
Peptide Inhibitors of Dengue-Virus Entry Target a Late-Stage Fusion Intermediate
The mechanism of membrane fusion by “class II” viral fusion proteins follows a pathway that involves large-scale domain rearrangements of the envelope glycoprotein (E) and a transition from dimers to trimers. The rearrangement is believed to proceed by an outward rotation of the E ectodomain after loss of the dimer interface, followed by a reassociation into extended trimers. The ∼55-aa-residue, membrane proximal “stem” can then zip up along domain II, bringing together the transmembrane segments of the C-terminus and the fusion loops at the tip of domain II. We find that peptides derived from the stem of dengue-virus E bind stem-less E trimer, which models a conformational intermediate. In vitro assays demonstrate that these peptides specifically block viral fusion. The peptides inhibit infectivity with potency proportional to their affinity for the conformational intermediate, even when free peptide is removed from a preincubated inoculum before infecting cells. We conclude that peptides bind virions before attachment and are carried with virions into endosomes, the compartment in which acidification initiates fusion. Binding depends on particle dynamics, as there is no inhibition of infectivity if preincubation and separation are at 4°C rather than 37°C. We propose a two-step model for the mechanism of fusion inhibition. Targeting a viral entry pathway can be an effective way to block infection. Our data, which support and extend proposed mechanisms for how the E conformational change promotes membrane fusion, suggest strategies for inhibiting flavivirus entry.
Published in the journal:
. PLoS Pathog 6(4): e32767. doi:10.1371/journal.ppat.1000851
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000851
Summary
The mechanism of membrane fusion by “class II” viral fusion proteins follows a pathway that involves large-scale domain rearrangements of the envelope glycoprotein (E) and a transition from dimers to trimers. The rearrangement is believed to proceed by an outward rotation of the E ectodomain after loss of the dimer interface, followed by a reassociation into extended trimers. The ∼55-aa-residue, membrane proximal “stem” can then zip up along domain II, bringing together the transmembrane segments of the C-terminus and the fusion loops at the tip of domain II. We find that peptides derived from the stem of dengue-virus E bind stem-less E trimer, which models a conformational intermediate. In vitro assays demonstrate that these peptides specifically block viral fusion. The peptides inhibit infectivity with potency proportional to their affinity for the conformational intermediate, even when free peptide is removed from a preincubated inoculum before infecting cells. We conclude that peptides bind virions before attachment and are carried with virions into endosomes, the compartment in which acidification initiates fusion. Binding depends on particle dynamics, as there is no inhibition of infectivity if preincubation and separation are at 4°C rather than 37°C. We propose a two-step model for the mechanism of fusion inhibition. Targeting a viral entry pathway can be an effective way to block infection. Our data, which support and extend proposed mechanisms for how the E conformational change promotes membrane fusion, suggest strategies for inhibiting flavivirus entry.
Introduction
Membrane fusion is a critical step for infectious entry of enveloped viruses into cells [1]. A viral “fusion protein” facilitates this process, generally in response to molecular cues specific for the cellular compartment in which viral penetration occurs. For example, dengue and other flaviviruses penetrate from endosomes, following uptake by clathrin-mediated endocytosis [2],[3], and proton binding is the immediate fusion trigger [4].
The flaviviruses are insect-borne agents with positive-strand RNA genomes packaged into compact particles, about 500 Å in diameter [5]. Their fusion protein, known as E, is the principal external protein of the virion. It is made as part of a polyprotein, which includes a “chaperone” protein, designated prM (precursor of M). Cleavage of prM during viral maturation releases most of its ectodomain and promotes formation of a well-ordered lattice of 90 E dimers on the virion surface [6],[7]. When the pH drops below about 6.2, E undergoes a large-scale conformational rearrangement that includes dissociation of the dimer and reconfiguration of the subunits into trimers (Fig. 1A,B) [8]. At an intermediate stage in this complex molecular reorganization, a hydrophobic “fusion loop” at one end of the extended E subunit inserts into the outer leaflet of the target membrane bilayer [9],[10],[11],[12]. Further rearrangement then draws together the fusion loop and the transmembrane segment anchoring E in the viral membrane. The latter step forces the two membranes together, allowing fusion to ensue.
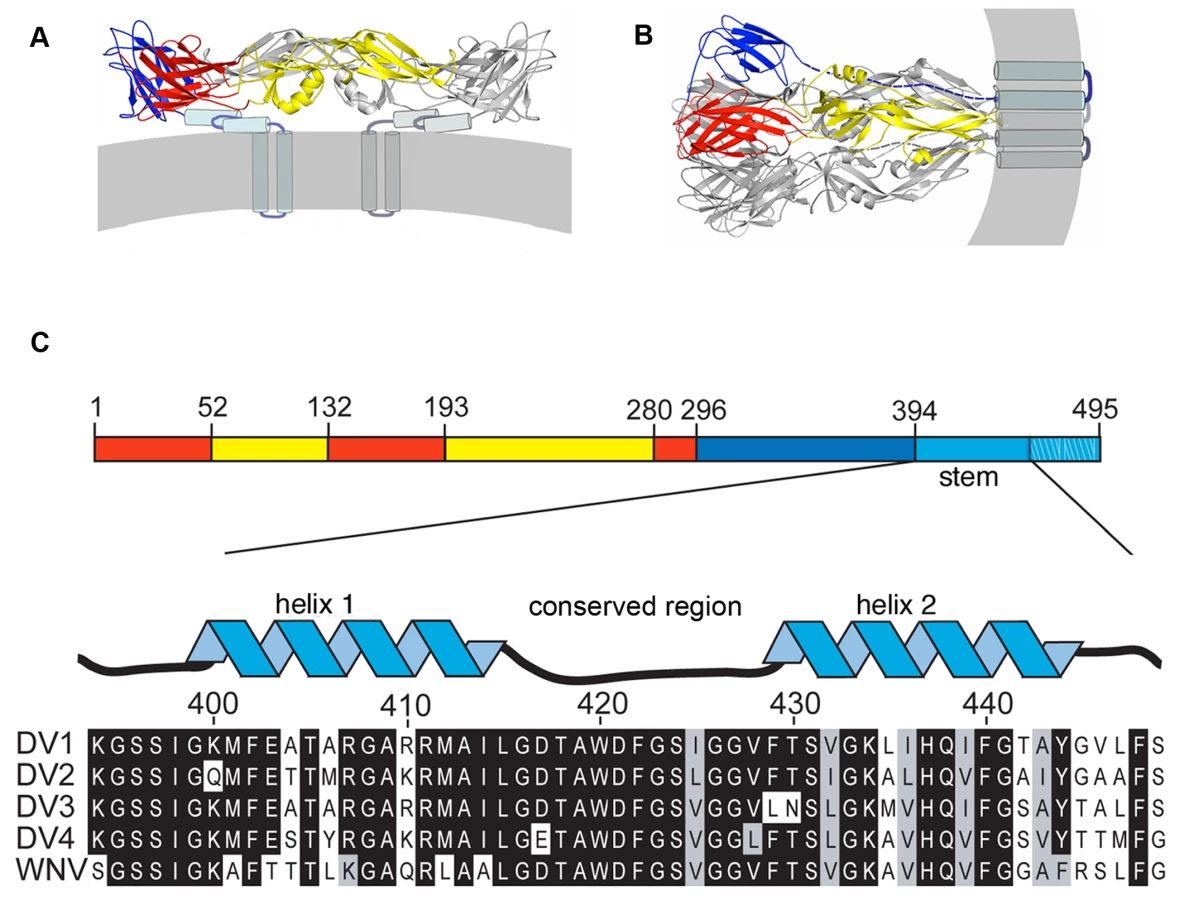
This molecular description of flavivirus fusion derives in part from crystal structures of several flavivirus E proteins in conformations corresponding to prefusion dimers and to postfusion trimers (Fig. 1A) [9],[11],[12],[13],[14]. These structure determinations were made possible by C-terminal truncation of the E ectodomain, through proteolytic treatment or recombinant expression, to increase protein solubility and biochemical stability [15]. These soluble forms of E (“sE”) include only the first 395 of the approximately 445 ectodomain residues; they lack a membrane-proximal region called the “stem” (Fig. 1C) that is substantially conserved among flaviviruses. Intermediate-resolution cryoEM reconstructions show that in the prefusion dimer, the stem segment folds into a helix-loop-helix sandwiched between the rest of the ectodomain and the membrane [7],[16]. In the postfusion conformation, it has been inferred that the stem extends along the edge of domain II, which bears the fusion loop at its tip (Fig. 1) [11].
In the postfusion conformation of E, the fusion loops and the C-terminal transmembrane anchors of all three subunits cluster at one end of the rearranged protein (Fig. 1B). The driving force for pinching together of the two membranes appears to come from contacts made by domain III, as it folds back against domain I at the base of the hairpin, and by the stem with domain II, as it “zips” up along domain II. A consequence of this picture is that interfering with either of these interfaces – for example, by a soluble form of domain III or of the stem – will probably prevent viral fusion. A well-known precedent is T-20/entfuvirtide, a peptide inhibitor of HIV-1 infectivity [17],[18],[19],[20]. T-20 is a soluble fragment of gp41 that blocks the analogous zipping-up step in gp41 refolding. Kielian and co-workers have shown that soluble dengue E-protein domain III can inhibit entry of dengue virus bound at the cell surface when fusion is triggered by transient acidification, and they have made similar observations for the related alphavirus fusion protein, E1 [21]. The likely mechanism involves binding of the soluble domain to the extended intermediate structure, thereby aborting the internal fold-back. One report also suggests that a stem peptide can inhibit dengue virus infection in a conventional infectivity assay, but with no mechanistic analysis [22]. The observation is puzzling, because we might expect a site for specific stem-peptide binding to be present only after induction of a conformational transition by the low pH of an endosome and an externally applied peptide to be inactive unless carried into the endosome by some non-specific process.
In the work reported here, we show that peptides derived from the dengue E protein stem indeed bind the trimeric, postfusion sE conformer, but not the prefusion dimer. The sequence specificity of this interaction is consistent with our prediction, based on the trimeric sE structure, that such peptides should be able to dock against domain II. We also show that the same stem peptides can inhibit viral infectivity by binding the virion at neutral pH before cell attachment and, presumably, by accompanying the virus particles into endosomes. Hydrophobic interactions between the C-terminal segment of the peptide and the viral membrane contribute to virion binding and hence to inhibiting infection; sequence-specific contacts may have a role in initial binding, in addition to their principal inhibitory contribution when internalized peptide binds rearranging E protein, after the virus has encountered a lowered pH.
Results
Binding of stem peptides with sE
The order of conformational changes in E outlined in the Introduction suggests that stem peptides might bind the trimer-clustered domain-II regions of sE. That is, because the entire stem is missing from the sE fragment, the sE trimer in effect represents an intermediate state in the conformational transition, just before the stem zips up along domain II. Fig. 1C shows the stem sequences from DV serotypes as well as the closely related West Nile Virus (WNV) and illustrates their nearly complete conservation. We made a set of synthetic peptides, all based on the DV2 consensus sequence, for affinity measurements. The peptides were conjugated to FITC at their N-termini, and their affinities for prefusion sE dimer and postfusion sE trimer were measured with a fluorescence polarization assay.
We produced the trimeric postfusion form of sE following the protocol of Modis et al (2004), which derived in turn from earlier work [23] on tick-borne encephalitis (TBE) virus. Acidification of sE dimer in the presence of liposomes leads to dimer dissociation, insertion of the fusion loop into the outer liposome leaflet, and catalysis of trimer formation by the enforced alignment of monomers on the membrane surface. The sE remains trimeric after neutralization; it is soluble in the presence of non-ionic detergents.
We measured binding isotherms by adding increasing amounts of either the prefusion or the postfusion conformer of sE to a constant concentration of FITC-tagged peptide. Dissociation constants (Kd) were calculated as described in Methods. Peptide DV2419–447 binds selectively to the postfusion conformer of sE; a scrambled version of DV2419–447 (DV2419–447(scram)), used as a specificity control, binds neither conformer (Fig. 2A). Results from this series of experiments are summarized in Figure 2B. Peptides containing the C-terminal half of the dengue virus type 2 stem bind the sE postfusion trimer, while those derived exclusively from the N-terminal half do not. Peptide 419–447 (DV2419–447) has the lowest Kd (∼150 nM), and we have chosen to use it for many of the subsequent studies reported below.

We confirmed the specificity of stem peptides for the postfusion sE conformer by showing that a biotinylated derivative of DV2419–447 stably associates with trimeric sE, using the “pull-down” protocol described in Methods. As shown in Fig. S1, biotinyl-DV2419–447 associates tightly with sE trimer but not with sE dimer. As a control for non-specific interaction of the biotinylated peptide with a detergent-solubilized protein, we used an irrelevant membrane protein (the mitochondrial uncoupling protein, UCP) and found no evidence for any association (data not shown).
Stem peptides inhibit viral fusion
If peptide binding to sE trimer mimics an association of stem with domain II during the fusion-promoting conformational change, then the bound peptides should block the final “zipping-up” step and thereby prevent fusion. We used two methods to monitor directly the effects of stem-derived peptides on dengue-virus fusion with liposomes. One is a trypsin susceptibility assay, which detects exposure of the viral core protein to liposome-encapsulated protease as a measure of complete membrane fusion and content mixing. The other is a pyrene fluorescence assay that has been validated in many published studies of alphaviruses and flaviviruses as a sensitive measure of hemifusion, an obligatory intermediate step in the merging of two bilayers [24],[25],[26].
For the content-mixing assay, liposomes encapsulating trypsin were made by extrusion in the presence of the protease. Unincorporated trypsin was removed by size exclusion chromatography. Virions and trypsin-containing liposomes were incubated at 37°C at neutral and low pH, in the presence of DV2419–447 or DV2419–447(scram); reactions were back-neutralized and incubated for 1hr at 37°C. Fusion, triggered by exposure to low pH, will deposit the viral core into the liposome interior, exposing the capsid protein to protease. The results in Figure 3A show that in the presence of DV2419–447, there is no more capsid degradation than in the pH 8.0 control, whereas DV2419–447(scram) has no protective effect with respect to the pH 5.5 treated reaction. Complete lysis of the liposomes with TX100 demonstrates that there is sufficient trypsin encapsulated to digest the capsid protein completely. DV2419–447 inhibits membrane fusion in this model system at a step prior to content mixing.

For the pyrene fluorescence measurements, virus was grown in medium supplemented with pyrene-hexadecanoic acid, which incorporates into membrane lipids. The pyrene labels the tip of the fatty-acid chain near the bilayer midplane; an excited fluorophore in one leaflet can collide with an unexcited fluorophore in the other leaflet, generating an excimer with an altered emission spectrum [27]. Fusion with an unlabeled liposome dilutes the pyrene labeled lipids at the hemifusion stage, leading to a decrease in fluorescence at the wavelength of the excimer emission maximum. The data in Fig. 3B show that addition of 1µM DV2419–447 substantially reduced the extent of hemifusion with respect to the effects of DV2419–447(scram) and DMSO as controls.
Effect of stem peptides on viral infectivity
To examine the effect of the stem peptides on dengue virus infectivity, we infected cells in the presence of varying peptide concentrations and then measured viral yield 24 hours post-infection by a standard viral plaque assay (see Methods). Stock solutions of peptides were made in DMSO, and the final DMSO concentration was always less than 1% when the stock was diluted into medium. Peptides derived from the C-terminal part of the stem inhibit viral replication. As shown in Fig. 2, the extent of inhibition correlates strongly with the affinity of the peptide for the sE trimer, as determined by fluorescence polarization measurements. Peptides that include the entire “helix II” segment are particularly active. DV2413–447 and DV2419–447 are the strongest inhibitors; neither scrambled version, DV2413–447(scram) or DV2419–447(scram), has any effect on viral titre.
Effect of order of addition
At what stage in the infectious cycle do stem peptides interfere with viral replication? The results shown in Fig. 2 were obtained from experiments in which peptide and virus were preincubated before application of the inoculum to cells. Thus, the peptides could associate with virions prior to adsorption of the latter to cells and inhibit completion of the envelope protein conformational transition after virion uptake into endosomes. Alternatively, it is possible that the peptides concentrate in endosomes and associate with virions only in a low-pH environment. Either explanation appears to present a puzzle. At the 500 nM concentration sufficient, in the case of the most effective peptides, to inhibit viral yields, an early endosome of 1500 Å diameter would be expected to contain only 1–2 peptides in the absence of some mechanism to concentrate the peptide. This number is probably too small to inhibit viral entry, as estimates for the number of antibodies required to neutralize dengue virus range from 20 upwards, and it is likely that at least the same number of peptides would be needed [28],[29]. Yet, the inhibitory activity of the peptides correlates with their binding to sE trimer, and the trimer transition should occur only after endosomal acidification. The correlation also appears to rule out a third possibility – that the peptides stimulate or inhibit a cell-signaling pathway, rather than inhibiting infection through direct interaction with virions.
To resolve these apparent contradictions, we carried out a series of order-of-addition experiments using either DV2419–447 or DV2419–447(sol). The latter spans the same sequence (419–447), but has an additional “solubility tag”, of sequence RGKGR, at its C-terminus. The hydrophobicity of the stem peptide causes it to aggregate almost immediately upon dilution from DMSO into the medium, decreasing its effective concentration as the incubation proceeds (data not shown); the solubility tag reduces aggregation and stabilizes the peptide concentration. Pre-incubation of BHK-21 monolayers with 10 µM DV2419–447 for various times, followed by infection with DV2 at a multiplicity of infection (MOI) 1 after washing the cells free of the peptide, did not affect the viral yield (data not shown). Thus, the peptide has no lasting effect on the capacity of the cells to support viral replication. Preincubation of virus with peptide DV2419–447 for various times prior to application to the monolayer leads to the same inhibition of infectivity observed when the two are mixed and applied immediately (Fig. 4A). Adding peptide at the end of the 1 hr interval used for the infection had a small inhibitory effect, suggesting the presence of some adsorbed but not yet endocytosed particles, whereas adding peptide 2 hr after application of the virus had no effect at all. We conclude that inhibition of infectivity results from a direct interaction of the peptide with virions and that the virion-bound peptide blocks an early step in infection.

In the experiments of Fig. 4, excess peptide was present during the incubation of virus and cells. Rapid passage of the virus through a short size-exclusion matrix, to remove excess peptide prior to application of the inoculum to the monolayer, did not reduce the extent of inhibition relative to a control passed similarly through the matrix but with no added peptide (Fig. 4B). We verified that the spin columns used for this treatment were effective in removing unbound peptide by showing that no inhibitory activity was present in the flow-through fraction after applying a 10 µM peptide solution (but no virus) to the column. Thus, peptide appears either to associate tightly with virions or to induce an irreversible, inactivating effect. Experiments described below rule out the latter interpretation.
Reversibility of stem peptide inhibition
The experimental results in Fig. 4C show that inhibition of infectivity by stem peptides is reversible. We preincubated a viral inoculum with DV419–447 at 37°C, then added liposomes in varying molar excess. We could restore infectivity to levels approaching the control by adding a suitable excess of liposomes, into which the hydrophobic peptide could partition. The reversibility of inhibition by stem peptides suggests that non-specific membrane interactions may dominate the initial association of peptide and virion and that this sequestration may concentrate the peptide in the viral membrane for rapid association during the short lifetime of the E-protein fusion intermediate.
Effect of temperature
Together with the observation that stem peptides bind sE in its rearranged, trimeric conformation, but not the sE dimer, the data in the preceding section suggest that the peptide gains access to sites, either on domain II or on the viral membrane, that are occluded in the equilibrium structure deduced by fitting the dimer model into cryoEM reconstructions [7]. Indeed, when we carried out the peptide-virion incubations at 4°C rather than 37°C, followed by quick warming and immediate application to the BHK-21 cell monolayer, we found little or no effect on viral titre (Fig. 5A). The results suggest that dynamics of the virion surface permit access to transiently exposed sites on E, on the membrane, or on both.

Direct detection of peptide association with virions
We examined the interaction of stem peptides and virions by incubating biotinyl-DV2419–447 with virions at 37°C for 45 minutes and then adding streptavidin resin (blocked with 10% fetal bovine serum to minimize non-specific association) to bind the peptide and any associated virus, which we detected by immunoblotting as shown in Fig. 5B. Binding of virions was efficient at pH 8, well above the pH threshold for fusion. As the concentration of peptide increased, we could detect increasing amounts of virus in the streptavidin eluate. Incubation of the virus with biotinyl-peptide at 4°C resulted in a noticeably lower yield of retained virions, consistent with the neutralization experiments (Fig. 5B).
To confirm association of stem peptides with virions we used the fluorescence properties of pyrene labeled virions as a direct measure of membrane interaction. We observed that addition of DV2419–447 produces a concentration dependent loss in excimer intensity (Fig. 6). We interpret this effect as a measure of interaction of the peptide with the bilayer. Membrane binding of stem peptides will decrease lipid mobility and hence decrease the likelihood of excimer formation. Consistent with this interpretation is the observation that DV2419–447(scram), which is as hydrophobic as the unscrambled peptide, also reduces the excimer signal (Fig. 6). The loss of any regular pattern of amphipathicity in the scrambled peptide may account for the higher concentration required to achieve the same effect on the pyrene excimer as with wild-type peptide.

At the respective concentrations at which WT and scrambled peptides decrease the excimer intensity by 20%, the former decreases viral titre by 90%, while the latter has no effect. This comparison shows that loss of excimer signal is not due to a generalized membrane disruption that would inactivate the virus. (Even at saturating concentrations, the peptides do not eliminate excimer fluorescence completely, which we can do by adding TritonX-100.) The comparison also shows that association of a peptide with the membrane, as monitored by loss of excimer signal, is not sufficient for neutralization of infectivity. Membrane association appears to be necessary, however, as indicated by comparison of peptides DV2419–447 and DV2419–440 (Fig. 2). The latter lacks the hydrophobic segment between residues 441 and 447, but it binds soluble E trimer as tightly as does the former (Fig. 2). Only at concentrations at which the shorter peptide reduces the pyrene excimer intensity does it also neutralize (Fig. 2A and B). These results also suggest that in the conformation E reaches when fusion is complete, amino-acid residues in its stem may bind along domain II only about as far as 440. On a mature virion, residues between 441 and the transmembrane anchor probably interact with the viral membrane; following fusion, they may interact with the fused bilayers.
Discussion
We have found that peptides derived from the membrane proximal region of the dengue virus E protein bind specifically to the postfusion conformer of sE, which is a model for a fusion intermediate. These peptides also inhibit viral infectivity – an initially surprising observation because transition of E to its postfusion conformation occurs in the sequestered environment of an endosome. We have shown that the inhibition results from direct interaction of the peptides with virions at neutral pH and hence that the virus carries the peptide with it into the cell. The peptides do not associate with virions at 4°C, suggesting that binding depends on dynamic properties of the viral envelope. We have further shown, using a liposome fusion assay, that the stem-derived peptides inhibit viral fusion at a step prior to hemifusion and fusion-pore formation.
How do these stem peptides inhibit fusion if their specific binding sites are not exposed until acidification in an endosome? One possible model is that the peptides bind to transiently exposed sites as the E protein fluctuates among an ensemble of conformations. A similar explanation has been offered to explain neutralization of dengue virus by antibodies for which the epitopes are blocked (or partly blocked) on the virion as modeled from the cryoEM structure [30]. The antibodies can trap a small fluctuation of the protein from its equilibrium conformation. The absence of peptide binding to dengue virions at low temperature is consistent with this type of model. Nonetheless, it seems unlikely that E could spontaneously fluctuate at neutral pH toward a conformation close to that of the postfusion trimer (the excursion of the domains and the rearrangement of the E protein are too extensive), and to consider a fluctuation-binding model, it is probably necessary to assume that the peptide associates with a single domain II, rather than with two that are clustered with each other as in the trimer. A small fluctuation could readily make an edge of domain II accessible to peptide: we know domain II to be hinged with respect to domain I and to adopt somewhat different hinge angles in different crystal forms of the sE dimer, suggesting a potential source of conformational heterogeneity [31]. Residual, uncleaved prM, which is present to varying degrees in most dengue inocula, is also likely to cause perturbations in the packing of E dimers on the virion surface [6],[32]. The association of prM with E tilts the latter outward from the particle surface, and we would expect that a small number of prM-E heterodimers would introduce dislocations and local disorder into the otherwise tightly packed lattice of E dimers covering the virion.
The results of our membrane-fusion assays with pyrene-labeled virus particles suggest an alternative, two-step model: step 1 is an initial hydrophobic association of peptide and viral membrane, and step 2 is a specific association with E during the low-pH triggered conformational transition (Fig. 7). Even a modest fluctuation of the closely packed E protein on the viral surface would give an exogenous peptide access to the underlying membrane bilayer, of which about two-thirds is free of intimate protein contacts [7]. A peptide that could associate with the membrane, e.g., as an amphipathic helix, would be carried with the virus particle into an endosome and would be available to “spring into place” upon transient exposure of its site on domain II, thereby blocking the fusion-promoting zipping-up of the E-protein stem. Only peptides with the appropriate sequence would be effective, consistent with the properties of the DV2419–447(scram) peptide, which binds well to virions, but not to E trimer, and has no effect on viral infectivity. The two-step model is also consistent with the sequence dependence of peptide binding as measured by the effect of peptides on the intensity of the pyrene excimer in Fig. 6 and with the requirements for trimer binding in vitro as detected by fluorescence polarization (Fig. 2).

The last seven residues of DV2419–447 appear to be important for inhibitory activity. This membrane-proximal segment is the most uniformly hydrophobic sequence in the stem. Residues 441–447 contribute substantially to the neutralizing potency of the peptide (compare peptides in Fig. 2) but not to its affinity for the sE trimer. Moreover, reducing their hydrophobicity by substituting the corresponding residues from WNV greatly reduces inhibition of infectivity without affecting trimer binding (data not shown). Thus, these residues might be particularly important contributors to step 1 of the two-step model outlined above. If so, some specific peptide-protein contacts might occur in concert with peptide-lipid association. That is, residues 441–447, which do not contribute to E-protein binding, could associate with the membrane, while some part of the rest of the peptide made contact with its site on E, exposed by fluctuation or disorder as suggested in the single-step model. According to this combination of the two models above, non-specific membrane interactions and specific domain-II interactions would jointly recruit the peptides, rather than functioning only sequentially. Additional specific interactions could then form upon full exposure of domain II during the low-pH induced conformational transition.
Our data suggest strategies, which might otherwise have seemed unlikely, for inhibiting entry of viruses that fuse in endosomes or other internal compartments. Peptides from the membrane-proximal region of the HIV gp41 ectodomain are effective inhibitors of HIV entry, but fusion at the cell surface has made the transient intermediate in envelope refolding seem a more plausible target for HIV than for viruses with envelopes that expose an extended intermediate only after internalization [19],[20]. If peptides or small molecules can be concentrated in a viral membrane by tethering to a hydrophobic “tail”, then it may be possible to enhance the potency of inhibitors identified by a simple binding assay such as the one we have used here.
Materials and Methods
Peptide synthesis
Peptides were synthesized with standard Fmoc chemistry on ABI 431 Peptide Synthesizers at the Tufts University Core Facility (Boston, MA). They were purified using reverse phase HPLC and analyzed by mass spectrometry. Biotin and fluorescein-isothiocyanate (FITC) were conjugated to the N-terminus of the peptides via a beta-alanine linker.
Liposomes
Liposomes [1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC): 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) (Avanti Polar Lipids) and cholesterol (Sigma-Aldrich) at 1∶1∶2 molar ratio in TAN buffer (20mM triethanolamine, 100mM NaCl, pH 8.0)] were prepared by freeze-thaw extrusion through a 0.2 µ filter as described previously [11]. Liposomes were used within 2 days of preparation.
Preparation of sE trimers
The postfusion sE trimer was produced as described [11]. Briefly, purified sE (Hawai'i Biotech) was incubated in the presence of liposomes for 15 mins at 37C, acidified with MES or sodium acetate to a final pH of 5.5, incubated for an additional 30 mins at 37C and at then at room temperature 1hr. Liposomes were solubilized with n-octyl-β-D-glucoside (β-OG) and n-undecyl-maltopyranoside (UDM). The solution was applied to a MonoS column (GE Healthcare) in 25mM sodium citrate pH 5.2, 100mM NaCl, 4mM UDM. sE trimer was eluted with a 1–1.5M NaCl step gradient and further purified by size-exclusion chromatography on Superdex 200 (GE Healthcare) in 10mM TEA, 80mM NaCl and 0.7mM UDM. Protein was dialyzed extensively using a 50-kDa molecular-weight cutoff membrane (Spectrapor).
Fluorescence polarization
Binding experiments were carried out in Corning, low-volume 384 well microplates and analyzed in a PerkinElmer EnVisions instrument (excitation wavelength, 485nm; emission wavelength, 535 nm). Stock concentrations of FITC-conjugated peptide were made in DMSO and dissolved in assay buffer immediately before use. Increasing concentrations of protein were used with a constant, 20nM, concentration of peptide. Each well contained a final volume of 20uL in TAN buffer. Plates were incubated at 4C and data collected at 3 and 24 hours. Kd were estimated as the concentration of protein with half-maximal increase in fluorescence polarization.
Biotin-streptavidin pull-down
Recombinant sE (5µg) or purified virions (2.5µg) were incubated with biotinylated DV2419–447 or DV2419–447(scram) for 15 mins at 37°C. Streptavidin resin was incubated with 10% FBS at room temperature for 1 hr prior to use, to reduce non-specific binding. Protein and peptide mixtures were incubated with the treated resin for 30 mins at 4°C with constant rotation. Reactions were spun at 14,000rpm in a microcentrifuge at 4C for 10 mins, the supernatant aspirated, and the resin resuspended in 1 mL TAN buffer and spun again for 15 mins. After six further washes in 1 mL TAN buffer, the resin was resuspended in SDS-loading buffer, incubated for 15 mins at 100°C, and analyzed by SDS-PAGE (10 or 15%, precast gels, Biorad). Protein bands were detected by Coomassie blue staining or by immunoblotting, as indicated. Immunoblots were visualized by chemiluminescence (ECL reagents, Pierce).
Trypsin sensitivity (content-mixing) assay
Liposomes were prepared as above, after addition of trypsin at 10 mg/ml to the lipids [33]. Unencapsulated trypsin was removed by passage of the suspension through a Superdex 200 gel filtration column (GE Healthcare). Purified virions (0.5–1 µg) were added to trypsin-containing liposomes in a final working volume of 200µL in TAN buffer. DV2 stem peptides were added at appropriate concentrations from DMSO stocks. Reactions were incubated at 37°C for 15 mins, acidified with sodium acetate pretitrated to reach a final pH of 5.5, and further incubated at 37°C for 30 mins. Reactions were neutralized with 1M TEA to a final pH of 8.0. Trypsin digestion was allowed to proceed for 1hr at 37°C. Aliquots of the reaction were resuspended in SDS-loading buffer, incubated for 20 mins at 100°C, and analyzed by SDS-PAGE followed by immunoblotting with an anti-dengue capsid antibody.
Pyrene fluorescence fusion assay
Dengue virions containing ω-pyrene fatty acids in some of their phospholipids were produced. Briefly, BHK-21 cells were grown in the presence of 15 µg/ml of pyrene-hexadecanoic acid (Invitrogen) prior to infection with dengue virus serotype 2. Virus was harvested 24 hrs postinfection and purified as described above. Pyrene labeled virions were added to a quartz cuvette in the presence of 200 µM liposomes (prepared as described above) and brought to final volume with TAN buffer, pH 8.0. Reactions were continuously monitored with 343 nm and 470 nm as the excitation and emission wavelengths, respectively. Reactions were acidified with MES, pretitrated to yield a final pH of 5.5. Reactions were stopped by addition of 0.1% TritonX-100 to cause complete dilution of the probe. The fusion index was calculated as a percent of the loss of excimer intensity upon addition of TritonX-100.
Cells and viruses
C6/36 cells were maintained in L-15 medium supplemented with 10% fetal bovine serum as well as penicillin, streptomycin, and ketonocazole (Invitrogen). For viral plaque assays, BHK-21 cells were seeded (5×104 cells/well) in 24-well, treated tissue-culture plates in α-MEM supplemented with HEPES, pen/strep antibiotics, and 5% Fetal Bovine Serum (FBS). Cells were plated <12 hrs before use and stored at 37C with 5% CO2.
Dengue virus serotype 2 New Guinea Clone (NGC) was adsorbed to confluent layers of C6/36 cells for 1hr at 25°C with rocking every 15 mins. L-15 medium (Mediatech) was added, and cells were incubated at 25°C for 7 days until syncytia formation was observed. The supernatant was clarified by centrifugation at 1000 RPM for 15 mins at 4°C. Aliquots were stored at −80°C.
Plaque assay
BHK-21 cells were seeded as described above. Aliquots from infections were thawed at 37°C in a water bath. 10 fold dilutions in Earle's balanced salt solution (EBSS) were prepared in 96-well plates by transferring 20 µl from one well to 180µl EBSS in the next. 100 µl of each dilution were added to cells. Plates were incubated for 1hr at 37°C and rocked every 15 mins. Unadsorbed virus was removed by two washes with 1 ml PBS, after which 1ml of α-MEM supplemented with carboxymethylcellulose (CMC), pen/strep antibiotics, HEPES and 2%FBS, was added to each well and incubated at 37°C for 4 days. The CMC overlay was aspirated, and cells were washed 2× with 1mL PBS and stained with crystal violet. Plates were washed with water to remove excess crystal violet and dried overnight.
Plaque reduction inhibition assays
Cell supernatant was diluted in EBSS to a stock concentration that would allow for infection at MOI of 1, based on 50,000 seeded cells. Peptides (or carrier) were added to the inoculum as indicated for each experiment. Cells were infected for 1hr at 37°C with gentle rocking every 15 mins. Virus (or virus∶peptide mixtures) were washed from cells with 1mL of PBS (2×) and overlay medium (α-MEM supplemented with HEPES, pen/strep antibiotics and 2% FBS) added. Plates were incubated at 37°C for 24 hrs. Aliquots of the supernatant were withdrawn and stored at −80°C for plaque assay as described above.
For experiments to remove excess peptide, virus and peptide were incubated at 37°C for 15 mins and passed over a small G-50 desalting column (GE Healthcare) equilibrated with EBSS. 100 µl of the flow-through (which contained the virus∶peptide complexes) were used to infect 5×104 BHK cells (MOI ∼1) in 24-well plates for 1hr at 37°C. Cells were washed 2× with PBS and overlayed with α-MEM medium supplemented with 2.0% FBS. Supernatants were harvested 24 hrs later and analyzed by the plaque assay described above.
Virus purification
Virus was precipitated from C6/36 infected cell supernatant by addition of polyethylene glycol molecular weight 8000 (PEG8000, Sigma-Aldrich) and incubation overnight at 4°C with constant mixing. The virus was then pelleted by centrifugation for 30 mins at 10,000×g, 4°C, and the pellet was resuspended in TAN buffer and clarified by centrifugation at 5,000×g for 20 mins at 4°C. Virions were further purified on a 10% and 40% potassium tartrate with 7.5 and 30% glycerol, step gradient and centrifuged at 146,000×g for 2.5 hrs at 4°C. The gradient was harvested into 10 fractions and immunoblotted with antibodies against E (obtained from ATCC: [34]) or C (supplied by Dr. J.G. Asakov: [35]) to locate the virus. Those fractions were then pooled, concentrated and cleared of potassium tartrate and glycerol by exchange into TAN buffer using a 100K molecular weight cut-off concentrator (Amicon). Virus concentration was determined with the BCA assay (Pierce). Purified virus was stored at 4°C and used within one week of purification.
Supporting Information
Zdroje
1. HarrisonSC
2008 Viral membrane fusion. Nat Struct Mol Biol 15 690 698
2. van der SchaarHM
RustMJ
ChenC
van der Ende-MetselaarH
WilschutJ
2008 Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog 4 e1000244 doi:10.1371/journal.ppat.1000244
3. van der SchaarHM
RustMJ
WaartsBL
van der Ende-MetselaarH
KuhnRJ
2007 Characterization of the early events in dengue virus cell entry by biochemical assays and single-virus tracking. J Virol 81 12019 12028
4. FritzR
StiasnyK
HeinzFX
2008 Identification of specific histidines as pH sensors in flavivirus membrane fusion. J Cell Biol 183 353 361
5. LindenbachBDRC
2001 Flaviviridae: The viruses and their replication Philadelphia Lippincott Williams & Wilkins 991 1041
6. YuIM
ZhangW
HoldawayHA
LiL
KostyuchenkoVA
2008 Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319 1834 1837
7. ZhangW
ChipmanPR
CorverJ
JohnsonPR
ZhangY
2003 Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol 10 907 912
8. AllisonSL
SchalichJ
StiasnyK
MandlCW
KunzC
1995 Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J Virol 69 695 700
9. ReyFA
HeinzFX
MandlC
KunzC
HarrisonSC
1995 The envelope glycoprotein from tick-borne encephalitis virus at 2 A resolution. Nature 375 291 298
10. AllisonSL
SchalichJ
StiasnyK
MandlCW
HeinzFX
2001 Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J Virol 75 4268 4275
11. ModisY
OgataS
ClementsD
HarrisonSC
2004 Structure of the dengue virus envelope protein after membrane fusion. Nature 427 313 319
12. BressanelliS
StiasnyK
AllisonSL
SturaEA
DuquerroyS
2004 Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. Embo J 23 728 738
13. ModisY
OgataS
ClementsD
HarrisonSC
2003 A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A 100 6986 6991
14. KanaiR
KarK
AnthonyK
GouldLH
LedizetM
2006 Crystal structure of west nile virus envelope glycoprotein reveals viral surface epitopes. J Virol 80 11000 11008
15. HeinzFX
MandlCW
HolzmannH
KunzC
HarrisBA
1991 The flavivirus envelope protein E: isolation of a soluble form from tick-borne encephalitis virus and its crystallization. J Virol 65 5579 5583
16. MukhopadhyayS
KimBS
ChipmanPR
RossmannMG
KuhnRJ
2003 Structure of West Nile virus. Science 302 248
17. WildC
OasT
McDanalC
BolognesiD
MatthewsT
1992 A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc Natl Acad Sci U S A 89 10537 10541
18. WildC
DubayJW
GreenwellT
BairdTJr
OasTG
1994 Propensity for a leucine zipper-like domain of human immunodeficiency virus type 1 gp41 to form oligomers correlates with a role in virus-induced fusion rather than assembly of the glycoprotein complex. Proc Natl Acad Sci U S A 91 12676 12680
19. KilbyJM
HopkinsS
VenettaTM
DiMassimoB
CloudGA
1998 Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med 4 1302 1307
20. ChanDC
KimPS
1998 HIV entry and its inhibition. Cell 93 681 684
21. LiaoM
KielianM
2005 Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus membrane fusion. J Cell Biol 171 111 120
22. HrobowskiYM
GarryRF
MichaelSF
2005 Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol J 2 49
23. StiasnyK
AllisonSL
SchalichJ
HeinzFX
2002 Membrane interactions of the tick-borne encephalitis virus fusion protein E at low pH. J Virol 76 3784 3790
24. ThompsonBS
MoeskerB
SmitJM
WilschutJ
DiamondMS
2009 A therapeutic antibody against west nile virus neutralizes infection by blocking fusion within endosomes. PLoS Pathog 5 e1000453 doi:10.1371/journal.ppat.1000453
25. CorverJ
BronR
SnippeH
KraaijeveldC
WilschutJ
1997 Membrane fusion activity of Semliki Forest virus in a liposomal model system: specific inhibition by Zn2+ ions. Virology 238 14 21
26. BronR
WahlbergJM
GaroffH
WilschutJ
1993 Membrane fusion of Semliki Forest virus in a model system: correlation between fusion kinetics and structural changes in the envelope glycoprotein. Embo J 12 693 701
27. SomerharjuP
2002 Pyrene-labeled lipids as tools in membrane biophysics and cell biology. Chem Phys Lipids 116 57 74
28. PiersonTC
XuQ
NelsonS
OliphantT
NybakkenGE
2007 The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1 135 145
29. PiersonTC
DiamondMS
2008 Molecular mechanisms of antibody-mediated neutralisation of flavivirus infection. Expert Rev Mol Med 10 e12
30. LokSM
KostyuchenkoV
NybakkenGE
HoldawayHA
BattistiAJ
2008 Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat Struct Mol Biol 15 312 317
31. ZhangY
ZhangW
OgataS
ClementsD
StraussJH
2004 Conformational changes of the flavivirus E glycoprotein. Structure 12 1607 1618
32. LiL
LokSM
YuIM
ZhangY
KuhnRJ
2008 The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319 1830 1834
33. PohMK
YipA
ZhangS
PriestleJP
MaNL
2009 A small molecule fusion inhibitor of dengue virus. Antiviral Res 84 260 266
34. HenchalEA
GentryMK
McCownJM
BrandtWE
1982 Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am J Trop Med Hyg 31 830 836
35. BulichR
AsakovJG
1992 Nuclear localization of dengue 2 virus core protein detected with monoclonal antibodies. J Gen Virol 73 2999 3003
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 4
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
Nejčtenější v tomto čísle
- The Effect of Vaccination on the Evolution and Population Dynamics of Avian Paramyxovirus-1
- Reconstitution of SARS-Coronavirus mRNA Cap Methylation
- Increased Monocyte Turnover from Bone Marrow Correlates with Severity of SIV Encephalitis and CD163 Levels in Plasma
- Deficiencies in Jasmonate-Mediated Plant Defense Reveal Quantitative Variation in Pathogenesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy