
Micro RNAs of Epstein-Barr Virus Promote Cell Cycle Progression and Prevent Apoptosis of Primary Human B Cells
Cellular and viral microRNAs (miRNAs) are involved in many different processes of key importance and more than 10,000 miRNAs have been identified so far. In general, relatively little is known about their biological functions in mammalian cells because their phenotypic effects are often mild and many of their targets still await identification. The recent discovery that Epstein-Barr virus (EBV) and other herpesviruses produce their own, barely conserved sets of miRNAs suggests that these viruses usurp the host RNA silencing machinery to their advantage in contrast to the antiviral roles of RNA silencing in plants and insects. We have systematically introduced mutations in EBV's precursor miRNA transcripts to prevent their subsequent processing into mature viral miRNAs. Phenotypic analyses of these mutant derivatives of EBV revealed that the viral miRNAs of the BHRF1 locus inhibit apoptosis and favor cell cycle progression and proliferation during the early phase of infected human primary B cells. Our findings also indicate that EBV's miRNAs are not needed to control the exit from latency. The phenotypes of viral miRNAs uncovered by this genetic analysis indicate that they contribute to EBV-associated cellular transformation rather than regulate viral genes of EBV's lytic phase.
Published in the journal:
. PLoS Pathog 6(8): e32767. doi:10.1371/journal.ppat.1001063
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001063
Summary
Cellular and viral microRNAs (miRNAs) are involved in many different processes of key importance and more than 10,000 miRNAs have been identified so far. In general, relatively little is known about their biological functions in mammalian cells because their phenotypic effects are often mild and many of their targets still await identification. The recent discovery that Epstein-Barr virus (EBV) and other herpesviruses produce their own, barely conserved sets of miRNAs suggests that these viruses usurp the host RNA silencing machinery to their advantage in contrast to the antiviral roles of RNA silencing in plants and insects. We have systematically introduced mutations in EBV's precursor miRNA transcripts to prevent their subsequent processing into mature viral miRNAs. Phenotypic analyses of these mutant derivatives of EBV revealed that the viral miRNAs of the BHRF1 locus inhibit apoptosis and favor cell cycle progression and proliferation during the early phase of infected human primary B cells. Our findings also indicate that EBV's miRNAs are not needed to control the exit from latency. The phenotypes of viral miRNAs uncovered by this genetic analysis indicate that they contribute to EBV-associated cellular transformation rather than regulate viral genes of EBV's lytic phase.
Introduction
Thousands of microRNAs (miRNAs) have been identified so far (miRBase, release 14, Sept. 2009; http://www.mirbase.org), which are small noncoding single-stranded RNAs of about 21 to 25 nucleotides in length. They are found transcribed in all multicellular organisms and certain viruses and often are phylogenetically conserved across species [1]–[3]. The 5′-ends of miRNAs, the so-called seed sequences, recognize partially complementary mRNA targets usually within their 3′ untranslated regions and repress translational of these mRNAs [4]. In recent years, miRNAs have emerged as key regulators of a number of biological processes including developmental timing, differentiation and pattering, but also cellular proliferation, cell death, immune response, haematopoesis, and cellular transformation or oncogenesis [5]–[10]. Individual miRNAs can directly regulate the expression of hundreds of different mRNAs [11] and possibly influence the steady state levels of more than 30% of the proteins in mammalian cells [2], [12].
One standard approach to identify targets of miRNAs relies on computational algorithms that build on the thermodynamic stability of miRNA/mRNA complexes and the evolutionary conservation of miRNA seed sequences [13] because sequences of the (cellular) mRNA target molecules are frequently preserved across species [10]. One major disadvantage of this dual approach lies in a large number of false positive predictions because many putative mRNA target sites might not be accessible due to mRNA folding. In addition, as this computational approach eliminates potential targets that are not conserved between different species or related viruses, it is inadequate for predicting targets of herpesviral miRNAs because their evolutionary conservation is surprisingly low among members of the herpesvirus family [14]–[16].
An alternative approach uses microarray analyses of cellular mRNAs upon ectopic expression of individual or multiple miRNAs [5], [17], [18]. This approach is useful to reveal direct and indirect downstream targets of miRNAs but it may miss authentic targets if their mRNA levels are not sufficiently down-regulated for reliable detection by microarray analysis. In addition, antisense oligonucleotides [19], [20] or competitive inhibitors [21] have been used for the experimental identification and/or subsequent verifications of potential target genes.
The identification and functional assessment of miRNAs can reveal a rich biology. One prominent example is the human miR-155, the product of the bic gene [22]. miR-155 was found to be overexpressed in several types of B-cell lymphoma [23] and its transgenic expression in mice caused B-cell malignancies [24]. miR-155 is an orthologue of Kaposi sarcoma-associated herpesvirus (KSHV)-encoded miR-K12-11 [25], [26] and candidate target genes, identified by microarray analysis, were confirmed to be regulated similarly by miR-K12-11 [ibid and 17]. Beyond this prominent example, relatively few targets of viral miRNAs have been experimentally confirmed probably due to a large number of false positive predictions and poor evolutionary conservation of viral miRNAs [14].
We have generated recombinant EBVs modified in their capacity to encode EBV's miRNAs to probe their functions in the viral life cycle. We show that viral mutants deficient in BHRF1 miRNAs are dramatically reduced in their support of proliferation of infected B cells early after infection. B cell newly infected with EBV lacking the BHRF1 miRNAs progressed through the cell cycle less efficiently and died by apoptosis more often than cells infected identically with the parental EBV. Our phenotypic characterization revealed that EBV's miRNAs support EBV-mediated B-cell activation but play no apparent role in maintaining viral latency in contrast to the miRNAs of Kaposi's sarcoma-associated herpesvirus (KSHV) [27 and references therin] showing that the miRNAs of related human γ-herpesviruses evolved to perform divergent sets of functions.
Results
Functional deletion and reconstitution of viral miRNAs in B95.8-based mutant EBVs
We assessed the role of EBV's miRNAs in EBV-mediated B-cell activation, transformation and/or viral latency genetically. All EBV's miRNAs are clustered in two areas of the genome, the BHRF1 and BART genes. We generated two recombinant EBV mutants that carry inactivated alleles of the BHRF1 or BART miRNAs or both (Figure 1) on the basis of the E.coli-cloned genome of the B95.8 strain of EBV [28]. This EBV genome, designated 2089 and regarded as the recombinant version of prototypic EBV [29], encodes three BHRF1 and five BART pre-miRNAs, which are processed to four BHRF1 and nine BART mature miRNAs, respectively (Figure 1B). To replace the wild-type alleles by nonfunctional alleles of the viral miRNAs, we altered all eight pre-miRNAs from which the 13 mature miRNAs sequences of this EBV strain arise to computed, scrambled versions that are expected to interfere with Drosha processing (Table 1 and Supporting Figure S1). All EBV miRNAs are located in non-coding regions of the BHRF1 and BART transcripts and genetic modifications within these sequences are therefore not expected to affect the protein coding capacities of both genes. The scrambled primary RNA sequences were designed to maintain the wild-type nucleotide composition and the overall genomic architecture of the original EBV DNA but to be unable to fold into the specific hairpin structures of pri-miRNAs. As a consequence the nuclear RNaseIII enzyme Drosha would not process the scrambled RNAs and no mature functional miRNAs could form. We replaced EBV's pre-miRNAs with their scrambled mutant sequences (Figure 1B and Supporting Figure S1) using galK-mediated recombination [30] in four consecutive rounds of genetic manipulation in E. coli (see Material and Methods for experimental details). The two final EBV mutants were checked by detailed restriction enzyme analyses. DNA sequencing confirmed the intended genetic alterations of the viral pre-miRNAs and the integrity of the maxi-EBV genomes. ΔmirBHRF1 EBV lacks the coding capacity of the BHRF1 miRNA locus and ΔmirALL EBV is devoid of all viral miRNAs (Figure 1B). Both mutant EBVs are otherwise prototype 2089 without any further genetic alterations, additional marker genes, or their remnants.
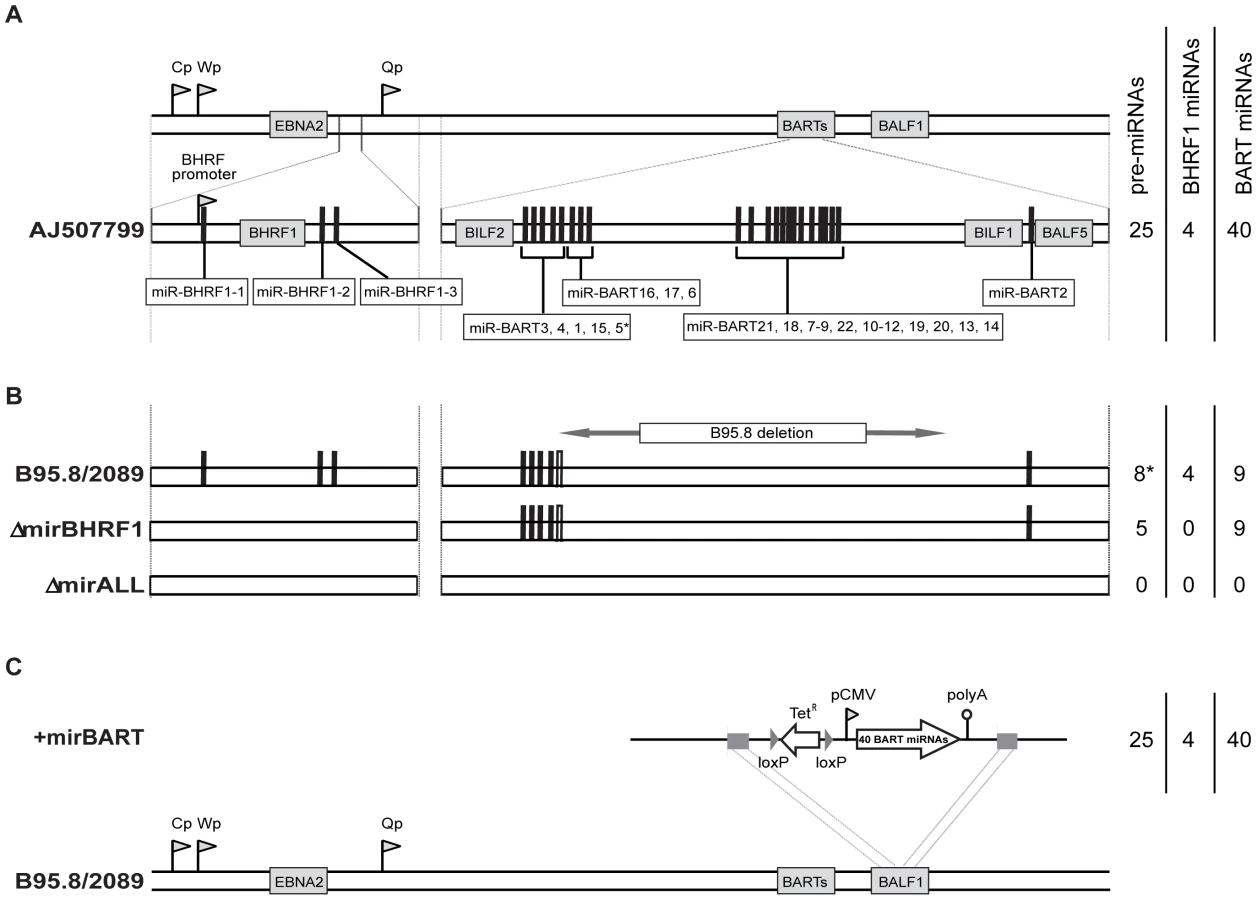

As shown in Figure 1A, EBV field strains other than the reference strain B95.8 encode up to 25 pre-miRNAs, which result in four mature BHRF1 miRNAs and 40 BART miRNAs. To examine the role of BART miRNAs that are not encoded in B95.8, we generated the reconstituted EBV mutant that ectopically expresses the full set of all BART miRNAs (+mirBART in Figure 1C). To construct this mutant, the BART miRNA cluster with twenty-two pre-miRNAs was assembled from sub- genomic fragments of the three distinct loci within the BART region (Figure 1A). The loci were PCR-amplified from Jijoye cell DNA and cloned into the expression vector pCDNA3. PCR primers were designed such that DNA stretches of at least 150bp in length flank each of the miRNA loci. Hence, all pre-miRNAs remain in their authentic sequence context minimizing the risk of aberrant RNA folding. This expression cassette was introduced into the BALF1 gene of prototype 2089 EBV (Figure 1C) as described in detail in the Material and Methods section. BALF1 encodes a viral homologue of the Bcl-2 family, and the insertion obliterates its coding capacity but BALF1 is a redundant gene and therefore dispensable for EBV's transforming functions [31].
Stocks of mutant viruses were generated in HEK293 cells stably transfected with maxi-EBV plasmid DNAs purified from E. coli [28]. Virus was produced after lytic cycle induction of the resulting HEK293 producer cell clones and quantified by infecting the B cell Raji cell line as described [32]. Because our recombinant EBVs encode green fluorescence protein (gfp), we could measure the concentration of GFP-transducing virions as “green Raji units” (GRU). We obtained virus stocks in the range of 104–105/ml GRUs similar to prototype 2089 EBV stocks [28].
We prepared primary human B cells from three samples of adenoid tissue and two samples of peripheral blood and infected them as described in detail in Material and Methods with prototype 2089 EBV or one of the three miRNA mutant EBVs. We obtained five sets of lymphoblastoid cell lines (LCLs), 20 in total, which we analyzed three to five months post infection (p.i.).
Steady state levels of selected BHRF-1 and BART miRNAs in established LCLs
We determined the steady state levels of two BHRF1 (Figure 2A, B) and five BART miRNAs (Figure 2C to G) in the established LCLs by quantitative real-time stem-loop PCR analyses. As a positive control, JM LCL was used, an LCL infected with an uncharacterized field strain of EBV that expresses all 44 viral miRNAs. The copy numbers of selected miRNAs per cellular transcriptome were determined with synthetic miRNA standards as references.

Prototype 2089 EBV-infected LCLs expressed BHRF1 miRNAs in the range of 8,000–12,000 copies per cell, which exceeded levels in JM LCL (Figure 2A, B). Expression levels of BHRF1 miRNAs in +mirBART EBV-infected LCLs were in the same range as in prototype 2089 EBV-infected LCL. As expected LCLs infected with ΔmirBHRF1 and ΔmiALL EBVs did not express the functionally deleted miRNAs.
We assessed the expression levels of two BART miRNAs of prototype 2089 EBV (Figure 2C, D) and three BART miRNAs absent in this EBV strain (Figure 2E to G). miR-BART1-5p and miR-BART2-5p were expressed at about 100–500 copies per cell. The relative low expression of BART miRNAs as compared to BHRF1 miRNAs is in accordance with the literature [33 and references therein] and was also observed in JM LCL cells infected with an uncharacterized field strain of EBV. LCLs infected with ΔmirBHRF1 EBV expressed these BART miRNAs at levels similar to prototype 2089 EBV-infected LCLs. +mirBART EBV infection mildly increased the levels of miR-BART1-5p and miR-BART2-5p (Figure 2C, D). Steady state levels of those miRNAs absent in B95.8-derived EBVs were considerably lower in +mirBART EBV-infected cells than in JM LCL cells (Figure 2E to G).
EBV's miRNAs have no discernable role in maintaining viral latency
Viral miRNAs have been implicated in maintaining herpesviral latency by inhibiting induction of the lytic cycle [34 for a recent review]. We therefore asked whether deleting EBV's miRNAs might lead to spontaneous induction of EBV's lytic phase in LCLs. We analyzed the expression of BZLF1 by semi-quantitative RT-PCR and the expression of BLLF1 by FACS in established LCLs. BZLF1 is the molecular switch gene of EBV, which can induce EBV's lytic phase, and BLLF1 codes for the late structural glycoprotein gp350/220 expressed on the surface of productively infected cells. The expression levels of BZLF1 transcripts did not consistently differ between B cells infected with prototype 2089 or miRNA mutant EBVs (Figure 3A). BLLF1 as a late lytic gene was also not detectably expressed in established LCLs infected with miRNA mutant EBVs (Figure 3B) indicating that EBV's miRNAs are not essential for maintaining herpesviral latency or inhibiting spontaneous reactivation of EBV's lytic phase in established cell lines.

We were surprised to learn that obliterating EBV's miRNAs did not lead to increased lytic reactivation given that other herpesvirus such as KSHV, HSV, and CMV have been reported to encode miRNAs, which maintain and stabilize latent infection [35]–[39]. We therefore analyzed the expression of BZLF1 mRNAs by semiquanitative RT-PCR in primary B cells from three different donors infected with the different mutant EBVs five and 15 days p.i.. Again, no discernable differences were seen indicating that BZLF1 transcripts are not regulated by EBV's miRNAs (Figure 3C). At early time points post infection BZLF1 is expressed at relative high levels [40], [41] but nevertheless cannot induce EBV's lytic phase [41]. Our recent findings together suggest that in contrast to other members of the herpesvirus family [34] EBV does not rely on its miRNAs but uses alternative means to control establishment or maintenance of latency.
Functional deletion of BHRF1 miRNAs causes minor alterations in cell cycle distribution but does not affect survival of established LCLs
While cultivating the twenty LCL lines for up to five months p.i. we noticed that LCLs infected with ΔmirBHRF1 and ΔmirALL proliferated slightly slower than LCLs infected with prototype 2089 or +mirBART EBVs (data not shown). We therefore analyzed the phenotypes of viral miRNA EBV mutants in established LCLs by monitoring their cell cycle distribution and the fraction of apoptotic cells.
The 20 LCL lines were plated at initial cell densities of 105 cells per ml and cultured for 2 days. Surface staining for Annexin-V, uptake of propidium iodine (PI), and FACS analyses of their DNA content after BrdU incorporation revealed the fraction of living cells and their cell cycle distributions, respectively. The proportions of cells that were double negative for Annexin-V and PI staining ranged between 75 to 85% for LCLs infected with either prototype 2089 or miRNA mutant EBVs (Figure 4A) with no discernable differences. LCLs infected with ΔmiBHRF1 and ΔmirALL mutant EBVs showed a slightly increased proportion of cells in G0/G1 with a reduction of cells in S phase when compared to prototype 2089 EBV-infected LCLs (Figure 4B). This tendency was mostly statistically significant (Supporting Figure S2) and suggested a possible role of EBV's BHRF1 miRNAs in controlling proliferation in latently infected cells.
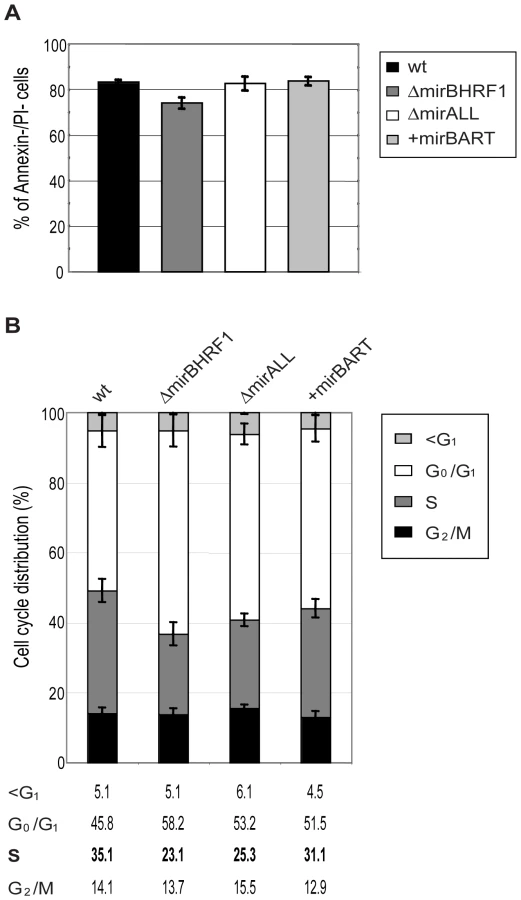
A previous, detailed genetic analysis of the BHRF1 gene, a viral homologue of the cellular Bcl-2 family, had not revealed a measurable phenotype in latently infected primary human B cells [31]. BHRF1 is a redundant gene because EBV carries two alleles of Bcl-2 family members, BHRF1 and BALF1, which are both highly expressed shortly after infection. Singly inactivated BALF1− or BHRF1− mutant EBVs were capable of yielding clonal LCLs but at a slightly higher dose than wild-type EBV. Only viral mutants with two inactivated Bcl-2 genes (i.e. both BALF1 and BHRF1) failed to rescue infected primary B lymphocytes from spontaneous apoptosis, prevented their cell cycle entry and did not generate LCLs [31]. Thus, either BHRF1 or BALF1 is dispensable for growth transformation by EBV because the two viral vBcl-2 members encode similar functions.
A recent publication indicated that BHRF1 is constitutively expressed as a latent protein in growth-transformed cells in vitro and may contribute to virus-associated lymphomagenesis in vivo [42]. Therefore, we were concerned that our current findings might result from reduced steady-state transcript levels of BHRF1 mRNA, which could be the consequence of the altered pre-miRNA sequences located in the 5′ and 3′ untranslated regions of that mRNA (Figure 1A). The alterations could adversely affect mRNA translation and reduce BHRF1 protein levels. Because antibodies that unambiguously detect BHRF1 protein early after infection or in strictly latently infected LCLs are not available, we assessed BHRF1 mRNA levels by quantitative RT-PCR analyses in established LCLs or primary B cells infected with prototype 2089 EBV or ΔmirBHRF1 mutant EBVs for five days, only (Supporting Figure S3). No discernable differences in LCLs were observed (Supporting Figure S3A and data not shown) but primary B cells infected with ΔmirBHRF1 revealed slightly enhanced (up to twofold) levels of BHRF1 mRNA early after infection (Supporting Figure S3B and data not shown). Our attempts to directly detect BHRF1 protein in newly infected primary B cells failed (data not shown). Low protein expression levels or the insufficient sensitivity of available antibodies prevent the detection of BHRF1 at early time points after infection but our quantitative RT-PCR results indicated that scrambling of the untranslated BHRF1 pre-miRNA sequences upstream and downstream of the BHRF1 coding sequence did not negatively affect the expression levels of this transcript. On the contrary the genetic alterations might even improve the expression or stability of the BHRF1 transcripts up to twofold (Supporting Figure S3B), which is expected to result in mildly enhanced protein levels that should counteract apoptosis in latently and newly infected primary B cells [31], [42 and references therein].
Functional deletion of BHRF1 miRNAs reduces early B-cell proliferation
Our studies with established LCLs infected with BHRF1 miRNA mutant EBVs indicated that these cells differed only in their cell cycle distribution but lacked other obvious phenotypes. We have found that the very early but transient expression of several lytic viral genes in primary human B cells is critical for their subsequent transformation and stable latent infection [31], [41]. BHRF1 is among the genes that are massively expressed initially after infection [31], [42]. It codes not only for one of the two viral Bcl-2 homologous but also for three pre-miRNAs that give rise to the four mature miRNAs of the BHRF1 locus (Figure 1A) [33 and references therein]. We suspected that the strong, initial expression of this gene might have implications for the expression and function of the encoded miRNAs and therefore examined their expression and the proliferation of primary B cells infected with prototype 2089 EBV or miRNA mutant EBVs at early time points post infection.
First, primary B cells prepared from adenoids were infected with prototype 2089 EBVs with a high multiplicity of infection (MOI) of 0.2 to ensure infection of many primary B cells. Quantitative stem-loop PCR analyses assessed the absolute levels of two miRNAs, miR-BHRF1-1 and miR-BHRF1-2-3p at day 5 p.i.. In primary cells miR-BHRF1-1 and miR-BHRF1-2-3p were expressed at about four- and twofold higher levels, respectively, early after infection (Supporting Figure S4; panels A and B) as compared to established LCLs infected with the same prototype 2089 EBV (Figure 2A, B). The levels early after infection exceeded the steady state levels of BHRF1 miRNAs seen in the reference JM LCL infected with an uncharacterized field strain of EBV (Supporting Figure S4; panels A and B). Similar findings apply to the expression levels of BART miRNAs early after infection (Supporting Figure S4), which were in a similar range or even exceeded those seen in JM LCL cells (miR-BART2-5p, miR-BART22, miR-BART1-5p; Supporting Figure S4C, D, F) with one exception (miR-BART8-5p; Supporting Figure S4E).
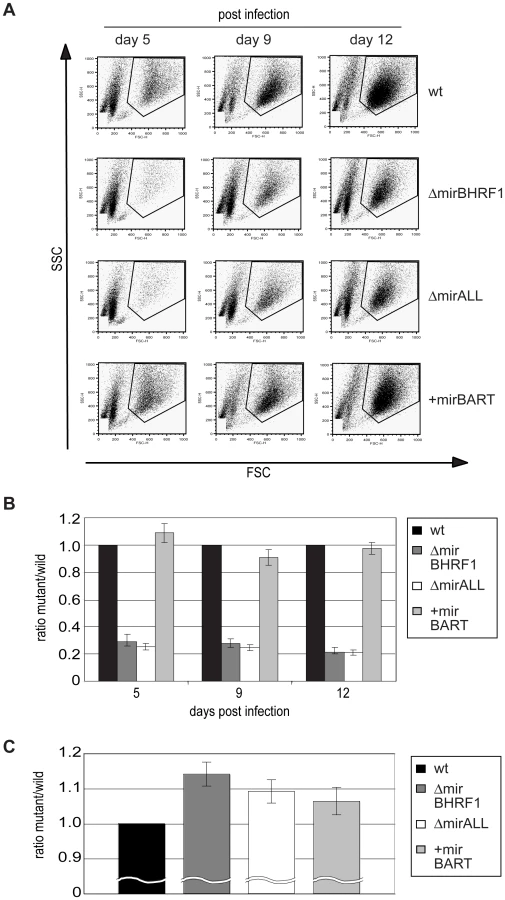
Next, primary B cells prepared from adenoids were infected with viral stocks having identical titers of the three different mutant EBVs, ΔmirBHRF1, ΔmirALL, and +mirBART EBV and prototype 2089 EBV with an multiplicity of infection (MOI) of 0.05 and a concentration of 4.5×105 cells per ml for 18hours. After collection and resuspension in fresh medium to the initial density, the infected cells were cultivated and analyzed by FACS at 5, 9, and 12 days p.i. (Figure 5A). Uninfected primary B cells showed the typical forward (FSC) and sideward (SSC) scatter characteristic of small and resting cells. Infected cells acquired typical lymphoblastic characteristics of activated cells, increased their forward and sideward scatter and were found in the defined LCL gate (Figure 5A, top panels). The absolute numbers of cells in this gate were determined by FACS counting with the aid of added APC-coupled calibration beads as a volume standard as described [31]. After infection with the two miR-BHRF1-negative EBVs, ΔmirBHRF1 and ΔmirALL, fewer cells were present in the LCL gate than after infection with prototype 2089 EBV as early as 5 days p.i. In contrast, the number of cells infected with +mirBART EBV was in a similar range as prototype 2089 EBV (Figure 5A). The inactivation of the BHRF1 miRNAs led to a four to five-fold reduction in outgrowth of B cells from three different donors (Figure 5B). The reduced numbers of growing cells infected with ΔmirBHRF1 or ΔmirALL EBVs were consistently observed over 12 days p.i.. Cells infected with ΔmirBHRF1 and ΔmirALL EBVs showed a slightly prolonged doubling time, which was not significantly different (p≥0.1, paired t test; data not shown) when compared to prototype 2089 or +mirBART EBV-infected cells (about 2 to 2.5 days/cell generation; Figure 5C). These combined observations showed that BHRF1 miRNAs are critical in primary B cells early after infection but largely dispensable in established LCLs.
BHRF1 miRNAs protect primary B cells from spontaneous apoptosis early after infection
Our initial findings indicated that BHRF1 miRNAs support B cells early after EBV infection but did not distinguish between BHRF1 miRNAs′ regulating cell cycle functions or counteracting the spontaneous apoptosis of primary B cells. To differentiate between these two roles, we first determined the proportion of viable cells in miRNA mutant or prototype 2089 EBV-infected B cells at different time points early after infection. Forward and sideward scatter analysis of primary B cells immediately after preparation showed mostly intact cells with a minor fraction of subcellular debris (Figure 6A, left panel). Uninfected primary B cells die rapidly in vitro [31], whereas EBV-infected cells become lymphoblastoid with characteristically increased forward and sideward scatter (Figure 6A, right panel). We deliberately chose a low MOI (0.05) in order to detect small changes in the fraction of infected and surviving B cells. As a consequence the majority of cells were uninfected, became highly granular or disintegrated in the course of infection. The gate in Figure 6 was set in order to include primary and activated, live and apoptotic cells but to exclude most of the subcellular debris. The cells in this gate were analyzed for the binding of Annexin-V and uptake of PI as indicators of early apoptosis and loss of membrane integrity, respectively.
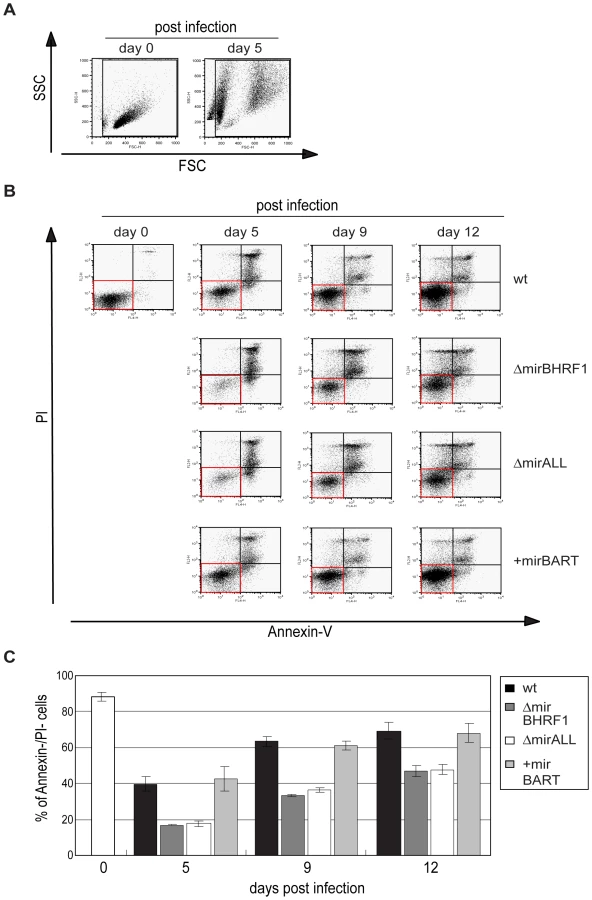
Measuring the percentage of cells, which were double-negative for Annexin-V and PI staining indicated that nearly 90% of the cells were alive on day 0 (Figure 6C). Infection with prototype 2089 EBV yielded up to 70% of cells, which were Annexin-V- and PI-negative twelve days p.i.. Both EBV mutants, ΔmirBHRF1 and ΔmirALL, also rescued the infected cells from cell death but considerably less efficiently than prototype 2089 EBV (Figure 6C). As already pointed out, we cannot unambiguously dissect BHRF1 protein-mediated effects from BHRF1 miRNA-mediated effects due to a lack of antibody reagents. However, the genetic and functional redundancy of EBV's anti-apoptotic Bcl-2 homologs [31] and the analysis of the levels of the BHRF1 transcript (Supporting Figure S3) clearly point to miRNAs of the BHRF1 cluster and their role in inhibiting apoptosis.
The larger fraction of apoptotic cells infected with the two mutant EBVs likely contributes to some of the reduction in numbers of proliferating cells in Figure 5 consistent with BHRF1 miRNAs contributing to cellular proliferation by supporting initial B-cell survival. We did not observe a clear difference between prototype 2089 EBV- and +mirBART EBV-infected cells, as would be expected from their similar growth characteristics (Figure 5).
BHRF1 miRNAs affect cell cycle distribution of infected B cells
Cells infected with ΔmirBHRF1 and ΔmirALL EBVs showed slightly prolonged doubling times (Figure 5C). We employed BrdU incorporation assays to compare the cell cycle distributions of the differently infected B cells early after infection as accurately as practical. The cell cycle status of uninfected (day 0) or infected cells on day 5, 9, and 12 p.i. with characteristics of resting lymphocytes and activated lymphoblasts in forward/sideward scatter analysis (Figure 7A) was analyzed by determining BrdU incorporation and 7-AAD uptake by FACS (Figure 7B). On average 7.2% of the uninfected cells were in S-phase immediately after isolation (Figure 7C). Whereas uninfected cells died rapidly, 39% or 35% of prototype 2089 or +mirBART EBV-infected cells were in S phase five days p.i.. In contrast, ΔmirBHRF1 and ΔmirALL EBV-infected cell cultures contained fewer cells in S-phase (21% or 17%, respectively) consistent with an increase in cells in G0/G1 and a higher proportion of apoptotic cells (Figure 7C). Thus, it appears that BHRF1 miRNAs both promote cell cycle entry or progression and block apoptosis in B cells early after their infection.
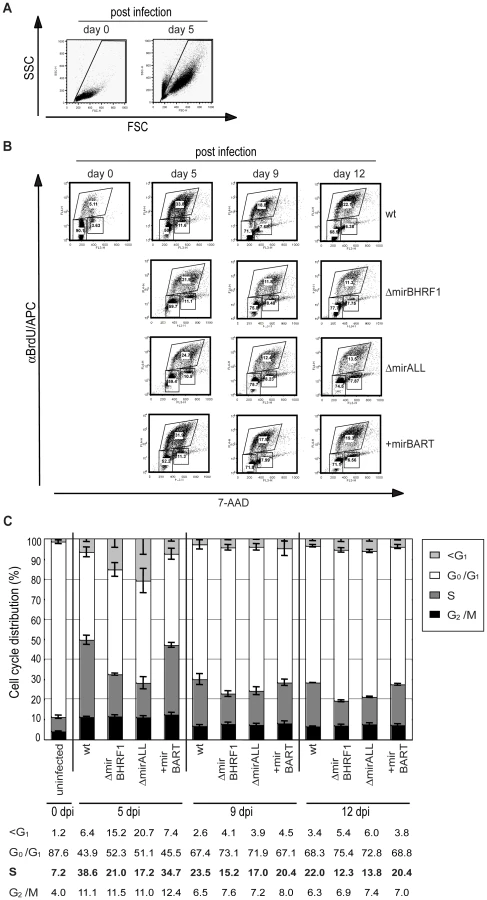
Discussion
EBV is now thought to encode more miRNAs than do other herpesviruses yet little has been established about the roles of these regulatory genes in EBV's life cycle. We have used genetic analysis to identify phenotypes mediated by the BHRF1 cluster of miRNAs. Derivatives of EBV lacking these miRNA genes yielded established B cell lines that behaved as did those infected with wild-type virus. However, careful scruting of B cells immediately following infection has uncovered two complementary functions that the BHRF1 miRNAs provided these cells. By five days post infection twice the numbers of cells infected with EBV lacking the BHRF1 miRNAs were undergoing apoptotic death as were those infected with EBV encoding these miRNAs. At this same time twice as many viable cells infected with EBV encoding the BHRF1 miRNAs were in S phase as were those infected with EBV lacking these miRNA genes. These findings indicate that the BHRF1 miRNAs inhibit apoptosis and promote proliferation during the early stages of infection. They are thus acting at a stage in the life cycle when EBV's multiple oncogenes are only beginning to function. The EBV-mediated differentiation of the resting B cell to the proliferating B cell blast requires multiple days following infection at low multiplicity, a scenario likely to reflect infections in vivo. It is under these circumstances that the BHRF1 miRNAs contribute substantially to promoting survival and proliferation of the infected B cell.
The functions of EBV's BHRF1 miRNAs differ from those characterized in KSHV's genome. In KSHV, an EBV-related human herpesvirus, several miRNAs counteract the spontaneous onset of KSHV's lytic cycle. Their expression promotes or maintains viral latency and shuts off viral lytic proteins. For example, two groups recently demonstrated that two different miRNAs of KSHV, miRK9* and miRK5, can down-regulate the expression of the viral transcription activator RTA [27], [35]. Another miRNA of KSHV, miR-K1, negatively regulates the IκBα protein level to increase NF-κB activity and indirectly inhibit viral lytic replication in certain cells [36]. It is tempting to speculate that the role of KSHV's miRNAs is not only to maintain latency but also to prevent the spontaneous expression of viral lytic genes, which, by analogy with EBV, might otherwise increase the susceptibility of virus-infected cells to T effector cells. Similar results have been obtained while studying the functions of miRNAs encoded in CMV and HSV, which also help maintain latent infection [38], [39].
A subset of BART miRNAs can negatively regulate the viral oncoprotein LMP1 (latent membrane protein 1) in nasopharyngeal carcinoma cells [43]. LMP1 has transforming activity but its high expression can cause growth inhibition and apoptosis. LMP1 regulates its level through its regulation of the Unfolded Protein Response (UPR) pathway and autophagy. EBV's miRNAs might limit inappropriately high LMP1 levels and thereby prevent apoptosis resulting from LMP1's regulation of the UPR [44]–[46]. It has also been shown that the pro-apoptotic protein PUMA (p53-upregulated modulator of apoptosis) is a target of miR-BART5 when expressed in epithelial cells, which prototype 2089 EBV does not encode (Figure 1). We investigated the expression levels of LMP1 and PUMA in 20 LCLs infected with prototype 2089 or miRNA mutant EBVs but did not observe a consistent correlation with the expression levels of LMP1 protein or PUMA mRNA (data not shown). The relative low expression levels of the BART miRNAs in the reconstituted EBV mutant +mirBART in established LCLs might not be sufficient to reveal phenotypes that correlate with the regulation of the two proteins in B cells with a latency III type program (Figure 2C–G). The low steady state levels might also mask other phenotypes that might be connected to viral reactivation or additional phenotypic effects not diclosed in this work. This caveat is of concern because the BART miRNAs are expressed at considerably higher levels in nasopharyngeal carcinoma cells [47]. Early after infection, these BART miRNAs (as well as BHRF1 miRNAs) are expressed at much higher levels (Supporting Figure S4). In fact, we observed under conditions of low cell density and reduced multiplicity of infection that the ectopically expressed BART miRNAs of EBV do promote proliferation of primary human B cells early after infection (Vereide et al., manuscript submitted).
It is likely that many targets of viral miRNAs remain to be identified because single miRNAs can target multiple mRNAs [11]. Computer algorithms based on the conservation of seed sequences between different species have been successfully used for the target prediction of cellular miRNAs, but this approach is hampered for the prediction of the targets of herpesviral miRNAs because they are evolutionarily poorly conserved [14] and do not share extended seed homology with cellular transcripts [9 and references therein]. Conversely, multiple miRNAs could simultaneously downregulate a single target gene even if the individual miRNAs are expressed at relatively low levels [48]. Therefore, the common experimental approach based on the ectopic expression or repression of individual miRNAs coupled to subsequent microarray analysis may prove inadequate to identify targets for herpesviral miRNAs. Given these difficulties it is essential to identify phenotypes mediated by EBV's miRNAs when they are expressed at physiological levels under normal conditions of infection as we have done. It is particularly intriguing that these genetic analyses show that EBV has evolved miRNAs to support its defining phenotype of transforming infected B cells.
Materials and Methods
Construction of miRNA mutant EBVs
EBVs used in this study were derived from p2089, which comprises the B95.8 EBV genome cloned onto an F-factor plasmid in E. coli [28]. The B95.8 EBV strain as well as p2089 encode a total of 13 known miRNAs, which are located in four clusters (Figure 1; miR-BHRF1-1, miR-BHRF1-2/3, miR-BART3/4/1/15/5, and miR-BART2). For their functional ablations, the wild-type miRNA sequences in each of the four clusters were replaced with computed, scrambled miRNA sequence (Table 1) in four consecutive rounds of homologous recombination with the galK-based recombineering system [30]. In a two-step approach this system allows modifying the p2089 genome via homologous recombinations in E. coli without permanently introducing selectable marker genes or cis-acting sequences (or their remnants) at the sites of genetic alterations.
Briefly, the recombineering E. coli strain SW105 has a deletion of the galactokinase (galK) gene and carries a lysogenic and temperature-sensitive λ prophage that makes recombination amenable. We introduced the p2089 plasmid into SW105 by electroporation.
In the first targeting step, we wanted to replace the miR-BHRF1-2/3 miRNAs in p2089 with their scrambled counterparts shown in Table 1. In the first step we inserted the galK gene into the miR-BHRF1-2/3 cluster deleting the entire locus. To achieve this step, the galK targeting cassette was PCR amplified with the pgalK plasmid as a template [30] with the following conditions: 94°C for 3min for initial denaturation, 94°C for 45sec, 54°C for 45sec, and 72°C for 2.5min in 15 cycles, followed by 20 cycles at 94°C for 45sec, 62°C for 45sec, and 72°C for 2.5min, and a final elongation step at 72°C for 3min. The PCR primers suitable for replacing each of the miRNA cluster with galK are listed in Supporting Table S1. After DpnI digestion, the gel-purified PCR fragment was electroporated into the heat-induced and therefore recombination-competent E. coli SW105 strain carrying the plasmid p2089. After selection for galK on minimal medium plates with galactose as the sole carbon source, plasmid DNAs were prepared from galK positive bacterial clones and carefully analyzed by restriction enzyme analysis. The resulting EBV plasmid p3994 was confirmed to carry galK replacing the BHRF1-2/3 miRNA cluster.
The second targeting step aimed at replacing galK with designed scrambled DNA sequences that maintain the original nucleotide composition but ablate the original pre-miRNA structures. The targeting constructs consisted of the scrambled pre-miRNA sequence as the core flanked by 150–200 bp long homologous arms on both sides for the efficient and precise replacement of galK. The targeting constructs were custom-made, synthetic DNA fragments cloned into pUC57 and obtained from a commercial service provider (Genscript Corporation). Four targeting constructs were ordered. To replace the galK gene inserted into the mir-BHRF1-2/3 locus the targeting p3969 plasmid was cut with appropriate restriction enzymes to liberate the synthetic DNA fragment. It was gel-purified and electroporated into recombination-competent E. coli SW105 cells carrying p3994, which were selected for loss of galK by growth on minimal plates containing 2-deoxy-galactose (DOG) and glycerol as carbon sources as described in detail [30]. Plasmid DNAs were prepared from galK negative bacterial clones and carefully analyzed by restriction enzyme analysis and extensive DNA sequencing covering at least two kbps of upstream and downstream flanking sequences in order to verify the correct insertion of the scrambled miRNA sequences.
We repeated the two-step approach and replaced the miR-BHRF1-1 cluster with scrambled sequences to generate the genomic EBV plasmid p4004, which lacks the four miRNAs of the BHRF1 locus. With this genomic EBV plasmid ΔmirBHRF1 EBV stocks were established and calibrated as described in detail [32].
On the basis of p4004, the two BART miRNA clusters in the cloned genome of B95.8 EBV were further replaced with the scrambled sequences shown in Table 1. The resulting genomic EBV plasmid p4027 lacks all functional viral miRNAs (ΔmirALL EBV). Restriction enzyme analysis and partial DNA sequencing as exemplified above verified the genetic compositions of the modified EBV genomes.
The B95.8 EBV strain and the derived genomic EBV plasmid p2089 encompass only 13 out of 44 miRNAs as compared to EBV field strains, which encode 31 additional BART miRNAs. To reconstitute an EBV genome that has the miRNA coding capacity of EBV field strains, we introduced an expression cassette, termed pCMV-miRBART, encompassing all known 22 BART pre-miRNAs driven by human CMV promoter into the BALF1 locus of p2089 by homologous recombination (Figure 1C). An expression cassette was assembled from three sub-genomic fragments containing the BART miRNAs that map to three distinct loci within the BART region (Figure 1A). These loci were PCR amplified from Jijoye cellular DNA and inserted into the expression vector pCDNA3 (Invitrogen), followed by sequencing to ensure accurate DNA amplification. Primers were designed such that DNA stretches of at least 150bp in length flank each of the miRNA loci. The expression cassette was termed pCMV-miRBART. The nucleotide positions of the amplified regions were as follows (all positions are given in reference to GenBank entry AJ507799): nucleotide coordinates #138803 to #140353 (containing miRs BART1, −3 to −6, −15 to −17), nucleotide coordinates #145331 to #149070 (miRs BART7 to −14, −12 to −14, −18 to −22) and nucleotide coordinates #152509 to #153034 (miR-BART2). In order to ensure proper function of the construct, we verified the expression of representative miRNAs from each of the inserted loci in transiently pCMV-miRBART-transfected cells by northern blot hybridization confirming similar relative expression levels as in wild-type EBV infected B-cell lines (data not shown).
To construct the maxi-EBV genome p4080 (+mirBART in Figure 1) a tetracycline resistance gene was introduced into the NruI site of pCMV-miRBART (p3971) to yield p4016. The final targeting plasmid p4079 was generated by inserting the SspI/DrdI fragment from p4016 cloned into the SmaI site of p2642, which contains the BALF1 gene to support its targeted homologous integration into the BALF1 locus as shown in Figure 1C. The targeting construct p4079 was linearized with BsrDI/BssHII digestion and electroporated into the SW105 strain carrying p2089. After tetracycline selection and restriction enzyme analysis, DNA sequencing confirmed the genomic EBV plasmid p4080 to contain the entire targeting construct at the desired location in the BALF1 locus.
Cells and culture
The EBV-positive Burkitt's lymphoma cell line Raji, the EBV-positive marmoset cell line B95.8, and HEK293 cells were maintained in RPMI 1640 medium (GIBCO). All media were supplemented with 10% FBS (PAA laboratories), penicillin (100 U/ml), and streptomycin (100µg/ml). Cells were cultivated at 37°C in a 5% CO2 incubator.
Preparation and quantification of infectious viral stocks
On the basis of HEK293 cells, virus producer cell lines were established after individual transfection of the genomic EBV plasmid DNAs and subsequent selection with hygromycin (80µg/ml). To obtain virus stocks, the producer cell lines were transiently transfected with expression plasmids encoding BZLF1 [49], BALF4 [50], and BRLF1 [51] to induce EBV's lytic cycle. Three days post transfection, supernatants were harvested and centrifuged at 3000rpm for 15min to remove cell debris. The titers of the different virus stocks were quantified and the concentrations of GFP-transducing virions expressed as “green Raji units” (GRUs) were determined as described previously [31]. Briefly, 105 Raji cells were incubated with serial dilutions of virus stocks at 37°C for 24hours. After an additional culture for 2 days, the percentage of GFP positive cells was determined by FACS using a FACS-Calibur instrument (Becton Dickinson).
Isolation, separation, and infection of human primary B lymphocytes
Human primary B cells from adenoids were separated from T cells by rosetting with sheep erythrocytes and purified by Ficoll-Hypaque density gradient centrifugation. B cells isolated from human peripheral blood mononuclear cells (PBMC) by Ficoll-Hypaque gradient centrifugation were purified using the B-cell isolation kit II (Miltenyi Biotec) and MACS separators (Miltenyi Biotec). For virus infection, primary B cells were incubated with each virus stock for 18 hrs. After replacement with fresh medium, the infected cells were seeded at an initial density of 4.5×105 cells per ml.
Anonymized adenoid tissue samples from routine adenoidectomies were provided by the Department of Otorhinolaryngology, Klinikum Grosshadern, Ludwig-Maximilians-University of Munich. The institutional review board, Ethikkommission of the Klinikum Grosshadern, approved of the study and did not require prior informed patient consent.
Analysis of cell proliferation, spontaneous apoptosis and cell cycle distribution of infected primary B cells
For each time point p.i., 1ml of each cell sample was collected by centrifugation and stained with 2µl of AnnexinV-Cy5 reagent (BioVision) and 50µg of propidium iodide (PI) in 250µl of Annexin-V binding buffer (BioVision) for 5 min at room temperature. As an internal FACS volume standard, 2×104 of APC-conjugated BD CaliBRITE beads (Becton-Dickinson) were added. The beads are small, resulting in a high intensity in the sideward scatter channel, and do not interfere with the cells to be analyzed. Cells were analyzed by FACS until 5,000 beads were counted and the numbers of the cells in the indicated gates were recorded.
For analysis of the cell cycle status, the infected cells were incubated with the thymidine analog BrdU for 2hrs prior to FACS analysis at each time point. The cells were stained with an APC-coupled BrdU-specific antibody after fixation and permeabilization, and the cellular DNA was counter-stained with the DNA intercalating dye 7-AAD according to the manufacturer's protocol (BrdU Flow Kit, BD Biosciences Pharmingen). 3×104 cells were acquired in FACS analysis. The FACS data were gated for cells in the G0/G1, S, G2/M phases of the cell cycle and for cells with a subG1 DNA content. The total of all events was set to 100%.
Absolute quantification of EBV-encoded microRNAs by stem-loop real-time PCR
Total RNA was extracted from infected cells using the Trifast reagent (peqGOLD). For cDNA synthesis, 500 ng of total RNA was reverse-transcribed using Superscript III reverse transcriptase (Invitrogen) with the mixture of miRNA specific stem-loop primers shown in Supporting Table S2. In the quantitative reconstruction experiments, total RNA from ΔmirALL EBV-infected LCLs was used as an EBV miRNA-negative but complex RNA sample in reverse transcription (RT) reactions. Details for reverse transcription reaction were described previously [52]. 4ng of cDNA aliquots from each sample were subjected to quantitative real-time PCR analysis. Each 10µl PCR reaction contained 0.5µM forward primer, 0.5µM reverse primer, and 1×SYBR green mix (Roche). The PCR reaction was performed in 96-well cluster plates at 95°C for 10min, followed by 45 cycles of 95°C for 15sec, 60°C for 1min with 10 initial cycles of touchdown steps (70–60°C). The absolute copy number of each miRNA in the test samples was reconstructed with the aid of standard curves generated with the serial dilution of synthetic miRNA (Metabion) as reference. The synthetic miRNAs were identical to the mature miRNA sequences as annotated in the microRNA database (miRBase, release 14, Sept. 2009; http://www.mirbase.org).
Supporting Information
Zdroje
1. KimVN
HanJ
SiomiMC
2009 Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10 126 139
2. BartelDP
2004 MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 281 297
3. LandgrafP
RusuM
SheridanR
SewerA
IovinoN
2007 A mammalian microRNA expression atlas based on small RNA library sequencing. Cell 129 1401 1414
4. BartelDP
2009 MicroRNAs: target recognition and regulatory functions. Cell 136 215 233
5. AmbrosV
2004 The functions of animal microRNAs. Nature 431 350 355
6. BushatiN
CohenSM
2007 microRNA functions. Annu Rev Cell Dev Biol 23 175 205
7. PfefferS
VoinnetO
2006 Viruses, microRNAs and cancer. Oncogene 25 6211 6219
8. VisoneR
CroceCM
2009 MiRNAs and cancer. Am J Pathol 174 1131 1138
9. GottweinE
CullenBR
2008 Viral and cellular microRNAs as determinants of viral pathogenesis and immunity. Cell Host Microbe 3 375 387
10. XiaoC
RajewskyK
2009 MicroRNA control in the immune system: basic principles. Cell 136 26 36
11. LewisBP
BurgeCB
BartelDP
2005 Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120 15 20
12. SelbachM
SchwanhausserB
ThierfelderN
FangZ
KhaninR
2008 Widespread changes in protein synthesis induced by microRNAs. Nature 455 58 63
13. RajewskyN
2006 microRNA target predictions in animals. Nat Genet 38 Suppl S8 13
14. WalzN
ChristallaT
TessmerU
GrundhoffA
2010 A global analysis of evolutionary conservation among known and predicted gammaherpesvirus microRNAs. J Virol 84 716 728
15. SchaferA
CaiX
BilelloJP
DesrosiersRC
CullenBR
2007 Cloning and analysis of microRNAs encoded by the primate gamma-herpesvirus rhesus monkey rhadinovirus. Virology 364 21 27
16. GrundhoffA
SullivanCS
GanemD
2006 A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 12 733 750
17. SamolsMA
SkalskyRL
MaldonadoAM
RivaA
LopezMC
2007 Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog 3 e65
18. ZiegelbauerJM
SullivanCS
GanemD
2009 Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet 41 130 134
19. KrutzfeldtJ
KuwajimaS
BraichR
RajeevKG
PenaJ
2007 Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 35 2885 2892
20. KrutzfeldtJ
RajewskyN
BraichR
RajeevKG
TuschlT
2005 Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438 685 689
21. EbertMS
NeilsonJR
SharpPA
2007 MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4 721 726
22. EisPS
TamW
SunL
ChadburnA
LiZ
2005 Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A 102 3627 3632
23. van den BergA
KroesenBJ
KooistraK
de JongD
BriggsJ
2003 High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer 37 20 28
24. CostineanS
ZanesiN
PekarskyY
TiliE
VoliniaS
2006 Pre-B cell proliferation and lymphoblastic leukemia/high-grade lymphoma in E(mu)-miR155 transgenic mice. Proc Natl Acad Sci U S A 103 7024 7029
25. GottweinE
MukherjeeN
SachseC
FrenzelC
MajorosWH
2007 A viral microRNA functions as an orthologue of cellular miR-155. Nature 450 1096 1099
26. SkalskyRL
SamolsMA
PlaisanceKB
BossIW
RivaA
2007 Kaposi's sarcoma-associated herpesvirus encodes an ortholog of miR-155. J Virol 81 12836 12845
27. DavidR
2010 miRNAs help KSHV lay low. Nature Reviews Microbiology 8 158 159
28. DelecluseHJ
HilsendegenT
PichD
ZeidlerR
HammerschmidtW
1998 Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc Natl Acad Sci U S A 95 8245 8250
29. BaerR
BankierAT
BigginMD
DeiningerPL
FarrellPJ
1984 DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 310 207 211
30. WarmingS
CostantinoN
CourtDL
JenkinsNA
CopelandNG
2005 Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33 e36
31. AltmannM
HammerschmidtW
2005 Epstein-Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis. PLoS Biol 3 e404
32. NeuhierlB
FeederleR
HammerschmidtW
DelecluseHJ
2002 Glycoprotein gp110 of Epstein-Barr virus determines viral tropism and efficiency of infection. Proc Natl Acad Sci U S A 99 15036 15041
33. PrattZL
KuzembayevaM
SenguptaS
SugdenB
2009 The microRNAs of Epstein-Barr Virus are expressed at dramatically differing levels among cell lines. Virology 386 387 397
34. CullenBR
2009 Viral and cellular messenger RNA targets of viral microRNAs. Nature 457 421 425
35. BellareP
GanemD
2009 Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6 570 575
36. LeiX
BaiZ
YeF
XieJ
KimCG
2010 Regulation of NF-kappaB inhibitor IkappaBalpha and viral replication by a KSHV microRNA. Nat Cell Biol 12 193 199
37. LuF
StedmanW
YousefM
RenneR
LiebermanPM
2010 Epigenetic Regulation of Kaposi's Sarcoma-Associated Herpesvirus Latency by Virus-Encoded MicroRNAs That Target Rta and the Cellular Rbl2-DNMT Pathway. J Virol 84 2697 2706
38. UmbachJL
KramerMF
JurakI
KarnowskiHW
CoenDM
2008 MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454 780 783
39. MurphyE
VanicekJ
RobinsH
ShenkT
LevineAJ
2008 Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci U S A 105 5453 5458
40. WenW
IwakiriD
YamamotoK
MaruoS
KandaT
2007 Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes. J Virol 81 1037 1042
41. KallaM
SchmeinckA
BergbauerM
PichD
HammerschmidtW
2010 AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc Natl Acad Sci U S A 107 850 855
42. KellyGL
LongHM
StylianouJ
ThomasWA
LeeseA
2009 An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog 5 e1000341
43. LoAK
ToKF
LoKW
LungRW
HuiJW
2007 Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc Natl Acad Sci U S A 104 16164 16169
44. LamN
SandbergML
SugdenB
2004 High physiological levels of LMP1 result in phosphorylation of eIF2 alpha in Epstein-Barr virus-infected cells. J Virol 78 1657 1664
45. LeeDY
SugdenB
2008 The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 111 2280 2289
46. LeeDY
SugdenB
2008 The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 27 2833 2842
47. CaiX
SchaferA
LuS
BilelloJP
DesrosiersRC
2006 Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog 2 e23
48. GrimsonA
FarhKK
JohnstonWK
Garrett-EngeleP
LimLP
2007 MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell 27 91 105
49. HammerschmidtW
SugdenB
1988 Identification and characterization of oriLyt, a lytic origin of DNA replication of Epstein-Barr virus. Cell 55 427 433
50. JanzA
OezelM
KurzederC
MautnerJ
PichD
2000 Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J Virol 74 10142 10152
51. FeederleR
KostM
BaumannM
JanzA
DrouetE
2000 The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J 19 3080 3089
52. Varkonyi-GasicE
WuR
WoodM
WaltonEF
HellensRP
2007 Protocol: a highly sensitive RT-PCR method for detection and quantification of microRNAs. Plant Methods 3 12
53. HofackerLL
FontanaW
StadlerPF
BonhoefferS
TackerM
1994 Fast Folding and Comparison of RNA Secondary Structures. Monatshefte f Chemie 125 167 188
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 8
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Jak souvisí postcovidový syndrom s poškozením mozku?
- Diagnostika virových hepatitid v kostce – zorientujte se (nejen) v sérologii
Nejčtenější v tomto čísle
- Contribution of Coagulases towards Disease and Protective Immunity
- Early Severe Inflammatory Responses to Uropathogenic Predispose to Chronic and Recurrent Urinary Tract Infection
- The Transcription Factor Rbf1 Is the Master Regulator for -Mating Type Controlled Pathogenic Development in
- Immune Modulation with Sulfasalazine Attenuates Immunopathogenesis but Enhances Macrophage-Mediated Fungal Clearance during Pneumonia
