
Identification of Host-Dependent Survival Factors for Intracellular through an siRNA Screen
The stable infection of host macrophages by Mycobacterium tuberculosis (Mtb) involves, and depends on, the attenuation of the diverse microbicidal responses mounted by the host cell. This is primarily achieved through targeted perturbations of the host cellular signaling machinery. Therefore, in view of the dependency of the pathogen on host molecules for its intracellular survival, we wanted to test whether targeting such factors could provide an alternate route for the therapeutic management of tuberculosis. To first identify components of the host signaling machinery that regulate intracellular survival of Mtb, we performed an siRNA screen against all known kinases and phosphatases in murine macrophages infected with the virulent strain, H37Rv. Several validated targets could be identified by this method where silencing led either to a significant decrease, or enhancement in the intracellular mycobacterial load. To further resolve the functional relevance of these targets, we also screened against these identified targets in cells infected with different strains of multiple drug-resistant mycobacteria which differed in terms of their intracellular growth properties. The results obtained subsequently allowed us to filter the core set of host regulatory molecules that functioned independently of the phenotypic variations exhibited by the pathogen. Then, using a combination of both in vitro and in vivo experimentation, we could demonstrate that at least some of these host factors provide attractive targets for anti-TB drug development. These results provide a “proof-of-concept” demonstration that targeting host factors subverted by intracellular Mtb provides an attractive and feasible strategy for the development of anti-tuberculosis drugs. Importantly, our findings also emphasize the advantage of such an approach by establishing its equal applicability to infections with Mtb strains exhibiting a range of phenotypic diversifications, including multiple drug-resistance. Thus the host factors identified here may potentially be exploited for the development of anti-tuberculosis drugs.
Published in the journal:
. PLoS Pathog 6(4): e32767. doi:10.1371/journal.ppat.1000839
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000839
Summary
The stable infection of host macrophages by Mycobacterium tuberculosis (Mtb) involves, and depends on, the attenuation of the diverse microbicidal responses mounted by the host cell. This is primarily achieved through targeted perturbations of the host cellular signaling machinery. Therefore, in view of the dependency of the pathogen on host molecules for its intracellular survival, we wanted to test whether targeting such factors could provide an alternate route for the therapeutic management of tuberculosis. To first identify components of the host signaling machinery that regulate intracellular survival of Mtb, we performed an siRNA screen against all known kinases and phosphatases in murine macrophages infected with the virulent strain, H37Rv. Several validated targets could be identified by this method where silencing led either to a significant decrease, or enhancement in the intracellular mycobacterial load. To further resolve the functional relevance of these targets, we also screened against these identified targets in cells infected with different strains of multiple drug-resistant mycobacteria which differed in terms of their intracellular growth properties. The results obtained subsequently allowed us to filter the core set of host regulatory molecules that functioned independently of the phenotypic variations exhibited by the pathogen. Then, using a combination of both in vitro and in vivo experimentation, we could demonstrate that at least some of these host factors provide attractive targets for anti-TB drug development. These results provide a “proof-of-concept” demonstration that targeting host factors subverted by intracellular Mtb provides an attractive and feasible strategy for the development of anti-tuberculosis drugs. Importantly, our findings also emphasize the advantage of such an approach by establishing its equal applicability to infections with Mtb strains exhibiting a range of phenotypic diversifications, including multiple drug-resistance. Thus the host factors identified here may potentially be exploited for the development of anti-tuberculosis drugs.
Introduction
Successful parasitization of macrophages by Mycobacterium tuberculosis (Mtb) reflects the equilibrium between host and pathogen, which is established and maintained through the modulation of macrophage-signaling mechanisms. This leads to the attenuation of several cellular processes that include fusion of phagosomes with lysosomes, antigen presentation, apoptosis, and the bactericidal responses initiated by the macrophage [1], [2], [3]. This attenuation represents the outcome of a dynamic process wherein bacterial molecules interfere with the signaling machinery of the host cell. Although a detailed picture is yet unavailable, many bacterial mediators of virulence have been identified and the strategies employed by them are currently being elucidated [4], [5], [6], [7], [8], [9]. The emerging theme, however, suggests that pathogen-directed manipulation of host processes is achieved through targeted perturbations in the host cell-signaling network [10], [11], [12], [13], [14]. It is these perturbations that then reorient the cellular response to support intracellular survival and growth of Mtb. A better understanding of the mechanisms involved in these perturbations should, therefore, greatly aid current efforts at developing new drugs for tuberculosis (TB).
We undertook this study to identify components of the host cell signaling machinery that are important for regulating an Mtb infection. For this, we performed an siRNA screen against all kinases and phosphatases, in mouse macrophages infected with a virulent strain of Mtb (H37Rv). Such experiments identified several host molecules that were involved either in facilitating, or suppressing, the intracellular infection. By then applying a filter of phenotypic variations, at the level of both drug resistance and intracellular growth properties, we could further distinguish those host molecules that regulated intracellular pathogen load in an Mtb strain-independent manner. A combination of in vitro and in vivo experimental approaches subsequently enabled us to establish that targeting such host factors indeed provides an attractive alternate strategy for the development of anti-TB drugs. Importantly, our results suggest that such an approach could also potentially address the problem of multiple drug resistance in TB infections.
Results
Identifying host proteins that regulate Mtb survival in the macrophage
The siRNA library employed in the primary screen consisted of a pool of two siRNAs per target gene, and the targets included 744 kinases and 288 phosphatases. Cells of the murine macrophage line, J774.1, were first infected with a virulent strain of Mtb (H37Rv), and then transfected with individual target-specific siRNA. The siRNA transfection was performed after infection of cells to ensure an un-hindered uptake of pathogen and, therefore, to select for only those host proteins that were involved in the maintenance of an established infection. The effect of specific silencing on intracellular mycobacterial load was then determined from cell lysates in terms of the colony forming units (CFU) subsequently obtained (Methods).
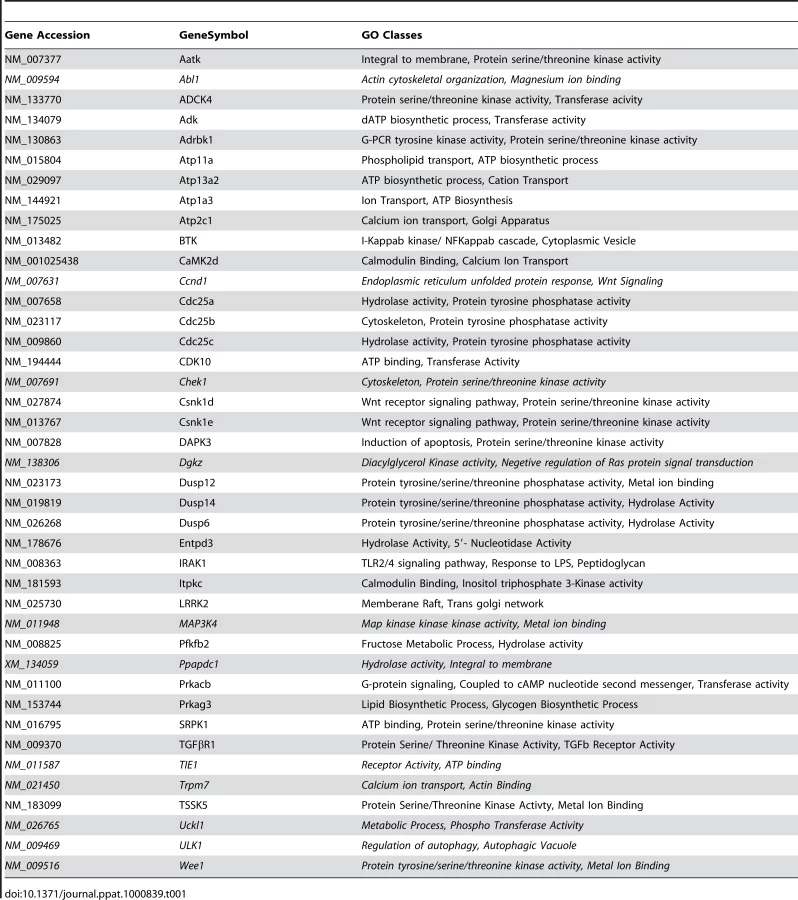
At one level, silencing of several macrophage proteins resulted in a significant decrease in titers of the intracellular bacteria obtained (Table S1). However, there were also many instances where a targeted protein-knockdown led to a marked increase in mycobacterial levels (Table S1). Selection of only those effects as significant, where intracellular bacterial load varied by greater than two standard deviations (2SD) of the mean CFU values obtained for the control wells (Methods), identified 203 target-specific siRNAs (Table S1). To next filter out any non-specificity arising from ‘off-target’ effects of the siRNA, we performed a second screen where the primary ‘hits’ were now silenced with an alternate pool of siRNAs (Methods). Although such an approach likely leads to an increased number of false negatives due to differences in silencing efficiency between the two target-specific siRNA pools, a high degree of confidence is – nonetheless - associated with any reproducible effects that are consequently obtained. This procedure led to a further reduction in the number of significant targets to forty one (Table S1). Then, as the final validation step, we tested whether these target-specific siRNA pools had any effect on the viability of either un-infected, or infected, J774.1 cells over the time course of our experiment (Text S1). In neither of these cases, however, did any of the siRNA pools negatively influence cell viability to an extent that was greater than 1.5 SD of the mean values obtained for the control wells (Table S2). Therefore, these forty one targets identified by the corresponding set of siRNA pools were taken as the validated target group. The effect of silencing of each of these on the intracellular bacterial load is shown in Figure 1A, whereas Figure 1B confirms that the siRNA pools used in the primary and in the validation exercise both yielded effects that were greater than 2SD of the mean CFU value obtained for the corresponding control wells. As is evident, the effects of target-suppression ranged from a near complete elimination of the infection, to a significant enhancement in intracellular bacterial titers (Fig. 1A). Thus, adaptation of mycobacteria in the intracellular milieu likely involves both facilitating and inhibitory contributions from some of the components of the host cell signaling machinery. The validated targets are listed in Table 1, which also includes a gene ontology-based classification of their functional roles. A more detailed description of these genes and their known association with disease and other cellular functions is provided in Table S3.
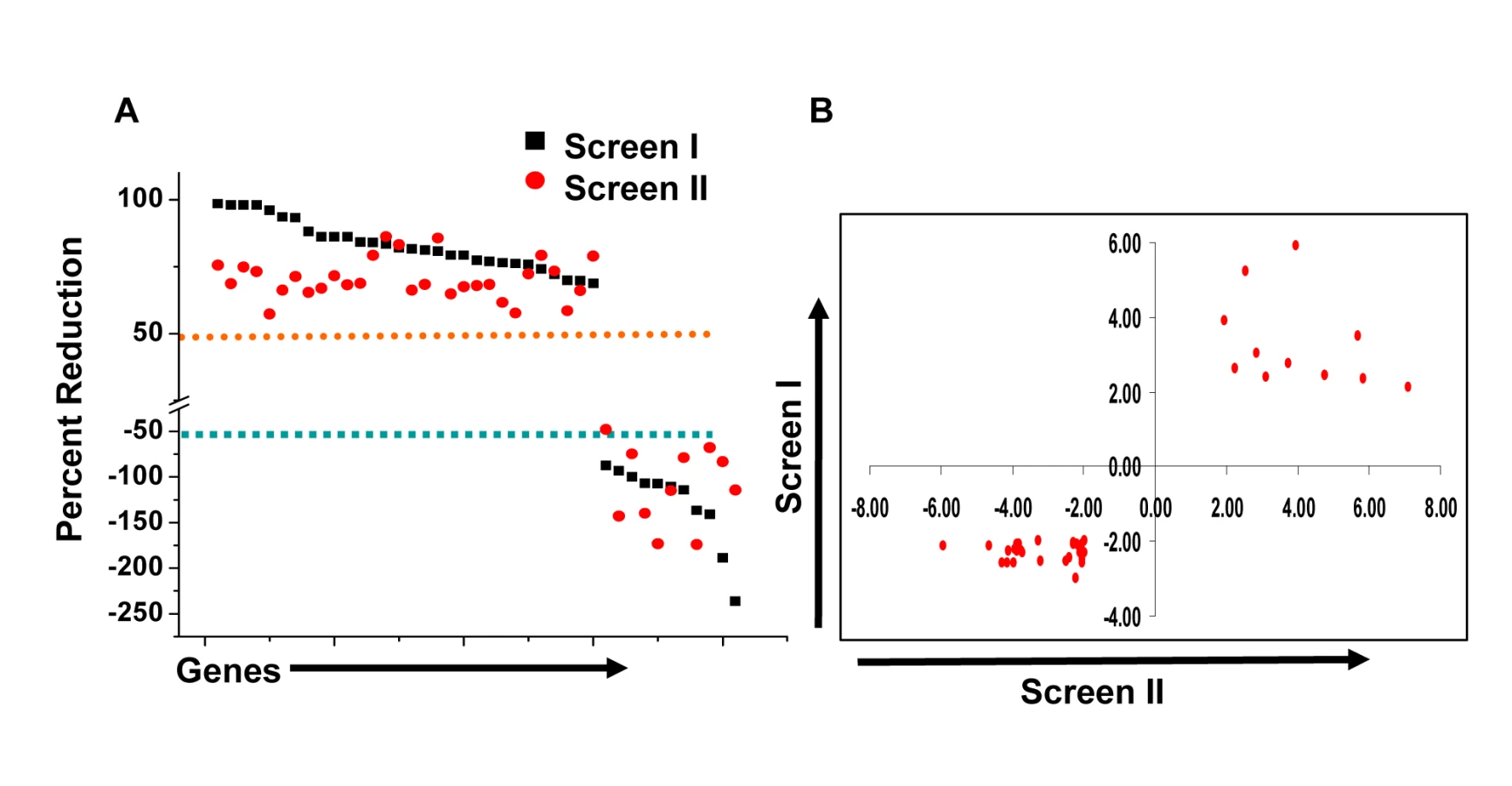
We next took four representative examples from each of the two distinct groups in Figure 1, and examined – by confocal microscopy - whether the observed modulation in CFU values also correlated with alterations in the level of mycobacteria localization, within the lysosome of the host cell. Interestingly, in infected cells treated with non-silencing siRNA, the co-localization of H37Rv with acidified lysosomes could be detected – with a mean overlap coefficient of 0.20 - in only a little over 50% of the cells, (Fig. 2). In contrast, those instances where silencing yielded a reduced CFU count in Figure 1 (Csnk1d, Adrbk1, Prkacb, and TgfβrI), showed a marked increase in mycobacterial co-localization with acidified lysozomes, both at the level of individual cells, as well as at that of the proportion of cells showing such co-localization (Fig. 2). On the other hand, values for both of these parameters were significantly reduced for the cases (Wee1, Abl1, Dgkz, and Chek1) where target gene silencing resulted in an increase in the CFU counts (Fig. 2, compare with Fig. 1).
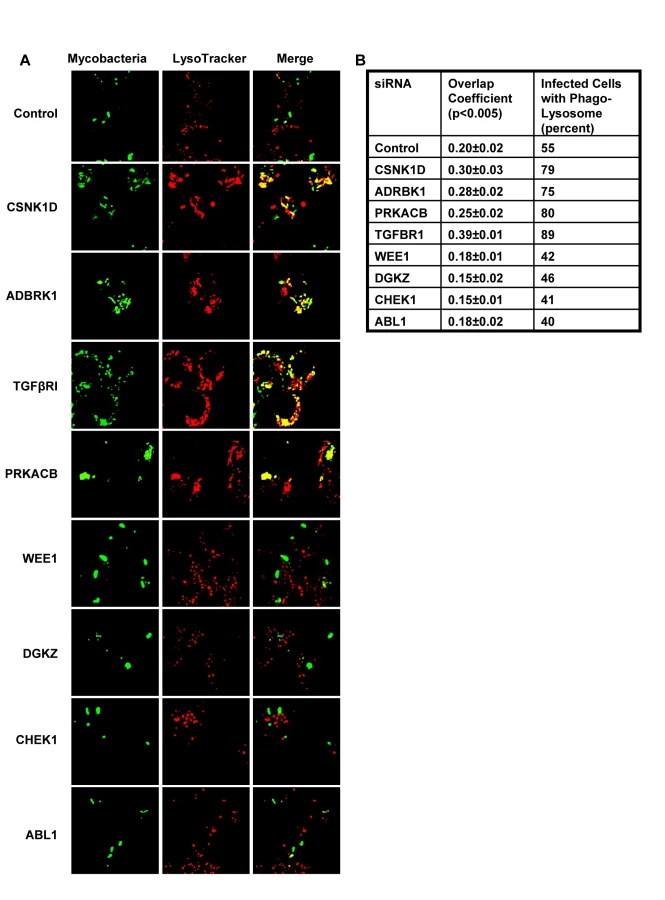
Thus, at least for the representative cases shown in Figure 2, the observed alterations in the CFU counts constitute a true reflection of shifts in the fate of the intracellular bacteria. Finally, an examination of the gene expression profiles both in un-infected J774.1 cells, and in cells infected with H37Rv either for 16h, 48h, or 96h, confirmed that the genes coding for all of the proteins listed in Figure 1 were indeed expressed in uninfected cells, albeit to different levels. Further, expression levels of thirty-four of these remained largely unaffected following infection of the cells (Table S4).
Identification of common host-derived protein targets for MDR-Mtb
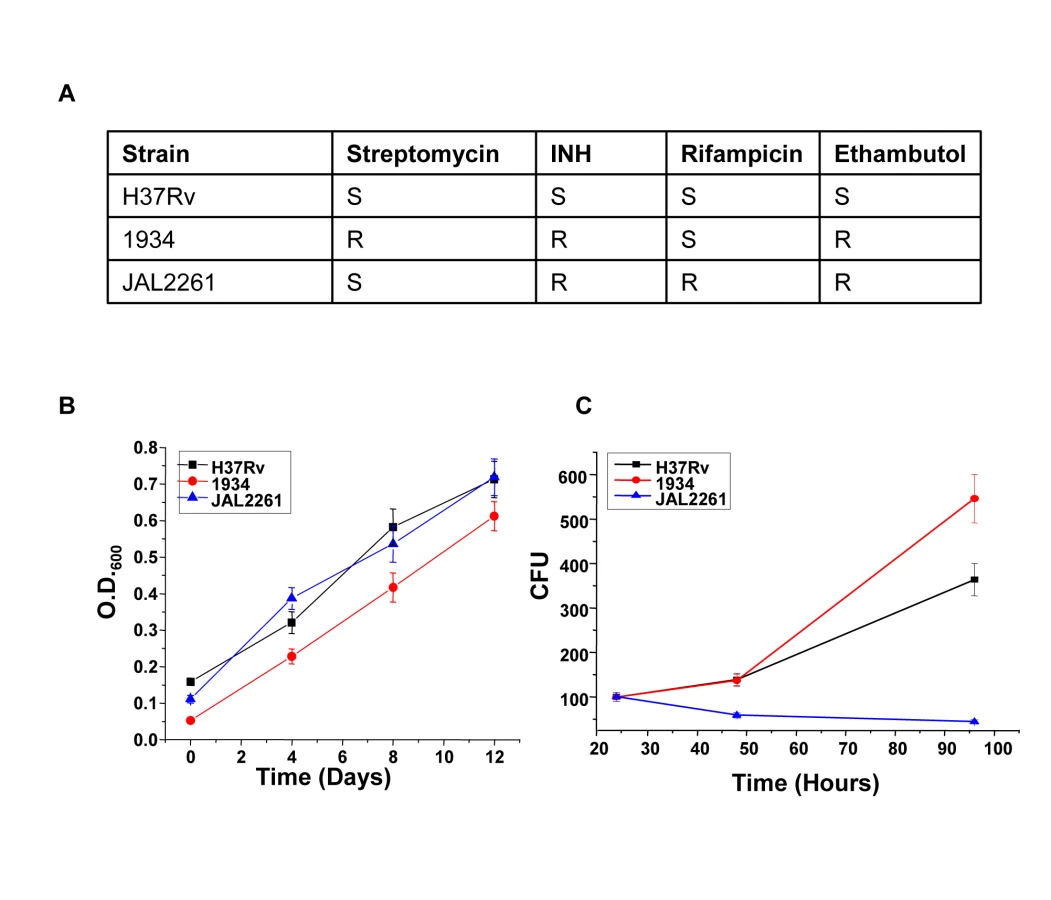
We next asked whether the proteins identified against H37Rv in Figure 1 would also be relevant for infections with field isolates of Mtb, particularly for those exhibiting multiple drug resistance (MDR). To address this we took two independent clinical isolates of MDR-Mtb, each displaying a distinct drug-sensitivity profile (Fig. 3A). The relative growth rates of these two isolates with, that of H37Rv, was first compared both in extracellular cultures, as well as in infected cells. Interestingly, while all the three isolates displayed similar growth properties under extracellular conditions (Fig. 3B) these, however, differed markedly when measured within infected J774.1 cells (Fig. 3C). In this latter experiment, a progressive increase in CFU counts was obtained for H37Rv over the time course studied whereas the MDR strain 1934 displayed an accelerated growth rate between the 48h and the 90h time points. In stark contrast, however, virtually no increase in bacterial titers could be observed for the other MDR strain, JAL2261, at any of the time points studied. A reduced intracellular growth rate for JAL2261, in comparison with the other two strains, was also similarly observed in primary murine macrophages (Fig. S1). Thus the three Mtb strains studied exhibit diverse growth properties within J774.1 cells.
We next infected J774.1 cells with each of the MDR-Mtb strains and tested for the effects of treatment with the siRNAs described in Figure 1. The results obtained are shown in Figure 4. While the overall profiles obtained for both 1934 and JAL2261 show distinctions from that for H37Rv, some overlaps were – nonetheless – clearly evident. This was particularly true in the case of 1934 where, of the 41 targets tested, comparable effects of target silencing (i.e. within a 30% deviation) on both 1934 and H37Rv were observed in 31 cases (Fig. 4). The majority of these instances (25 of 31) represented the group wherein siRNA treatment yielded a reduction in the intracellular bacterial load, whereas a relatively poorer correspondence was obtained for the group where increased bacterial titers were observed (6 out of 11; Fig. 4). Discrepant results were obtained for Trpm7 and Prkag3 although the cause for this is presently unclear.

In contrast to 1934, a comparison between the results obtained for JAL2261 and H37Rv revealed a significantly reduced overlap, with the similarity in effects being restricted to only 11 instances of siRNA treatment (Fig. 4). Further, all of these cases derived exclusively from the subset involving a consequent reduction in the level of the intracellular mycobacteria (Fig. 4). This reduced sensitivity of JAL2261, to perturbations in host protein levels, is probably consistent with the fact that it exists in a non-dividing, or slow-dividing, state within the host cells. Thus the results in Figure 4 identify 11 host targets, whose depletion uniformly led to a reduction in intracellular load of all the three Mtb strains tested.
Host factors regulating intracellular Mtb levels provide attractive targets for drug development
The identification of host factors commonly implicated in regulating intracellular levels of all the three Mtb strains tested raised the possibility that at least some of these may serve as targets for the development of drugs against both drug-sensitive and drug-resistant forms of Mtb infection. At least at the level of ‘proof of principle’, employing existing chemical inhibitors of any of these targets can readily test such a possibility. Therefore, for the present purposes, we chose to examine the effects of inhibition of TGF-β type-1 receptor (TGFβRI). Our choice of this target was based on the results of our microarray experiments which indicated that transcript levels of both this receptor, as well as that of its isoform TGF-β type-2 receptor (TGFβRII) were significantly increased in cells infected with Mtb for 16h, although these levels then returned to their basal values at the later times (Fig. 5A). Further, expression of the genes for the cognate ligands, TGFβI and TGFβII were also induced (Fig. 5A). Here, although the extent of induction of TGFβI was not significant (i.e. below the cut-off threshold) this, nonetheless, translated into a significant increase in the levels of this cytokine in the culture supernatants (Fig. 5B). It was, therefore, logical to infer that secretion of TGFβ by infected cells would lead to the corresponding activation of its receptor, either through autocrine or paracrine mechanisms. Further, at least based on the results of our siRNA screen, it seemed likely that this activation was somehow relevant for maintenance of the intracellular pathogen.
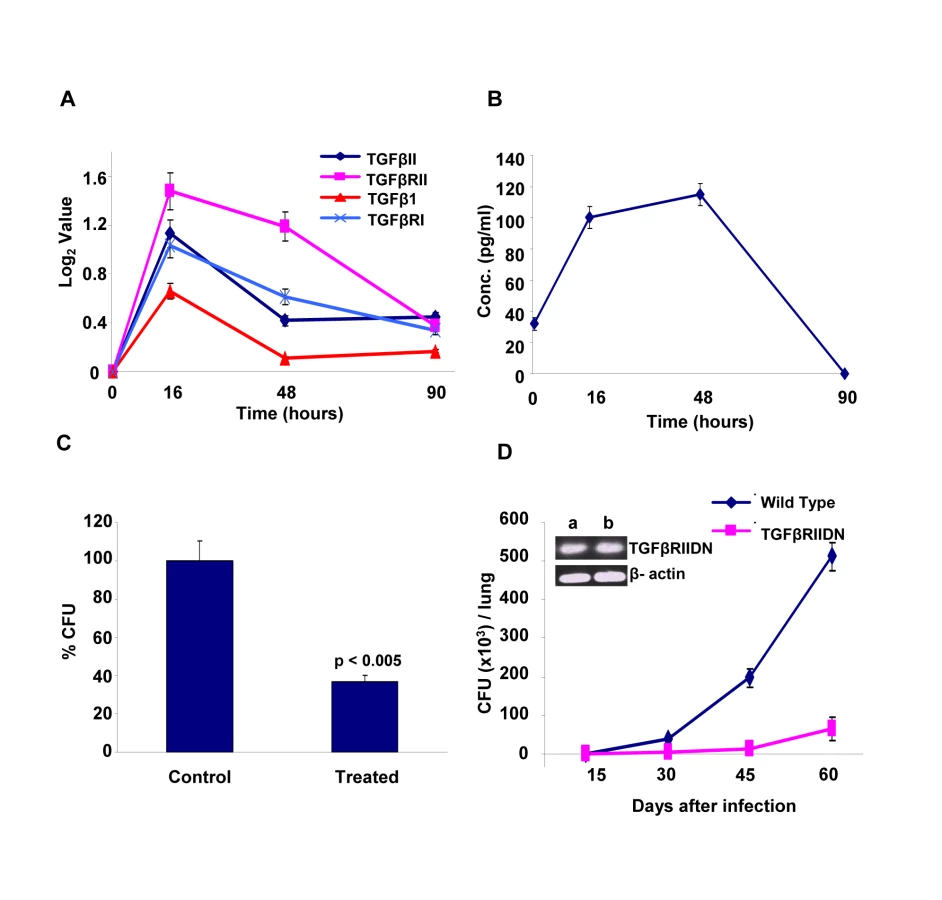
To experimentally verify the inferred role for TGFβ-dependent activation of its receptor, we added neutralizing anti-TGFβ antibodies to cultures of H37Rv-infected J774 cells and then determined its consequent effect in terms of the mycobacterial CFUs obtained. Figure 5C shows that addition of anti-TGFβ antibodies resulted in a substantial reduction in intracellular Mtb load, thus confirming the relevance of this cytokine in regulating intracellular Mtb. To further establish this, we also performed experiments in mice that were transgenic for the dominant negative form of the TGFβ receptor type II (TGFβRIIDN). Although our screen identified TGFβ receptor type-I as the hit, signaling through TGFβ1 involves the formation of an active heteromeric complex between the type-I and type-II receptors. Thus, binding of TGFβ to the type-II receptor leads to the recruitment and phosphorylation of the type-I receptor, with the subsequent activation of downstream pathways [15]. Consequently, TGFβ-dependent signaling would be similarly affected by either silencing the type-1 receptor, or over–expressing the dominant negative form of the type-II receptor. As shown in Figure 5D, TGFβRIIDN mice were significantly more resistant to an aerosol route of infection with H37Rv, than their wild type counterparts. This was consistent with the fact that macrophages isolated from the transgenic mice indeed showed over-expression of TGFβRIIDN (Fig. 5D). Collectively then, the results in Figure 5 provide strong experimental support for the likelihood that TGFβ-dependent activation of its receptor on macrophages is critical for the survival of intracellular Mtb.
To inhibit TGFβRI activation, we employed the compound 4-[4-(2,3-Dihydro-1,4-benzodioxin-6-yl)-5-(2-pyridinyl)-1H-imidiazol-2-yl]benzene (D4476). Our choice of this inhibitor was guided by the fact that, in addition to TGFβRI, this compound is also known to inhibit casein kinase 1 (CSNK1) [16], [17], another member of our validated target list in Figure 4. Interestingly, CSNK1 also represents a downstream intermediate in the TGFβR signaling pathway, and has been shown to regulate both ligand-independent (i.e. basal), as well as ligand-induced signaling processes [18]. Thus, it was anticipated that the simultaneous inhibition of TGFβRI and one of its downstream signaling intermediates would yield a more effective inhibition of TGFβ-dependent signaling and, therefore, a more potent effect on intracellular Mtb levels.
Cells infected either with H37Rv, 1934, or JAL2261 were treated with increasing doses of D4476, and the consequent effect on intracellular pathogen load was then determined as the resulting CFU values obtained (Fig. 6A). A dose-dependent decrease in mycobacterial CFUs, with increasing inhibitor concentrations, was clearly obtained for all the three isolates tested (Fig. 6A). Importantly, this effect was specific for the mycobacteria and this drug displayed no toxicity towards the host cell (Fig. 6B). In parallel studies employing confocal microscopy we observed that treatment with the inhibitor also led to a corresponding increase in localization of each of the mycobacterial isolates within acidified lysosomes of the cell (Fig. 6C). This further supports that treatment with the inhibitor leads to an enhanced clearance of the intracellular pathogen. Importantly, the effect of D4476 addition was specific for intracellular mycobacteria, with no significant effect on mycobacterial growth in extracellular cultures (Fig. 6D). This confirms that the inhibitor did not directly target Mtb. Rather it more likely interfered with the intracellular survival mechanisms of the pathogen. Importantly, from the standpoint of pharmacological intervention, that simultaneous inhibition of TGFβRI and its downstream signaling intermediate leads to a more potent effect on intracellular Mtb could be demonstrated by comparing the relative efficacy of D4476 with that of inhibitors specific for only either TGFβRI (LY364947 [19]) or CSNK1 (IC261, [17]) (Fig. 6E).
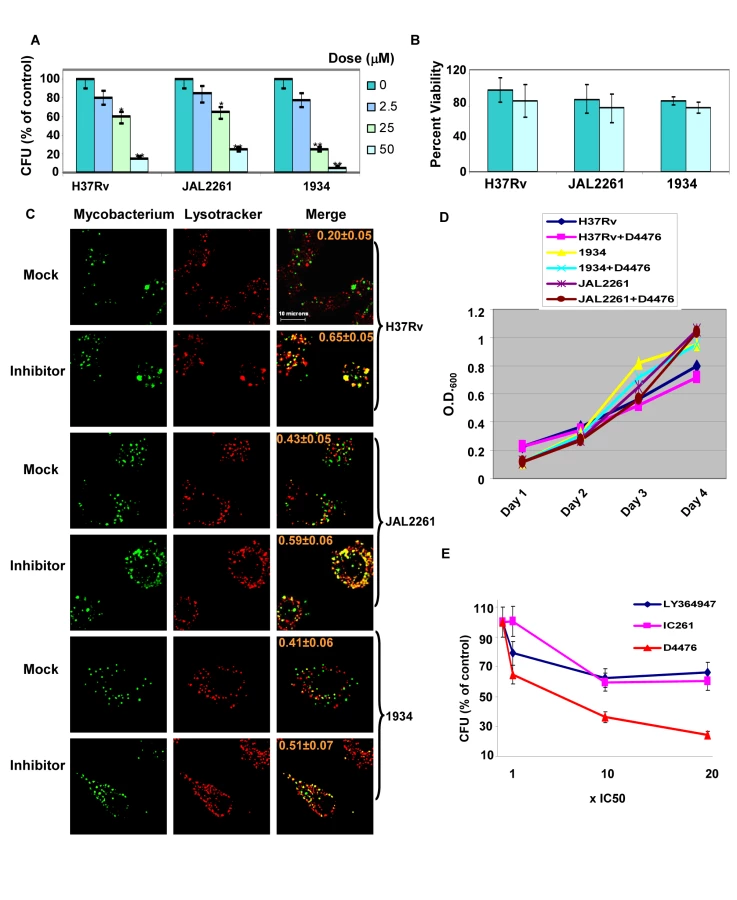
The inhibitory effect of D4476 on intracellular Mtb survival could also be demonstrated in primary murine macrophages infected with each of the three Mtb strains. Thus, consistent with the findings in J774 cells, addition of D4476 to infected primary macrophages resulted in enhanced co-localization of all three mycobacterial strains with the lysosome, and also a significant reduction in CFU count in each of these cases (Figure S1). Thus the cumulative results in Figure 6 confirm a key role for TGFβR signaling in regulating intracellular Mtb survival, but in a manner that is independent of at least the spectrum of phenotypic variations encompassed by this group of isolates.
Compound D4476 also eliminates Mtb from infected mice
To further validate the relevance of targeting host factors, we also examined the efficacy of D4476 in the murine model of TB infection. For this, BALB/c mice were intravenously infected with H37Rv and these mice were then treated with two different concentrations of D4476. Treatment was initiated at ten days after the infection, and included six administrations of the relevant dose of the inhibitor (Text S1). As shown in Figure 7A, a clear reduction in CFU counts was obtained from the lungs of infected mice treated with D4476. Further, the magnitude of this inhibition was sensitive to the dose of the compound with a dose of 4 nmol/g of body weight yielding a nearly 80% reduction in CFU counts. Similar results were also obtained in mice infected through the aerosol route (Fig. 7B), confirming that the efficacy of D4476 was independent of the route of infection employed. D4476-dependent clearance of infection was also evidenced through recovery from the infection-induced splenomegaly (Fig. 7C). Further, histochemical staining of lung sections also revealed a significant reduction in the number of epitheloid cell granulomas, and in that of acid-fast staining bacilli, in treated versus the untreated mice (Figure 7D).
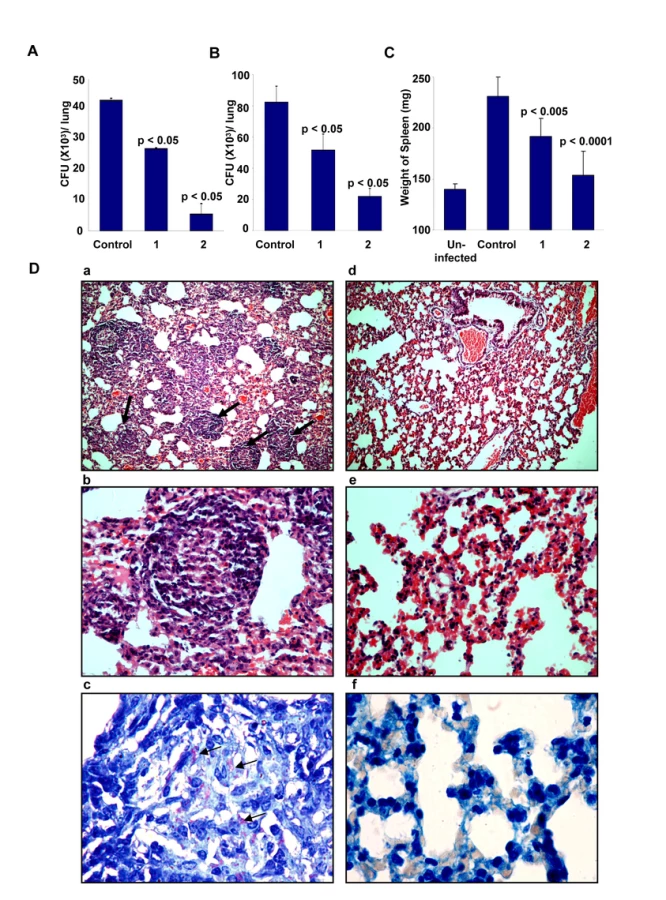
The collective results in Figure 7, therefore, demonstrate the in vivo efficacy of D4476 in terms of eliminating an Mtb infection. While its potency may be considered to be low we emphasize, however, that our objective here was not to propose this compound as a drug against TB. Rather, these and the preceding experiments were merely intended to reveal and highlight the feasibility of targeting relevant host factors, as an alternate strategy for the chemotherapy of TB.
Discussion
Recent years have witnessed a resurgence of efforts directed at tuberculosis (TB) drug research and development. This has been spurred by the increasing incidence of drug resistance in the infected population, engendering an urgent need for the development of improved regimens for TB treatment. Although drug resistant - particularly multi- and extremely-drug resistant - TB poses a daunting challenge, the situation is further complicated by the predominance of latent and/or persistent forms of TB infection in the population [20]. These latter infections are characterized by phenotypic resistance - or tolerance - to drugs, despite being genotypically sensitive to them [20]. Indeed, latent/persistent infection constitutes over ninety nine percent of the TB infected population worldwide [20].
Conventional approaches to drug research have targeted unique processes or enzymes of the pathogen. However, despite its many successes, this approach suffers from the risk of generating newer variants exhibiting drug resistance [21]. Further, this strategy is also limited by its inability to address infection states where the pathogen is metabolically less active such as in persistent or latent infections. This issue is especially relevant in the case of Mtb infections. An alternate paradigm for drug discovery has recently emerged, at least for intracellular pathogens [22]. This is based on the fact that the survival of an intracellular pathogen in the hostile cellular environment requires it to exploit and subvert various host factors. Therefore, identifying - and then targeting – such host factors should provide an additional route for the therapeutic management of these infectious diseases. Here, the anticipations also are that such a strategy will be less likely to induce microbial resistance [22].
To explore the possibilities offered by this avenue, siRNA-based screens are now being widely employed to identify such host factors for a range of viral, bacterial, and parasitic infections [23], [24], [25], [26], [27], [28], [29]. In this connection, a recent screen of the human kinome identified a kinase cluster around protein kinase B (PKB) as being obligatory for the intracellular survival of Salmonella typhimurium [30]. Interestingly, while inhibitors of PKB were effective against this pathogen, they were also capable of partially inhibiting infection of human macrophages with a strain of MDR-Mtb. We, therefore, undertook the present study to further explore the potential of such an approach in the specific context of Mtb.
Our siRNA screen successfully identified several host factors involved in the regulation of H37Rv survival within the milieu of the host macrophage. Interestingly, this group of target proteins included those that facilitated, as well as those that impeded Mtb survival. While a mechanistic resolution of the biochemical pathways involved is clearly needed, this identification of molecular components with opposing functional roles strongly supports the existence of a host-specified axis that regulates the fate of intracellular Mtb. Such an axis probably reflects the level of equilibrium achieved between the intracellular processes initiated to eliminate the infection, and the pathogen-mediated manipulation of the host machinery in its own favor [31]. In this connection, it is pertinent to note that previous studies have identified several host proteins that play an important role during the course of an Mtb infection. However, with the exception of a few examples [32], the majority of these proteins are involved in processes that occur either during, or soon after, the endocytic uptake of Mtb. Thus, for example, the role of the calcium signaling pathway- involving PI3K, PKB, CaMKII and Coronin 1 – in regulating endocytosis and subsequent fusion of phagosomes with lysosomes has been well studied [1], [12]. Similarly, there is also information available on the biochemical pathways that mediate microbicidal responses of the infected macrophage [33]. In contrast to these early responses, however, our screen was designed to specifically capture those host molecular components that are involved in the later time window of the infection process. That is, in the window where the pathogen is either in the process of establishing, or has established, a dynamic equilibrium with the intracellular machinery of the host cell. Consequently, our present study examined a relatively less explored facet of macrophage infection by Mtb and, therefore, identified several novel host molecules whose role in regulating intracellular Mtb has not been hitherto suspected. From the standpoint of targeting host factors as a drug development strategy for TB, we believe that it is such factors that regulate the maintenance of an established infection that would be more relevant.
An important aspect of our studies was the additional filtration, of the identified H37Rv-specific host factors, against MDR-Mtb variants that not only exhibited altered drug-sensitivity profiles, but also altered properties of intracellular growth. Interestingly, a significant proportion of these factors could be validated against the other rapidly growing strain, 1934. In contrast, only a small subset of these targets retained efficacy against JAL2261, a strain whose levels did not significantly increase within infected cells. These differences are consistent with at least a priori expectations that the extent of cross-regulatory interactions between host and pathogen would correlate directly with state of replication activity of the pathogen.
The net outcome of our experiments was the identification of a core list of host targets that were involved in regulating survival of Mtb, independent of variations in either drug sensitivity profiles or growth properties in the host cell. Further, we could also demonstrate, at the level of ‘proof of principle’, that at least some of these host factors may provide novel targets for the development of anti-TB drugs. This was exemplified by our findings that the simultaneous inhibition of TGFβRI and CSNK1 substantially inhibited intracellular survival of both drug-sensitive and multiple drug-resistant strains of MTB. In addition to J774.1 cells, this effect was also observed in primary murine macrophages. More importantly though, our experiments involving D4476 administration to H37Rv-infected mice also provided in vivo validation for the possibility of targeting host factors as a possible approach for TB therapy. Here, it will be important to test the potential applicability of the remaining host factors identified by our screen in this regard. Further, the mechanism by which inhibition of TGFβRI and CSNK1 induces elimination of the infection is also of interest.
Thus, in summary, our present results highlight the existence of host factors that regulate the intracellular survival of Mtb in a manner that is insensitive to variations in either the drug-sensitivity profile, or the intracellular growth properties. In addition, we also provide proof-of-concept demonstration that targeting at least some of these molecules can provide an alternate approach for the chemotherapy of TB. Again as demonstrated here, a highlight of such a strategy would be that it holds the promise of eventually being able to develop suitable drugs that function in a manner that is independent of the phenotypic and genotypic diversification exhibited by Mtb in the field [20]. However, further validation in the context of human infections will be necessary before such a promise can be realized. Further, the issue of possible toxicity to the host cell - as a result of inhibition of key host molecules - may also require to be addressed.
Methods
A detailed description of all the experimental protocols employed, and the standardization procedures are provided in the Text S1.
Ethics statement
All animal experiments were carried out in accordance with guidelines approved and created by the ICGEB animal ethics committee.
Animals
Female BALB/c mice 4–6 wk of age kept in pathogen free environment. TGFβRDN (TGFβ receptor dominant negative, BALB/C background) mice, 6 to 8 weeks of age, were purchased from Jackson Laboratory, Bar Harbor, Maine. These mice were maintained and breed in a specific-pathogen-free biosafety level-3 facility.
Cells and culture
Murine macrophage cell line J774.1 (American Type Culture Collection) was used in this study. J774.1 cells were cultured in RPMI 1640 (Gibco Laboratories) supplemented with 10% FCS (Hyclone) and were maintained between 2 and 10×105 cells per mL at 37°C in a humidified, 5% CO2 atmosphere. Before infection cells were plated in 96 well plates at 2.5×104 cells per well overnight.
siRNA library
Mouse Phosphatase siRNA set V 1.0 and Mouse kinase siRNA set V 1.0 library from Qiagen (two siRNAs/target) were used for the study. For validation experiments siRNAs against the kinases were obtained from Sigma siRNA (MISSION siRNA Mouse Kinase Panel, three siRNAs/target), whereas the phosphatase-specific siRNAs were procured from Dharmacon (SMARTpool) (four siRNAs/target).
Infection of cells and the siRNA screen
J774.1 cells (25,000 cells/well in 96-well plates) were infected with mycobacteria at an MOI ∼10 (10 bacteria /cell, Figure S2). After 4h, infected cells were washed twice with warm RPMI and treated with gentamicin (100 µg/ml) for 2h to remove any remaining extra cellular bacteria, and then in complete RPMI containing 10 µg/mL gentamicin for the rest of the experiment. These cells were then transfected with siRNA at a final concentration of 100nM using hiperfect transfection reagent (Qiagen) according to manufacturer's protocol. At 48h a second siRNA treatment was performed (see Text S1), and the cells cultured for an additional 36h (i.e. a total culture period of 90h after initiation of infection). At this point cells were solubilized in 50 µL of 0.06% SDS (in 7H9 medium), and CFUs determined using either the undiluted lysate, or from lysate dilutions of 1∶10, or 1∶100. Each siRNA pool was evaluated in duplicate and the mean of the CFU values, obtained at the two cell lysate dilutions was determined. Each 96-well plate also included six negative control wells. In two of these cells were treated with scrambled siRNA following infection, whereas another two included infected cells that were transfected with GFP-specific siRNA. In the remaining two wells, infected cells were treated only with the transfection reagent (Hiperfect). The mean CFU count obtained from all of these six wells, in each 96-well plate, was taken as the control value for that plate for determining the effects of target-specific siRNAs. The standard deviation (SD) for the control values was also calculated. A cut off of two SD from the mean value of control wells was employed to designate the effects of a given siRNA treatment as significant. The ‘hits’ obtained from this primary screen were then further validated with siRNA pools obtained from alternate sources as described above. Here, ‘hits’ were confirmed on the basis of the percent change in CFUs from the mean control value, in addition to an SD cut off value of two. That is, an increase or decrease of 50% from the control value for an siRNA with the additional caveat that this deviation from the mean control value was greater than 2SD was selected as validated (see Fig. 1). The validation exercise involved two separate experiments, whereas the subsequent comparison of the effects of the validated siRNA pools in cells infected with H37Rv, with that in cells infected with 1934 and JAL2261 was also ascertained in two additional independent experiments.
RNA isolation and microarray analysis
In two separate experiments, cells were plated in six-well plate (2×106 cells per well) and infected with H37Rv as described above. At 16h, 48h, and 96h later these, and uninfected, cells were lysed and RNA was isolated with trizol. One-color microarray-based gene expression analysis was performed by hybridizing against a mouse whole genome array consisting of probes for 44,000 genes (Agilent).
Confocal microscopy
Bacteria were stained using the membrane stain PKH67 (Sigma) according to the manufacturer's protocol. J774.1 cells were seeded onto #1 thickness, 12 mm diameter glass cover-slips pre-coated with fibronectin in 24-well tissue culture plates at a density of 0.07×106 cells per cover-slip, respectively and infected with stained bacteria using the protocol as described above. Cells were incubated with 100nM Lysotracker during the last hour of the 72 hr chase at 37°C and then fixed with 4% para-formaldehyde (Sigma). The cover slips were washed thoroughly with PBS and were mounted on slides with Antifade (Biorad). Stained cells were observed with a Nikon TE 2000E laser scanning confocal microscope equipped with 60×/1.4 NA PlanApochromat DIC objective lens, and the extent of bacterial co-localization with acidified lysosomes was determined as the Overlap Coefficient (see Text S1).
Infection of mice
Groups of naive mice (Female BALB/c mice 4–6 wk of age at 4/group) were infected with 1×106 M. tuberculosis H37Rv via the tail vein. One group of mice was sacrificed 24 h later and lung homogenates were plated onto 7H11 agar plates for confirming infection. At ten days post infection, compound D4476 a final concentration of either 25 µM or 50 µM was injected into the tail vein of mice for treatment. The i.v. injection was repeated on seventh day of treatment. In addition the inhibitor was also given intraperitoneally on the third, fifth, tenth and twelfth day after initiation of treatment. At fourteen days following treatment, mice were sacrificed by carbon dioxide narcosis.
For experiments involving the aerosol route of infection, mice (8 per group) were infected with H37Rv by delivering between 100–150 bacteria per lung – as determined by the culture of lung homogenates at 24 h later - during 30 min of exposure. At the relevant times after infection, mice were sacrificed by carbon dioxide narcosis. The lungs were perfused and removed aseptically and weighed. The lungs were then homogenized and dilutions of these homogenates were plated on 7H11 agar plates for subsequent enumeration of the CFU.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KoulA
HergetT
KleblB
UllrichA
2004 Interplay between mycobacteria and host signalling pathways. Nat Rev Microbiol 2 189 202
2. KaufmannSH
ColeST
MizrahiV
RubinE
NathanC
2005 Mycobacterium tuberculosis and the host response. J Exp Med 201 1693 1697
3. FlynnJL
ChanJ
2001 Immunology of tuberculosis. Annu Rev Immunol 19 93 129
4. ColeST
BroschR
ParkhillJ
GarnierT
ChurcherC
1998 Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393 537 544
5. GehringAJ
RojasRE
CanadayDH
LakeyDL
HardingCV
2003 The Mycobacterium tuberculosis 19-kilodalton lipoprotein inhibits gamma interferon-regulated HLA-DR and Fc gamma R1 on human macrophages through Toll-like receptor 2. Infect Immun 71 4487 4497
6. RaghavanS
ManzanilloP
ChanK
DoveyC
CoxJS
2008 Secreted transcription factor controls Mycobacterium tuberculosis virulence. Nature 454 717 721
7. RengarajanJ
BloomBR
RubinEJ
2005 Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A 102 8327 8332
8. WalburgerA
KoulA
FerrariG
NguyenL
Prescianotto-BaschongC
2004 Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304 1800 1804
9. EhrtS
SchnappingerD
2007 Mycobacterium tuberculosis virulence: lipids inside and out. Nat Med 13 284 285
10. MacMickingJD
TaylorGA
McKinneyJD
2003 Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302 654 659
11. KelleyVA
SchoreyJS
2003 Mycobacterium's arrest of phagosome maturation in macrophages requires Rab5 activity and accessibility to iron. Mol Biol Cell 14 3366 3377
12. JayachandranR
SundaramurthyV
CombaluzierB
MuellerP
KorfH
2007 Survival of mycobacteria in macrophages is mediated by coronin 1-dependent activation of calcineurin. Cell 130 37 50
13. ViaLE
DereticD
UlmerRJ
HiblerNS
HuberLA
1997 Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem 272 13326 13331
14. FrattiRA
ChuaJ
VergneI
DereticV
2003 Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci U S A 100 5437 5442
15. ShiY
MassagueJ
2003 Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113 685 700
16. RenaG
BainJ
ElliottM
CohenP
2004 D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep 5 60 65
17. BainJ
PlaterL
ElliottM
ShpiroN
HastieCJ
2007 The selectivity of protein kinase inhibitors: a further update. Biochem J 408 297 315
18. WaddellDS
LiberatiNT
GuoX
FrederickJP
WangXF
2004 Casein kinase Iepsilon plays a functional role in the transforming growth factor-beta signaling pathway. J Biol Chem 279 29236 29246
19. SawyerJS
BeightDW
BrittKS
AndersonBD
CampbellRM
2004 Synthesis and activity of new aryl- and heteroaryl-substituted 5,6-dihydro-4H-pyrrolo[1,2-b]pyrazole inhibitors of the transforming growth factor-beta type I receptor kinase domain. Bioorg Med Chem Lett 14 3581 3584
20. GinsbergAM
SpigelmanM
2007 Challenges in tuberculosis drug research and development. Nat Med 13 290 294
21. WalshC
2000 Molecular mechanisms that confer antibacterial drug resistance. Nature 406 775 781
22. SchwegmannA
BrombacherF
2008 Host-directed drug targeting of factors hijacked by pathogens. Sci Signal 1 re8
23. AgaisseH
BurrackLS
PhilipsJA
RubinEJ
PerrimonN
2005 Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science 309 1248 1251
24. BrassAL
DykxhoornDM
BenitaY
YanN
EngelmanA
2008 Identification of host proteins required for HIV infection through a functional genomic screen. Science 319 921 926
25. KonigR
ZhouY
EllederD
DiamondTL
BonamyGM
2008 Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 135 49 60
26. KrishnanMN
NgA
SukumaranB
GilfoyFD
UchilPD
2008 RNA interference screen for human genes associated with West Nile virus infection. Nature 455 242 245
27. PhilipsJA
RubinEJ
PerrimonN
2005 Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science 309 1251 1253
28. PielageJF
PowellKR
KalmanD
EngelJN
2008 RNAi screen reveals an Abl kinase-dependent host cell pathway involved in Pseudomonas aeruginosa internalization. PLoS Pathog 4 e1000031 doi:10.1371/journal.ppat.1000031
29. PrudencioM
RodriguesCD
HannusM
MartinC
RealE
2008 Kinome-wide RNAi screen implicates at least 5 host hepatocyte kinases in Plasmodium sporozoite infection. PLoS Pathog 4 e1000201 doi:10.1371/journal.ppat.1000201
30. KuijlC
SavageND
MarsmanM
TuinAW
JanssenL
2007 Intracellular bacterial growth is controlled by a kinase network around PKB/AKT1. Nature 450 725 730
31. YoungD
StarkJ
KirschnerD
2008 Systems biology of persistent infection: tuberculosis as a case study. Nat Rev Microbiol 6 520 528
32. GanH
LeeJ
RenF
ChenM
KornfeldH
2008 Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat Immunol 9 1189 1197
33. WarnerDF
MizrahiV
2007 The survival kit of Mycobacterium tuberculosis. Nat Med 13 282 284
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 4
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Diagnostika virových hepatitid v kostce – zorientujte se (nejen) v sérologii
Nejčtenější v tomto čísle
- The Effect of Vaccination on the Evolution and Population Dynamics of Avian Paramyxovirus-1
- Reconstitution of SARS-Coronavirus mRNA Cap Methylation
- Deficiencies in Jasmonate-Mediated Plant Defense Reveal Quantitative Variation in Pathogenesis
- A Timescale for Evolution, Population Expansion, and Spatial Spread of an Emerging Clone of Methicillin-Resistant