
Shedding Light on the Elusive Role of Endothelial Cells in Cytomegalovirus Dissemination
Cytomegalovirus (CMV) is frequently transmitted by solid organ transplantation and is associated with graft failure. By forming the boundary between circulation and organ parenchyma, endothelial cells (EC) are suited for bidirectional virus spread from and to the transplant. We applied Cre/loxP-mediated green-fluorescence-tagging of EC-derived murine CMV (MCMV) to quantify the role of infected EC in transplantation-associated CMV dissemination in the mouse model. Both EC- and non-EC-derived virus originating from infected Tie2-cre+ heart and kidney transplants were readily transmitted to MCMV-naïve recipients by primary viremia. In contrast, when a Tie2-cre+ transplant was infected by primary viremia in an infected recipient, the recombined EC-derived virus poorly spread to recipient tissues. Similarly, in reverse direction, EC-derived virus from infected Tie2-cre+ recipient tissues poorly spread to the transplant. These data contradict any privileged role of EC in CMV dissemination and challenge an indiscriminate applicability of the primary and secondary viremia concept of virus dissemination.
Published in the journal:
. PLoS Pathog 7(11): e32767. doi:10.1371/journal.ppat.1002366
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002366
Summary
Cytomegalovirus (CMV) is frequently transmitted by solid organ transplantation and is associated with graft failure. By forming the boundary between circulation and organ parenchyma, endothelial cells (EC) are suited for bidirectional virus spread from and to the transplant. We applied Cre/loxP-mediated green-fluorescence-tagging of EC-derived murine CMV (MCMV) to quantify the role of infected EC in transplantation-associated CMV dissemination in the mouse model. Both EC- and non-EC-derived virus originating from infected Tie2-cre+ heart and kidney transplants were readily transmitted to MCMV-naïve recipients by primary viremia. In contrast, when a Tie2-cre+ transplant was infected by primary viremia in an infected recipient, the recombined EC-derived virus poorly spread to recipient tissues. Similarly, in reverse direction, EC-derived virus from infected Tie2-cre+ recipient tissues poorly spread to the transplant. These data contradict any privileged role of EC in CMV dissemination and challenge an indiscriminate applicability of the primary and secondary viremia concept of virus dissemination.
Introduction
Human Cytomegalovirus (HCMV), a member of the betaherpesvirus subfamily, represents an important opportunistic viral pathogen in the immune compromised host. Fetuses, AIDS patients, and recipients of both bone marrow and solid organ transplants are at high risk for the development of debilitating and potentially life-threatening CMV disease. Depending on the risk constellation and immunosuppressive regimen, CMV disease can occur in up to 60% of heart or kidney transplant recipients. Therefore, HCMV is the most important viral pathogen especially during the first six months after transplantation [1], [2]. The large variety of symptoms results from the broad cell and organ tropism of the virus [3], [4]. In addition, the virus is able to disseminate via blood [5]. According to Fenner (1949) a virus enters - after initial replication at the entry site (epithelia or transplant) - the blood stream and disseminates throughout the body to distal organs via a so-called primary viremia, which was confirmed to apply also to HCMV and MCMV [6], [7]. It is proposed that progeny virus from such organs can re-enter the blood circulation leading to a secondary viremia [6], [7] thus increasing the risk for widespread dissemination.
Leukocyte depletion of blood products derived from seropositive donors prior to transfusion efficiently prevents transfer of CMV to seronegative recipients [8], [9] indicating that virus present in blood is predominantly cell associated. The cell types responsible for this dissemination are of particular interest. Three kinds of cells have been suggested to be involved in virus dissemination via blood. All of them have been shown to be able to transfer infectious virus ex vivo: polymorphonuclear leukocytes (PMNL), monocytes/macrophages, and detached infected vascular endothelial cells (EC). Although PMNL are thought to be only abortively infected, they might still function as vehicles for infectious virus [10]. Circulating infected monocytes become permissive upon differentiation into tissue macrophages and may then release infectious progeny within target organs [11]. For example, rat CMV was transferred via in vitro infected granulocytes or monocytes [12]. Vascular EC are suggested to play an important role in CMV dissemination, and unique genetic features govern the CMV - EC interaction [13]. EC support productive infection and may detach upon infection thus serving as shuttles for the virus to other organs via the blood stream [14], [15], [16]. EC are permissive for HCMV in vitro [3] and are commonly found to be infected in tissue samples from both immune compromised patients [17] and mice [18]. In addition, EC support latent infection with the potential to reactivate CMV [19] and to start a new episode of infection. Notably, HCMV infection is a risk factor for restenosis after coronary atherectomy [20] and accelerates atherosclerosis following cardiac transplantation [21]. The anatomical position of EC lining blood vessels implies a bidirectional role in virus entry into and exit from the blood circulation and therefore might define the ability of viruses in general to disseminate via blood. In fact, HCMV-infected EC can protrude from the wall into the lumen of the blood vessels in patients with active cytomegalovirus infection [16]. Furthermore, circulating giant endothelial cells were found in blood samples of transplant patients [14] suggesting detachment of infected EC from the vessel wall and dissemination of HCMV via EC throughout the body.
Despite the undisputed and unique potential of EC in CMV infection and pathogenesis, it is still unknown whether infected EC are responsible for systemic virus dissemination during primary infection, contribute to this process, or merely represent an epiphenomenon with no causal involvement in the pathogenesis of organ disease [22]. Quantitative aspects of the contribution of infected EC to virus dissemination in the transplant situation are scarce and the presence of infected EC in the circulating blood does not prove that infected EC or HCMV produced by EC contribute or even govern virus dissemination from one site or organ to another.
To quantify and to address the fate of virus produced by specific cells, we developed a Cre/loxP mediated approach to label virus in defined cell types in vivo and then trace the viral progeny of that cell type [23], [24]. Cre recombinase recognizes two adjacent loxP sites and deletes the intervening DNA sequence. This reaction can remove a transcriptional stop signal between promoter and coding sequence resulting in gene expression. To study the role of EC in MCMV replication an MCMV mutant was used that contains a Cre-inducible egfp expression cassette (MCMV-flox). Mice expressing Cre recombinase under control of either the Tie2 or the Tek promoter, which is selectively expressed in vascular EC (Tie2-cre and Tek-cre mice), were infected with MCMV-flox. In this in vivo infection model MCMV-flox is efficiently recombined resulting in MCMV-rec only during virus replication in EC. It is important to note that Cre-mediated recombination of MCMV-flox is equally efficient in Tie2- and Tek-cre mice and only mediated by EC - as shown using bone marrow chimeras - thus providing highly concordant results by both mouse strains [24]. The resulting recombination is then stably maintained in the viral genome of the virus progeny.
Vascular EC are present in all organs. A way to study the role of EC in virus dissemination from one organ to another is to either introduce organs from an EC cre-negative donor mouse into an EC cre-positive host or vice versa. Here, we investigated export of EC-derived virus from heart and kidney transplants to recipients as well as import of EC-derived virus from recipients into heart transplants. This was achieved by counting and comparing the contribution of EGFP-positive EC-derived progeny to the total virus load of organs and tissues.
EC-derived virus from infected heart or kidney transplants readily disseminated to organs of MCMV-naïve recipients. The bulk of virus produced in and disseminated from heart is EC-derived, whereas in kidneys infected EC only provide a minor contribution. Yet, we found no evidence for any preferential dissemination of EC-derived virus from both types of transplants to other organs. The heart transplant was also tested as a target organ of EC-derived virus produced in recipient tissues. To our surprise, in contrast to the strong dissemination of virus originating from an infected transplant there was only minimal seeding of host EC-derived virus progeny to the transplant. Interestingly, this was independent of whether transplantation was performed prior to or after systemic host infection. In summary, our data argue against a privileged role of EC in virus dissemination.
Results
Virus dissemination from infected hearts into non-infected recipients
Transplantation of organs from HCMV seropositive donors to seronegative recipients (D+/R-) is a known situation in transplantation medicine and represents the “high risk constellation” because up to 60% of the recipients can develop CMV disease [25]. In this D+R- setting CMV disease is caused by dissemination of HCMV from the transplanted organ to the recipient causing systemic symptoms with multiple organs being involved. The cellular source of disseminated virus has not been addressed, yet virus dissemination from infected heart transplants has also been described in the mouse model [26], [27]. To investigate whether and to which extent virus derived from EC of the transplant disseminates to organs of uninfected recipients, hearts from acutely infected Tie2-cre mice were transplanted heterotopically into non-infected syngeneic C57BL/6 mice (Fig. 1A). Four days after transplantation mice were sacrificed and organs collected to determine the amounts of non-recombined (non-EC-derived) and recombined (EC-derived) virus. In the heart transplant, high virus loads (∼105 PFU/g organ) of predominantly recombined virus (∼85%) were observed, confirming that the transplantation procedure itself did not affect MCMV replication in general and demonstrating a very high recombination efficiency (Fig. 1B). This is in accordance with high recombination efficiency observed previously for heart and lungs of Tie2-cre mice [24]. Virus titers in different organs of mice infected via the heart transplant were 10 to 10,000-fold lower than generally seen following systemic (i.v.) infection with ∼1×106 PFU [24]. The relative amounts of MCMV-rec and MCMV-flox in the recipient organs, however, essentially reflected the situation in the heart transplant, with some minor variance. Thus, EC-derived virus virtually disseminated equally well as non-EC-derived virus from the heart transplant.

Virus dissemination from infected kidneys into non-infected recipients
Next, we studied dissemination of MCMV following kidney transplantation. Kidneys represent the majority of transplanted organs in medicine. Similar to heart transplantation, the transplantation of kidneys from seropositive donors to seronegative recipients is associated with a high risk to develop CMV-related complications [28], [29], [30]. Four days after heterotopic transplantation of infected kidneys of Tie2-cre mice into non-infected C57BL/6 mice, recipient organs were analyzed for the presence of disseminated virus (Fig. 2A). In contrast to the heart, only about 20% of virus within transplanted kidneys was recombined (Fig. 2B). This low contribution of EC-derived virus to total virus load in kidney is in line with previous observations [24]. As recombination rates were similar in Tie2-cre and Tek-cre mice, we believe that the low proportion of MCMV-rec in Tie2-cre kidneys does not necessarily indicate a low recombination efficiency in renal EC but may rather result from an alternative mode of virus entry into kidney tissue bypassing the vascular endothelium for replication in other cell types, one candidate being kidney epithelial cells. The relative levels of virus titers in liver, spleen, and lungs were comparable to those observed following heart transplantation. Yet, the percentage of recombined, EC-derived virus in most organs essentially mirrored the situation in the transplanted kidney, and there was no preferential dissemination of EC-derived virus (∼20%). Collectively, the findings after transplantation of two different organs did not support the hypothesis of a predominant role of EC in virus dissemination during the first four days of infection. Only in blood a significantly higher proportion of MCMV-rec was found on day four post kidney transplantation in 3 out of 4 mice. However, as the absolute virus titers were close to the detection limit, any interpretation has to be seen with caution.

Minor contribution of EC-derived virus from the transplanted heart to virus dissemination in the systemically infected host
In the preceding experiments, the systemic infection originated from a pre-infected transplanted organ. Next, we studied the contribution of EC of a transplanted Tie2-cre+ heart to virus dissemination during the situation of systemic infection of C57BL/6 recipients. Under these conditions, all organs, including the transplanted heart, become infected simultaneously. Thus, MCMV-rec, wherever found, must have originated from ECs of the transplant. Note that under such conditions the infection of the transplant does not have a head start. Four days after transplantation mice were systemically (i.v.) infected with MCMV-flox and four days later they were sacrificed and virus titers determined (Fig. 3A). As expected, the majority of virus in the transgenic heart transplant was found to be recombined (Fig. 3B). Despite this, we observed only very little dissemination of EGFP+ EC-derived MCMV from the transplant to infected recipient organs. In the lungs, some MCMV-rec was found at very low numbers, four orders of magnitude lower than MCMV-flox, whereas in other organs MCMV-rec was at or below detection limit. This result is in stark contrast to the dissemination from the infected transplant (Fig. 1) where 70-90% of virus progeny in recipient organs were EC-derived. It is important to note that total virus titers in both the endogenous and transplanted heart were very similar (Fig. 3B), indicating efficient vascularization of the heterotopic heart transplant after surgery. This excludes an impaired blood flow as a presumed reason for the observed poor dissemination of recombined virus. We thus conclude that virus dissemination from the heart plays a negligible role during systemic infection.

Another striking difference between virus dissemination from both transplanted heart and kidney as compared to systemic (i.v.) infection was the extent of virus production in different organs. In contrast to i.v. infection, which resulted in peak titers in the lung and high titers in heart, kidney, liver, spleen and adipose tissue (Fig. 3), virus dissemination from transplanted heart and kidney (Figs. 1 and 2) resulted in peak titers in spleen but significantly lower titers in liver and lung, and in almost no virus detectable in the endogenous heart, kidneys, and adipose tissue. This cannot simply be explained by organ specific differences in virus production kinetics [24] but rather indicates a qualitative difference in virus dissemination between systemic (i.v.) infection (free virus) and transplant-mediated infection.
Limited colonization of the heart by EC-derived virus via secondary viremia
During systemic infection following transplantation of a cre-positive heart to a cre-negative mouse no significant contribution of virus dissemination from the heart transplant to other organs was observed. To study not only cardiac EC but EC in general as a source of virus dissemination, Tie2-cre or Tek-cre recipient mice received a non-transgenic heart. Recipients were then infected i.v. with MCMV-flox (Fig. 4A). As expected, the host organs showed the previously described organ-specific contribution of EC to total virus load [24]. Specifically, in liver the bulk of virus is derived from hepatocytes as we recently showed using Alb-cre mice selectively expressing Cre recombinase in hepatocytes [24], whereas the cell type producing the bulk of virus in kidney remains to be determined. In all other organs, >60% of virus proved to be EC-derived. Yet, although the transplanted heart contained a total virus load comparable to that of the endogenous heart, there was only a minute (about 1%) contribution of recombined virus to the amount of virus in the transplant (Fig. 4B). We repeated the experiments in Tek-cre mice, another mouse line transgenic for cre in EC, and obtained essentially the same results (Fig. 4C). To confirm that this small contribution of MCMV-rec to the infection of a heart transplant was truly due to virus seeding to the organ and not just reflected virus present in the circulation, organ perfusion was performed in order to flush out blood cells prior to analysis (Fig. 4C). In any case, the data revealed an only minute dissemination of EC-derived virus via secondary viremia following systemic infection.

Dissemination of EC-derived MCMV from an infected host to a transplant
The low degree of dissemination of MCMV-rec into the heart could be the result of two scenarios. We expected that the immune response induced by systemic infection actively prevented secondary import of EC-derived virus into the transplant. Alternatively, after initial virus seeding by systemic (i.v.) infection, local virus production might simply outnumber secondary import of EC-derived virus. To address this issue and to initiate the activation of immune functions, systemic infection was performed prior to transplantation. Specifically, Tie2-cre or Tek-cre mice were first i.v. infected with MCMV-flox and only then received heart transplants of non-infected C57BL/6 mice either 20 h or 3 days after infection (Fig. 5A/B). Strikingly, systemic infection prior to transplantation increased the relative contribution of EC-derived virus in the transplant from ∼5% (Fig. 4B) to ∼60% independent of the time delay between infection and transplantation (Fig. 5A/B). This average of about 60% MCMV-rec reflects the average contribution of MCMV-rec in the organism in general. However, total virus titers in the heart transplant were 100- to 1000-fold lower than in both the endogenous heart exposed to i.v. infection as well as the hearts transplanted prior to i.v. infection (Fig. 4B/C). It is important to note that the absolute amounts of recombined virus in the heart transplants (grey circles in Fig. 5A/B) were on the same level with those observed following i.v. infection after heart transplantation (grey circles in Fig. 4B/C). Similar results were obtained after perfusion of recipient organs thus demonstrating that the detected virus was not blood-borne but was indeed produced within the transplanted organ.
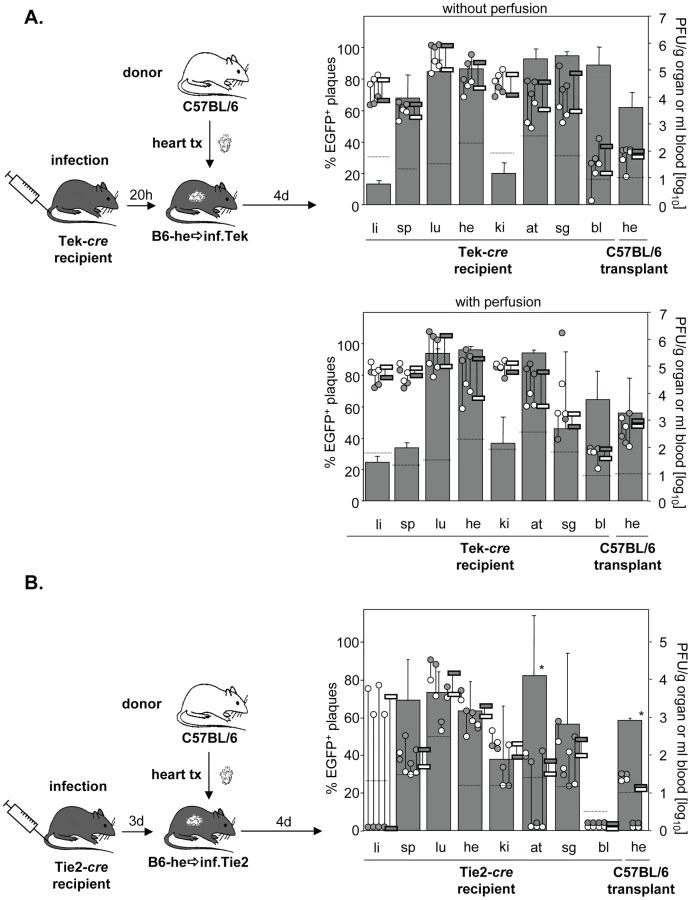
Total titers in the transplant decreased when transplantation was delayed from 20 h to 3 days after infection, reflecting the situation at day 5 and 7 p.i., respectively. In two animals transplanted three days after infection, virus titers in the heart transplant even fell below the detection limit of 10 PFU/g organ, probably reflecting enhanced control by the host immune system at day 7. This is supported by the relatively low virus titers in spleen, kidneys and adipose tissues as well as by the lack of detectable virus in blood (Fig. 5B).
In conclusion, we were surprised to see that ongoing virus replication and the accompanying immune response in the transplanted heart did obviously not alter the absolute amount of EC-derived virus originating from recipients' tissues by secondary viremia. These data demonstrate that virus dissemination between organs – originating from both endothelial and non-endothelial cells – has only minor effects on organ viral load following systemic infection.
Discussion
One hallmark of CMV infection is the ability of the virus to infect many cell types and tissues from which again the virus may spread. Apparently, immune control defines to which extent this potential is realized in a given scenario. Therefore, the various clinical conditions need to be considered to explain CMV pathogenesis. Blood specimens play an important role in CMV diagnostics. Proper usage of the information gained by this analysis should monitor or even predict events that happen in organs. However, it is currently unclear under which conditions CMV is spread via blood. Fenner et al. were the first to propose a two-step dissemination model for systemic virus infections. Primary viremia transports the virus from the site of entry to liver and spleen where the virus replicates. Secondary viremia then causes dissemination from liver and spleen throughout the body [31]. This model became widely accepted for many viruses to this day, including CMV [6], [32]. Yet, the original model was developed prior to any knowledge on innate immunity control functions and did thus not consider major factors in virus host defense. Recently, we challenged this view for CMV infection in the mouse model with respect of the role of the liver. Virus produced in hepatocytes is locally disseminated to other cell types but is not distributed from the liver to other organs via secondary viremia [24].
In the present study the vascular EC were analyzed for their claimed role in contributing to the CMV load in organs, and in disseminating the virus via primary or secondary viremia. Our salient findings are as follows: EC-derived virus significantly, ∼50% of the body virus pool, contributes to total virus load during acute infection. This contribution was quantified for the first time for the major organs. Yet, there was obviously no preference for dissemination of EC-derived virus over virus produced by other cell types. In addition, and similar to hepatocyte-derived virus, EC-derived virus was poorly disseminated via secondary viremia. These data raise doubts on the indiscriminate applicability of the primary and secondary viremia concept to virus infections in general.
Properties of EC have enticed scientists to consider them as key production sites for virus dissemination, as they may release free virus particles directly into the blood stream or may detach from the vessel wall and transfer virus to other organs via the blood stream [14], [15]. Moreover, EC could transfer the virus by contact to other cell types such as monocytes or granulocytes [33], [34], which would then disseminate the EC-derived virus to other organs [12], [35]. On the other hand, EC-derived virus may also spread to underlying parenchyma and leave the organs via the draining lymph nodes to eventually reach the blood circulation via the thoracic duct. As heterotopic, abdominal transplants are not connected to lymph vessels, exiting virus would enter the peritoneal cavity that is drained by the mediastinal lymph nodes. This lymphatic dissemination route was recently described after intraperitoneal MCMV infection [36] and is also generally accepted as dissemination route after local infections, including intraplantar infection with MCMV [37].
Here, we provide the first quantitative analysis of organ- and cell type-specific virus dissemination. From an infected organ EC-derived virus readily disseminated to the other, uninfected organs. In the specific cases shown here, the infected transplanted organ (heart or kidney) created the condition of a primary viremia initiating from a defined source. EC-derived virus remained a stable fraction in both heart (∼80%) and kidney (∼20%) throughout the first week of infection [24], thereby providing a constant supply of virus. Yet, the percentage of EC-derived virus that disseminated to other organs essentially mirrored the relative contribution of EC in the transplanted organ. Thus there was no preferential seeding of EC-derived virus.
Infected EC might detach from the vessel wall and circulate. In fact, HCMV-infected EC were considered as a parameter for the diagnosis of HCMV organ involvement and for the study of the pathogenesis of disseminated infection [16]. This conclusion was originally based on the finding of two symptomatic patients with a high load of infected circulating EC, but experimental evidence for EC-derived virus colonizing other organs was missing so far. In the mouse model we now provide a nuanced view on the role of EC in virus dissemination. If the infected heart transplant is the source of primary infection, then EC-derived virus is readily disseminated, but without preference. During secondary viremia, however, there is only negligible import of EC-derived virus into the transplant as well as export from the transplant, and this is apparently independent of the extent of ongoing virus replication, associated inflammation, and immune control.
Do our findings formally exclude any prominent role of EC-derived virus? The answer is both yes and no. Yes, we can exclude this role in the mouse model and for the temporal conditions of our experiments. Unfortunately, the more time passes after initial infection of the animal the definition of virus as being EC-derived virus becomes more and more indirect. EC-derived virus progeny keeps the marker independent of the cell type in which the virus replicates in further replication rounds. Thus, with our experimental setup we cannot study later phases of infection when other conditions of virus productivity and dissemination may prevail. However, according to our previous experience, second and third replication rounds contribute less and less to the viral load in the immune competent host due to the onset of immune control [24]. We have not yet studied the situation of the immune deficient host for the EC progeny. For the hepatocyte-derived progeny, however, we know that immune suppressive regimens, even if combined, do not lift the strong dissemination block [24].
Nevertheless, by comparing the virus titers in different organs following transplant-derived and i.v. infection, we observed striking differences. Systemic (i.v.) infection with tissue culture produced virus preparations resulted in a uniform distribution of virus to many organs, whereas transplant-derived virus appeared to preferentially colonize spleen, lung and liver but not heart, adipose tissues and kidneys. This cannot be explained by known differences in organ specific virus kinetics. Therefore, cell-free virus, which is usually used for experimental infection, is apparently able to efficiently colonize all organs, whereas virus leaving an infected organ via a natural route reveals a different kind of spread. What could be the cause of the difference between i.v. infection with a solution enriched in isolated virions and the spread of infection from an infected organ? The most plausible explanation is that the virus leaving an infected organ during systemic infection is predominantly transported in a cell-associated manner. Yet, this difference in organ and tissue distribution shows no preference for EC-derived virus and is altogether marginal with respect to total virus load in an organ.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the recommendations and guidelines for the care and use of laboratory animals according to Tierschutzgesetz (TierSchG, BGBI S. 1105; 25.05.1998). All animal experiments were approved by the responsible state office (Regierung von Oberbayern) under permit number 55.2-1-54-2531-19-07.
Cells and mice
M2-10B4 (CRL-1972; ATCC) and BALB/c-derived mouse embryo fibroblasts (MEF) were grown as described previously [38]. Transgenic Tie2-cre [39] and Tek-cre [11] mice were housed at the animal facility of the Max von Pettenkofer-Institute under specified-pathogen-free (SPF) conditions. Cre-transgenic mouse strains were maintained on the C57BL/6J background. Experiments were performed with gender matched pairs of mice at 3 to 12 months of age. C57BL/6J mice were obtained from Janvier. Tek-cre mice were obtained from Jackson Laboratories (nr. 4128).
Viruses and infection of mice
All viruses were derived from the molecular clone pSM3-fr [40]. Mutant virus (MCMV-flox) was generated as described [24]. Viruses were propagated on M2-10B4 cells and purified as described [41]. Virus quantification was done by standard plaque titration assay on MEF. Mice were infected intravenously (i.v.; into a tail vein) with 8×105 PFU in a volume of 300 µl.
Organ transplantations
Syngeneic transplantations of hearts or kidneys were performed between C57BL/6 mice and Tie2-cre or Tek-cre mice that were maintained on the genetic background of C57BL/6 mice.
Heart transplant model: Abdominal-heterotopic cardiac transplants were performed, as previously described [42]. Briefly, the ascending aorta of the graft was anastomosed to the abdominal aorta of the recipient and the pulmonary artery to the inferior vena cava while the pulmonary veins were ligated. The graft function was assessed by daily palpation.
Kidney transplant model: The murine kidney transplantation was performed as described previously [43]. Briefly, the left kidney of the donor was harvested and transplanted into the recipient. The kidneys of the recipients were not removed. A bladder patch was anastomosed to the recipient's bladder. No signs of rejection due to Cre expression by EC of the transplants or by EC of the recipient were seen throughout the experiments excluding host versus graft or graft versus host reactions, respectively.
Virus determination in organs
Virus load in organs was determined by plaque assay as described previously [24] with the modification that blood samples were sonicated before they were added to MEF in a volume of 10 µl per well. The numbers of MCMV-rec and MCMV-flox plaque forming units (PFU) were determined from organ homogenates after 4 days and from blood after 5 days using a fluorescence microscope (Olympus). Only plaques visible in bright field were considered for the calculation. PFU were calculated per ml of blood or g of organ.
Perfusion of recipients and heart transplants
Mice were anaesthetized and the peritoneal cavity was opened. After injection of 50 µl of heparin into the inferior vena cava, abdominal aorta and vena cava were cut cranially of the transplant. After all organs were perfused with 5 ml PBS via the vena cava the heart transplant was removed and perfused separately with 3 ml PBS.
Statistical analysis
The percentage of MCMV-rec compared to total virus organ load per group, mean values, and standard deviations were determined.
Zdroje
1. FishmanJARubinRH 1998 Infection in organ-transplant recipients. N Engl J Med 338 1741 1751
2. Pouteil-NobleCEcochardRLandrivonGDonia-MagedATardyJC 1993 Cytomegalovirus infection—an etiological factor for rejection? A prospective study in 242 renal transplant patients. Transplantation 55 851 857
3. PlachterBSinzgerCJahnG 1996 Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res 46 195 261
4. SinzgerCDigelMJahnG 2008 Cytomegalovirus cell tropism. Curr Top Microbiol Immunol 325 63 83
5. ManezRKusneSRinaldoCAguadoJMSt GeorgeK 1996 Time to detection of cytomegalovirus (CMV) DNA in blood leukocytes is a predictor for the development of CMV disease in CMV-seronegative recipients of allografts from CMV-seropositive donors following liver transplantation. J Infect Dis 173 1072 1076
6. CollinsTMQuirkMRJordanMC 1994 Biphasic viremia and viral gene expression in leukocytes during acute cytomegalovirus infection of mice. J Virol 68 6305 6311
7. GriffithBPChenMIsomHC 1990 Role of primary and secondary maternal viremia in transplacental guinea pig cytomegalovirus transfer. J Virol 64 1991 1997
8. EisenfeldLSilverHMcLaughlinJKlevjer-AndersonPMayoD 1992 Prevention of transfusion-associated cytomegalovirus infection in neonatal patients by the removal of white cells from blood. Transfusion 32 205 209
9. GilbertGLHayesKHudsonILJamesJ 1989 Prevention of transfusion-acquired cytomegalovirus infection in infants by blood filtration to remove leucocytes. Neonatal Cytomegalovirus Infection Study Group. Lancet 1 1228 1231
10. GrefteAHarmsenMCvan der GiessenMKnollemaSvan SonWJ 1994 Presence of human cytomegalovirus (HCMV) immediate early mRNA but not ppUL83 (lower matrix protein pp65) mRNA in polymorphonuclear and mononuclear leukocytes during active HCMV infection. J Gen Virol 75 Pt 8 1989 1998
11. IbanezCESchrierRGhazalPWileyCNelsonJA 1991 Human cytomegalovirus productively infects primary differentiated macrophages. J Virol 65 6581 6588
12. van der StrateBWHillebrandsJLLycklama a NijeholtSSBeljaarsLBruggemanCA 2003 Dissemination of rat cytomegalovirus through infected granulocytes and monocytes in vitro and in vivo. J Virol 77 11274 11278
13. AdlerBSinzgerC 2009 Endothelial cells in human cytomegalovirus infection: one host cell out of many or a crucial target for virus spread? Thromb Haemost 102 1057 1063
14. GrefteABlomNvan der GiessenMvan SonWTheTH 1993 Ultrastructural analysis of circulating cytomegalic cells in patients with active cytomegalovirus infection: evidence for virus production and endothelial origin. J Infect Dis 168 1110 1118
15. GrefteAvan der GiessenMvan SonWTheTH 1993 Circulating cytomegalovirus (CMV)-infected endothelial cells in patients with an active CMV infection. J Infect Dis 167 270 277
16. PercivalleERevelloMGVagoLMoriniFGernaG 1993 Circulating endothelial giant cells permissive for human cytomegalovirus (HCMV) are detected in disseminated HCMV infections with organ involvement. J Clin Invest 92 663 670
17. SinzgerCGrefteAPlachterBGouwASTheTH 1995 Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J Gen Virol 76 Pt 4 741 750
18. KoffronAJHummelMPattersonBKYanSKaufmanDB 1998 Cellular localization of latent murine cytomegalovirus. J Virol 72 95 103
19. SeckertCKRenzahoATervoHMKrauseCDeegenP 2009 Liver sinusoidal endothelial cells are a site of murine cytomegalovirus latency and reactivation. J Virol 83 8869 8884
20. ZhouYFLeonMBWaclawiwMAPopmaJJYuZX 1996 Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N Engl J Med 335 624 630
21. KoskinenPKNieminenMSKrogerusLALemstromKBMattilaSP 1993 Cytomegalovirus infection and accelerated cardiac allograft vasculopathy in human cardiac allografts. J Heart Lung Transplant 12 724 729
22. SalzbergerBMyersonDBoeckhM 1997 Circulating cytomegalovirus (CMV)-infected endothelial cells in marrow transplant patients with CMV disease and CMV infection. J Infect Dis 176 778 781
23. SacherTJordanSMohrCAVidyAWeynAM 2008 Conditional gene expression systems to study herpesvirus biology in vivo. Med Microbiol Immunol 197 269 276
24. SacherTPodlechJMohrCAJordanSRuzsicsZ 2008 The major virus-producing cell type during murine cytomegalovirus infection, the hepatocyte, is not the source of virus dissemination in the host. Cell Host Microbe 3 263 272
25. ChouSWNormanDJ 1988 The influence of donor factors other than serologic status on transmission of cytomegalovirus to transplant recipients. Transplantation 46 89 93
26. RubinRHWilsonEJBarrettLVMedearisDN 1984 Primary cytomegalovirus infection following cardiac transplantation in a murine model. Transplantation 37 306 310
27. ShanleyJDBillingsleyAMShelbyJCorryRJ 1983 Transfer of murine cytomegalovirus infection by heart transplantation. Transplantation 36 584 586
28. HoMSuwansirikulSDowlingJNYoungbloodLAArmstrongJA 1975 The transplanted kidney as a source of cytomegalovirus infection. N Engl J Med 293 1109 1112
29. SagedalSNordalKPHartmannADegreMHolterE 2000 A prospective study of the natural course of cytomegalovirus infection and disease in renal allograft recipients. Transplantation 70 1166 1174
30. ToupanceOBouedjoro-CamusMCCarquinJNovellaJLLavaudS 2000 Cytomegalovirus-related disease and risk of acute rejection in renal transplant recipients: a cohort study with case-control analyses. Transpl Int 13 413 419
31. FennerF 1949 Mouse-pox; infectious ectromelia of mice; a review. J Immunol 63 341 373
32. SaederupNAguirreSASparerTEBouleyDMMocarskiES 2001 Murine cytomegalovirus CC chemokine homolog MCK-2 (m131-129) is a determinant of dissemination that increases inflammation at initial sites of infection. J Virol 75 9966 9976
33. GrundyJELawsonKMMacCormacLPFletcherJMYongKL 1998 Cytomegalovirus-infected endothelial cells recruit neutrophils by the secretion of C-X-C chemokines and transmit virus by direct neutrophil-endothelial cell contact and during neutrophil transendothelial migration. J Infect Dis 177 1465 1474
34. WaldmanWJKnightDAHuangEHSedmakDD 1995 Bidirectional transmission of infectious cytomegalovirus between monocytes and vascular endothelial cells: an in vitro model. J Infect Dis 171 263 272
35. StoddartCACardinRDBonameJMManningWCAbenesGB 1994 Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J Virol 68 6243 6253
36. HsuKMPrattJRAkersWJAchilefuSIYokoyamaWM 2009 Murine cytomegalovirus displays selective infection of cells within hours after systemic administration. J Gen Virol 90 33 43
37. BöhmVSimonCOPodlechJSeckertCKGendigD 2008 The immune evasion paradox: immunoevasins of murine cytomegalovirus enhance priming of CD8 T cells by preventing negative feedback regulation. J Virol 82 11637 11650
38. Cicin-SainLPodlechJMesserleMReddehaseMJKoszinowskiUH 2005 Frequent coinfection of cells explains functional in vivo complementation between cytomegalovirus variants in the multiply infected host. J Virol 79 9492 9502
39. ConstienRFordeALiliensiekBGroneHJNawrothP 2001 Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 30 36 44
40. WagnerMJonjicSKoszinowskiUHMesserleM 1999 Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J Virol 73 7056 7060
41. MenardCWagnerMRuzsicsZHolakKBruneW 2003 Role of murine cytomegalovirus US22 gene family members in replication in macrophages. J Virol 77 5557 5570
42. CorryRJWinnHJRussellPS 1973 Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation 16 343 350
43. RussellPSChaseCMColvinRBPlateJM 1978 Kidney transplants in mice. An analysis of the immune status of mice bearing long-term, H-2 incompatible transplants. J Exp Med 147 1449 1468
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 11
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Diagnostika virových hepatitid v kostce – zorientujte se (nejen) v sérologii
Nejčtenější v tomto čísle
- Multiple Candidate Effectors from the Oomycete Pathogen Suppress Host Plant Immunity
- The Splicing Factor Proline-Glutamine Rich (SFPQ/PSF) Is Involved in Influenza Virus Transcription
- A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis
- SH3 Domain-Mediated Recruitment of Host Cell Amphiphysins by Alphavirus nsP3 Promotes Viral RNA Replication