SRA-domain proteins required for DRM2-mediated de novo DNA methylation. PLoS Genet 4 e1000280
23. KannoT
BucherE
DaxingerL
HuettelB
BohmdorferG
2008 A structural-maintenance-of-chromosomes hinge domain-containing protein is required for RNA–directed DNA methylation. Nat Genet 40 670 675
24. KannoT
HuettelB
MetteMF
AufsatzW
JaligotE
2005 Atypical RNA polymerase subunits required for RNA–directed DNA methylation. Nat Genet 37 761 765
25. KannoT
MetteMF
KreilDP
AufsatzW
MatzkeM
2004 Involvement of putative SNF2 chromatin remodeling protein DRD1 in RNA–directed DNA methylation. Curr Biol 14 801 805
26. LahmyS
PontierD
CavelE
VegaD
El-ShamiM
2009 PolV(PolIVb) function in RNA–directed DNA methylation requires the conserved active site and an additional plant-specific subunit. Proc Natl Acad Sci U S A 106 941 946
27. OnoderaY
HaagJR
ReamT
NunesPC
PontesO
2005 Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell 120 613 622
28. PontierD
YahubyanG
VegaD
BulskiA
Saez-VasquezJ
2005 Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes Dev 19 2030 2040
29. ZilbermanD
CaoX
JohansenLK
XieZ
CarringtonJC
2004 Role of Arabidopsis ARGONAUTE4 in RNA–directed DNA methylation triggered by inverted repeats. Curr Biol 14 1214 1220
30. Bies-EtheveN
PontierD
LahmyS
PicartC
VegaD
2009 RNA–directed DNA methylation requires an AGO4-interacting member of the SPT5 elongation factor family. EMBO Rep 10 649 654
31. HeXJ
HsuYF
ZhuS
LiuHL
PontesO
2009 A conserved transcriptional regulator is required for RNA–directed DNA methylation and plant development. Genes Dev 23 2717 2722
32. HeXJ
HsuYF
ZhuS
WierzbickiAT
PontesO
2009 An effector of RNA–directed DNA methylation in arabidopsis is an ARGONAUTE 4- and RNA–binding protein. Cell 137 498 508
33. HerrAJ
JensenMB
DalmayT
BaulcombeDC
2005 RNA polymerase IV directs silencing of endogenous DNA. Science 308 118 120
34. SmithLM
PontesO
SearleI
YelinaN
YousafzaiFK
2007 An SNF2 protein associated with nuclear RNA silencing and the spread of a silencing signal between cells in Arabidopsis. Plant Cell 19 1507 1521
35. CaoX
AufsatzW
ZilbermanD
MetteMF
HuangMS
2003 Role of the DRM and CMT3 methyltransferases in RNA–directed DNA methylation. Curr Biol 13 2212 2217
36. CaoX
JacobsenSE
2002 Locus-specific control of asymmetric and CpNpG methylation by the DRM and CMT3 methyltransferase genes. Proc Natl Acad Sci U S A 99 Suppl 4 16491 16498
37. ChanSW
HendersonIR
ZhangX
ShahG
ChienJS
2006 RNAi, DRD1, and histone methylation actively target developmentally important non-CG DNA methylation in arabidopsis. PLoS Genet 2 e83
38. KanedaM
OkanoM
HataK
SadoT
TsujimotoN
2004 Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429 900 903
39. OkanoM
BellDW
HaberDA
LiE
1999 DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99 247 257
40. Bourc'hisD
BestorTH
2004 Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431 96 99
41. Bourc'hisD
XuGL
LinCS
BollmanB
BestorTH
2001 Dnmt3L and the establishment of maternal genomic imprints. Science 294 2536 2539
42. GowherH
LiebertK
HermannA
XuG
JeltschA
2005 Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol Chem 280 13341 13348
43. SuetakeI
MorimotoY
FuchikamiT
AbeK
TajimaS
2006 Stimulation effect of Dnmt3L on the DNA methylation activity of Dnmt3a2. J Biochem 140 553 559
44. KaretaMS
BotelloZM
EnnisJJ
ChouC
ChedinF
2006 Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J Biol Chem 281 25893 25902
45. JiaD
JurkowskaRZ
ZhangX
JeltschA
ChengX
2007 Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449 248 251
46. OoiSK
QiuC
BernsteinE
LiK
JiaD
2007 DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 448 714 717
47. AravinAA
HannonGJ
2008 Small RNA silencing pathways in germ and stem cells. Cold Spring Harb Symp Quant Biol 73 283 290
48. AravinAA
SachidanandamR
Bourc'hisD
SchaeferC
PezicD
2008 A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell 31 785 799
49. CarmellMA
GirardA
van de KantHJ
Bourc'hisD
BestorTH
2007 MIWI2 is essential for spermatogenesis and repression of transposons in the mouse male germline. Dev Cell 12 503 514
50. DengW
LinH
2002 miwi, a murine homolog of piwi, encodes a cytoplasmic protein essential for spermatogenesis. Dev Cell 2 819 830
51. Kuramochi-MiyagawaS
KimuraT
IjiriTW
IsobeT
AsadaN
2004 Mili, a mammalian member of piwi family gene, is essential for spermatogenesis. Development 131 839 849
52. Kuramochi-MiyagawaS
WatanabeT
GotohK
TotokiY
ToyodaA
2008 DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev 22 908 917
53. FreitagM
LeeDW
KotheGO
PrattRJ
AramayoR
2004 DNA methylation is independent of RNA interference in Neurospora. Science 304 1939
54. CaoX
SpringerNM
MuszynskiMG
PhillipsRL
KaepplerS
2000 Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc Natl Acad Sci U S A 97 4979 4984
55. JeddelohJA
BenderJ
RichardsEJ
1998 The DNA methylation locus DDM1 is required for maintenance of gene silencing in Arabidopsis. Genes Dev 12 1714 1725
56. SazeH
KakutaniT
2007 Heritable epigenetic mutation of a transposon-flanked Arabidopsis gene due to lack of the chromatin-remodeling factor DDM1. Embo J 26 3641 3652
57. WooHR
DittmerTA
RichardsEJ
2008 Three SRA-domain methylcytosine-binding proteins cooperate to maintain global CpG methylation and epigenetic silencing in Arabidopsis. PLoS Genet 4 e1000156
58. LiCF
PontesO
El-ShamiM
HendersonIR
BernatavichuteYV
2006 An ARGONAUTE4-containing nuclear processing center colocalized with Cajal bodies in Arabidopsis thaliana. Cell 126 93 106
59. SoppeWJ
JacobsenSE
Alonso-BlancoC
JacksonJP
KakutaniT
2000 The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol Cell 6 791 802
60. HendersonIR
ZhangX
LuC
JohnsonL
MeyersBC
2006 Dissecting Arabidopsis thaliana DICER function in small RNA processing, gene silencing and DNA methylation patterning. Nat Genet 38 721 725
61. XieZ
JohansenLK
GustafsonAM
KasschauKD
LellisAD
2004 Genetic and functional diversification of small RNA pathways in plants. PLoS Biol 2 E104
62. GasciolliV
MalloryAC
BartelDP
VaucheretH
2005 Partially redundant functions of Arabidopsis DICER-like enzymes and a role for DCL4 in producing trans-acting siRNAs. Curr Biol 15 1494 1500
63. KozlovG
NguyenL
LinT
De CrescenzoG
ParkM
2007 Structural basis of ubiquitin recognition by the ubiquitin-associated (UBA) domain of the ubiquitin ligase EDD. J Biol Chem 282 35787 35795
64. MuellerTD
FeigonJ
2002 Solution structures of UBA domains reveal a conserved hydrophobic surface for protein-protein interactions. J Mol Biol 319 1243 1255
65. KimSA
VacratsisPO
FiresteinR
ClearyML
DixonJE
2003 Regulation of myotubularin-related (MTMR)2 phosphatidylinositol phosphatase by MTMR5, a catalytically inactive phosphatase. Proc Natl Acad Sci U S A 100 4492 4497
66. WillertEK
FitzpatrickR
PhillipsMA
2007 Allosteric regulation of an essential trypanosome polyamine biosynthetic enzyme by a catalytically dead homolog. Proc Natl Acad Sci U S A 104 8275 8280
67. ShabalinaSA
KooninEV
2008 Origins and evolution of eukaryotic RNA interference. Trends Ecol Evol 23 578 587
68. LiCF
HendersonIR
SongL
FedoroffN
LagrangeT
2008 Dynamic regulation of ARGONAUTE4 within multiple nuclear bodies in Arabidopsis thaliana. PLoS Genet 4 e27
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 10
- Souvislost haplotypu M2 genu pro annexin A5 s opakovanými reprodukčními ztrátami
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Příjem alkoholu a menstruační cyklus
Nejčtenější v tomto čísle
- Genome-Wide Identification of Targets and Function of Individual MicroRNAs in Mouse Embryonic Stem Cells
- Common Genetic Variants and Modification of Penetrance of -Associated Breast Cancer
- Allele-Specific Down-Regulation of Expression Induced by Retinoids Contributes to Climate Adaptations
- Simultaneous Disruption of Two DNA Polymerases, Polη and Polζ, in Avian DT40 Cells Unmasks the Role of Polη in Cellular Response to Various DNA Lesions