
The Origin and Nature of Tightly Clustered Deletions in Precursor B-Cell Acute Lymphoblastic Leukemia Support a Model of Multiclonal Evolution
Recurrent submicroscopic deletions in genes affecting key cellular pathways are a hallmark of pediatric acute lymphoblastic leukemia (ALL). To gain more insight into the mechanism underlying these deletions, we have studied the occurrence and nature of abnormalities in one of these genes, the B-cell translocation gene 1 (BTG1), in a large cohort of pediatric ALL cases. BTG1 was found to be exclusively affected by genomic deletions, which were detected in 65 out of 722 B-cell precursor ALL (BCP-ALL) patient samples (9%), but not in 109 T-ALL cases. Eight different deletion sizes were identified, which all clustered at the telomeric site in a hotspot region within the second (and last) exon of the BTG1 gene, resulting in the expression of truncated BTG1 read-through transcripts. The presence of V(D)J recombination signal sequences at both sites of virtually all deletions strongly suggests illegitimate RAG1/RAG2-mediated recombination as the responsible mechanism. Moreover, high levels of histone H3 lysine 4 trimethylation (H3K4me3), which is known to tether the RAG enzyme complex to DNA, were found within the BTG1 gene body in BCP-ALL cells, but not T-ALL cells. BTG1 deletions were rarely found in hyperdiploid BCP-ALLs, but were predominant in other cytogenetic subgroups, including the ETV6-RUNX1 and BCR-ABL1 positive BCP-ALL subgroups. Through sensitive PCR-based screening, we identified multiple additional BTG1 deletions at the subclonal level in BCP-ALL, with equal cytogenetic distribution which, in some cases, grew out into the major clone at relapse. Taken together, our results indicate that BTG1 deletions may act as “drivers” of leukemogenesis in specific BCP-ALL subgroups, in which they can arise independently in multiple subclones at sites that are prone to aberrant RAG1/RAG2-mediated recombination events. These findings provide further evidence for a complex and multiclonal evolution of ALL.
Published in the journal:
. PLoS Genet 8(2): e32767. doi:10.1371/journal.pgen.1002533
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002533
Summary
Recurrent submicroscopic deletions in genes affecting key cellular pathways are a hallmark of pediatric acute lymphoblastic leukemia (ALL). To gain more insight into the mechanism underlying these deletions, we have studied the occurrence and nature of abnormalities in one of these genes, the B-cell translocation gene 1 (BTG1), in a large cohort of pediatric ALL cases. BTG1 was found to be exclusively affected by genomic deletions, which were detected in 65 out of 722 B-cell precursor ALL (BCP-ALL) patient samples (9%), but not in 109 T-ALL cases. Eight different deletion sizes were identified, which all clustered at the telomeric site in a hotspot region within the second (and last) exon of the BTG1 gene, resulting in the expression of truncated BTG1 read-through transcripts. The presence of V(D)J recombination signal sequences at both sites of virtually all deletions strongly suggests illegitimate RAG1/RAG2-mediated recombination as the responsible mechanism. Moreover, high levels of histone H3 lysine 4 trimethylation (H3K4me3), which is known to tether the RAG enzyme complex to DNA, were found within the BTG1 gene body in BCP-ALL cells, but not T-ALL cells. BTG1 deletions were rarely found in hyperdiploid BCP-ALLs, but were predominant in other cytogenetic subgroups, including the ETV6-RUNX1 and BCR-ABL1 positive BCP-ALL subgroups. Through sensitive PCR-based screening, we identified multiple additional BTG1 deletions at the subclonal level in BCP-ALL, with equal cytogenetic distribution which, in some cases, grew out into the major clone at relapse. Taken together, our results indicate that BTG1 deletions may act as “drivers” of leukemogenesis in specific BCP-ALL subgroups, in which they can arise independently in multiple subclones at sites that are prone to aberrant RAG1/RAG2-mediated recombination events. These findings provide further evidence for a complex and multiclonal evolution of ALL.
Introduction
Acute lymphoblastic leukemia (ALL) is the most common form of cancer in children, and can be subdivided in T-lineage leukemia (T-ALL) and B-cell precursor leukemia (BCP-ALL). BCP-ALL makes up about 80% of all pediatric ALL cases, and comprises genetically distinct subtypes. These ALL subtypes are characterized by specific genetic abnormalities, including aneuploidies (hyperdiploidy and hypodiploidy) and chromosomal translocations leading to ETV6-RUNX1, E2A-PBX1, BCR-ABL1, and MLL gene fusions [1]. An additional layer of complexity was recently revealed by the identification of recurrent copy number alterations (CNAs), including single gene deletions [2]–[4], some of which are strongly associated with disease progression and outcome [5]–[7]. Despite the high number of CNAs that has been identified in BCP-ALL, the total number of structural genetic lesions in each individual leukemia sample is usually limited to less than 10 per case. A subset of these genetic lesions, predominantly single gene deletions, may act as ‘drivers’ in the development of BCP-ALL. Genes commonly affected by these driver deletions function in key pathways such as cell cycle regulation (CDKN2A, CDKN2B, RB1), lymphoid development (PAX5, EBF1, IKZF1, RAG1/2), and nuclear hormone receptor signaling (NR3C1, TBL1XR1).
One of the newly identified players in BCP-ALL is the B cell translocation gene-1 (BTG1) [4], [8]. BTG1 belongs to the BTG/TOB family of anti-proliferative genes (BTG1-BTG4, TOB1 and TOB2) and expression of their gene products is altered in a variety of different cancers [9]. The BTG1 gene has been reported to be affected by deletions in approximately 10% of the BCP-ALL cases [2], [3], and to be targeted by nonsense and missense point mutations in diffuse large B-cell lymphoma [10]. BTG1 was shown to directly interact with multiple transcription factors and modulators involved in the regulation of crucial cellular processes including mRNA turnover and histone modification [11]–[14]. Furthermore, we recently demonstrated that BTG1 regulates the glucocorticoid receptor (GR)-dependent transcriptional response in leukemic cells [8], which turns this gene into an interesting target for modulating therapy response.
Several recurrent chromosomal abnormalities in leukemia show a clustering of their breakpoints, suggesting that features intrinsic to the DNA may underlie their origin. A common mechanism underlying these abnormalities is non-homologous end joining (NHEJ). NHEJ is employed by human cells to repair DNA double strand breaks caused by external damage or by intermediate steps of V(D)J recombination and class switch recombination (CSR) events during lymphocyte development [15]–[17]. The RAG complex mediates V(D)J recombination through the recognition of recombination signal sequences (RSS) located directly adjacent to the V, D, or J segments, thereby generating the variability of immunoglobulins (Ig) as well as B- and T-cell receptors. Illegitimate RAG-mediated recombination has been proposed to be involved in the translocation process of the LMO2 and TAL1 proto-oncogenes with TCRδ [18], and appears to be the cause of recurrent intragenic deletions of the IKZF1 gene in BCP-ALL and lymphoid blast crisis in chronic myeloid leukemia (CML) [19], [20].
Comprehensive genomic analyses of relapsed BCP-ALL have revealed that diagnosis and relapse samples are clonally related, but may exhibit subtle differences, frequently involving lesions detected at the time of diagnosis that are absent at the time of relapse [5], [21], [22]. Furthermore, diagnosis and relapse samples may carry alternative lesions affecting genes like e.g. CDKN2A and PAX5, suggesting that these lesions occur repeatedly during clonal evolution of BCP-ALL [5]. These findings are in line with the generally accepted concept that each leukemic outgrowth comprises a heterogeneous population of leukemic cells of which only a subset has the potential to drive progression of the disease, survive therapy, and/or induce relapse development [23]. Recent studies in BCP-ALL have revealed that this multiclonal architecture supports a complex model of multiclonal evolution [24], [25]. During disease progression and after post-treatment relapse, shifts can occur in the dominance of leukemic subclones, most likely triggered by new selective bottlenecks, whereas the subclonal diversity appears to be maintained.
In the present study, we have characterized the origin and nature of BTG1 deletions in a cohort of 831 pediatric ALL cases. We demonstrate that BTG1 deletions are exclusively found in BCP-ALL, both in the predominant clone and in (multiple) minor subclones, and are strongly associated with ETV6-RUNX1 and BCR-ABL1 positive ALL, but rare in hyper-diploid cases. Furthermore, we show that these deletions most likely arise from illegitimate RAG-mediated recombination, are tightly clustered, and virtually all share their telomeric breakpoint within the second exon of BTG1, resulting in the expression of a read-through transcript that gives rise to highly instable truncated BTG1 protein. These results provide important insight into the mechanism underlying the occurrence of BTG1 deletions in BCP-ALL and support the recently observed complexity of multiclonal evolution of ALL.
Results
BTG1 Is Targeted by Deletions in Specific Subsets of BCP-ALL
We screened 831 pediatric ALL diagnosis samples for copy number aberrations in BTG1 (Entrez Gene: 694) using multiplex ligation-dependent probe amplification (MLPA). In 722 BCP-ALL samples, we found a total of 65, predominantly monoallelic, deletions (9%), whereas no BTG1 copy number loss was detected in any of the T-ALL samples (n = 109; P = 0.001; Table 1). In addition, MLPA performed on genomic DNA derived from BCP-ALL (n = 10) and T-ALL (n = 5) cell lines revealed BTG1 deletions in the BCP-ALL cell lines REH, SUP-B15, MUTZ5 and 380, while none could be detected in the T-ALL cell lines (see Materials and Methods). Sequencing of the entire gene in 135 BCP-ALL cases and of exon 2, which contains the major body of the open reading frame, in 158 additional BCP-ALL cases and 44 T-ALL cases revealed no mutations. Furthermore, bisulfite sequence analysis showed absence of BTG1 promoter hypermethylation in 20 BCP-ALL and 5 T-ALL cases.
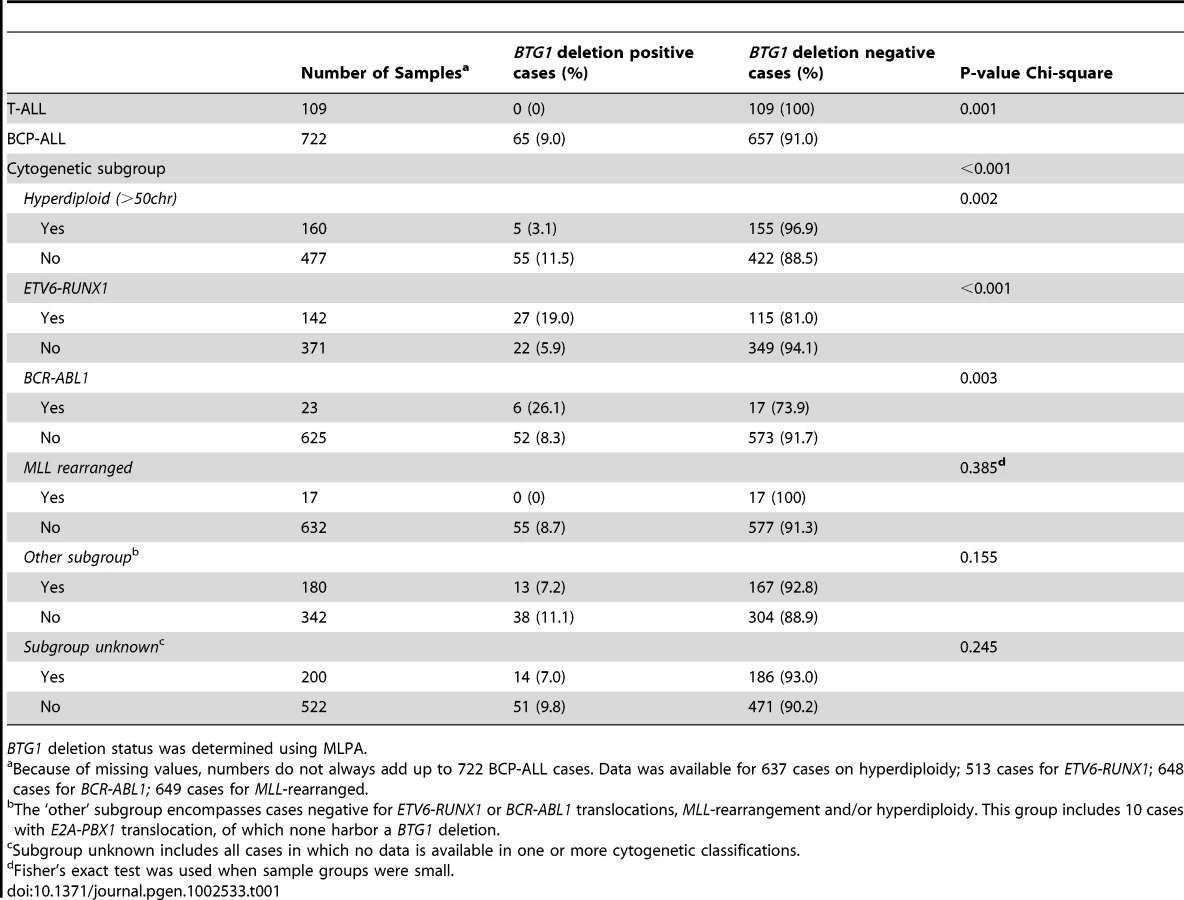
BTG1 deletions were found to be unevenly distributed between the different cytogenetic subgroups, being enriched in ETV6-RUNX1 (TEL-AML1) and BCR-ABL1 positive cases, and less frequent in hyperdiploid cases (P<0.001, P = 0.003 and P = 0.002, respectively; Table 1). In addition, targeted copy number analysis of recurrently affected genes in ALL revealed that cases with BTG1 deletions more frequently harbored deletions of ETV6, RB1 and EBF1 (P = 0.007, P<0.001 and P<0.001 respectively; Table S1). Together, our data indicate that BTG1 is affected by deletions in specific subsets of pediatric BCP-ALL patients.
A Deletion Breakpoint Hotspot Maps within the Second Exon of BTG1
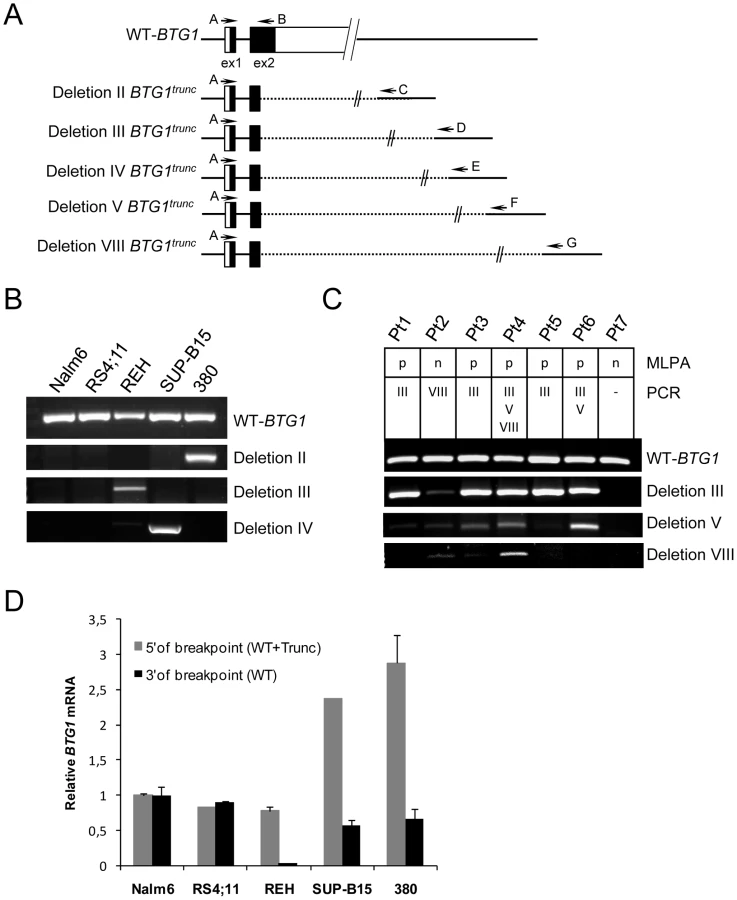
To further define the size and location of the BTG1 deletions, SNP-based genomic profiling was performed on 24 BTG1 deletion-positive BCP-ALL diagnosis samples and the BCP-ALL derived cell lines REH, SUP-B15, and 380. In nearly all cases, the telomeric deletion breakpoint was located within the BTG1 gene. Subsequent PCR-based fine-mapping and direct sequencing revealed that the intragenic breakpoints tightly clustered within a region of 33 base pairs in the second (and last) exon of BTG1, with the majority (75%) of the breakpoints being located within a stretch of 10 base pairs (Figure 1A, Table S2). The centromeric breakpoints mapped to 8 different positions downstream of the BTG1 gene, resulting in deletions ranging in size from 101 to 557 kb (deletions I-VIII). This highly specific clustering of deletion breakpoints allowed us to perform a PCR-based screening of all 65 BCP-ALL diagnosis samples. Hence, we were able to exactly map the deletions in 52 of the 65 cases and found that deletion III was most prevalent, comprising almost half of all lesions identified (49%). Deletions V and VIII were found in 15 and 17% of the cases, respectively (Figure 1B). These findings illustrate the high degree of clustering of BTG1 deletion breakpoints in BCP-ALL.

Subclones with Distinct BTG1 Deletions Frequently Arise in BCP-ALL Subtypes
Remarkably, while validating the BTG1 deletions with breakpoint-spanning PCRs, multiple BTG1 deletions were identified in 18 of the 65 deletion positive cases (27%), all of which turned out to be monoallelic losses using MLPA. The individual deletions appeared to be unique in their exact breakpoints and flanking interstitial sequences (Table S2), thus indicating the presence of multiple subclones which independently acquired BTG1 deletions. In one patient, as many as 4 unique deletions were identified (Figure 2A, Table S3). Triggered by this finding we performed a similar PCR-based BTG1 deletion screening for the three most common deletions (III, V, and VIII) in 89 deletion negative BCP-ALL cases as scored by MLPA or SNP array and were able to confirm the presence BTG1 deletions in minor subclones in 16 cases (18%), which is substantially more frequent than the occurrence of this abnormality in the predominant leukemic clone (Figure 2B). Similar to the previously observed clonal BTG1 deletions, these subclonal events were detected in several ETV6-RUNX1–positive cases and not in the hyperdiploid subgroup (Figure 2B, Table S4). Furthermore, no subclonal BTG1 deletions were detected in 77 T-ALL cases nor in 26 bone marrow samples from healthy volunteers. Together, we conclude that BTG1 deletions are associated with specific BCP-ALL subtypes where they can repeatedly arise in independent subclones.
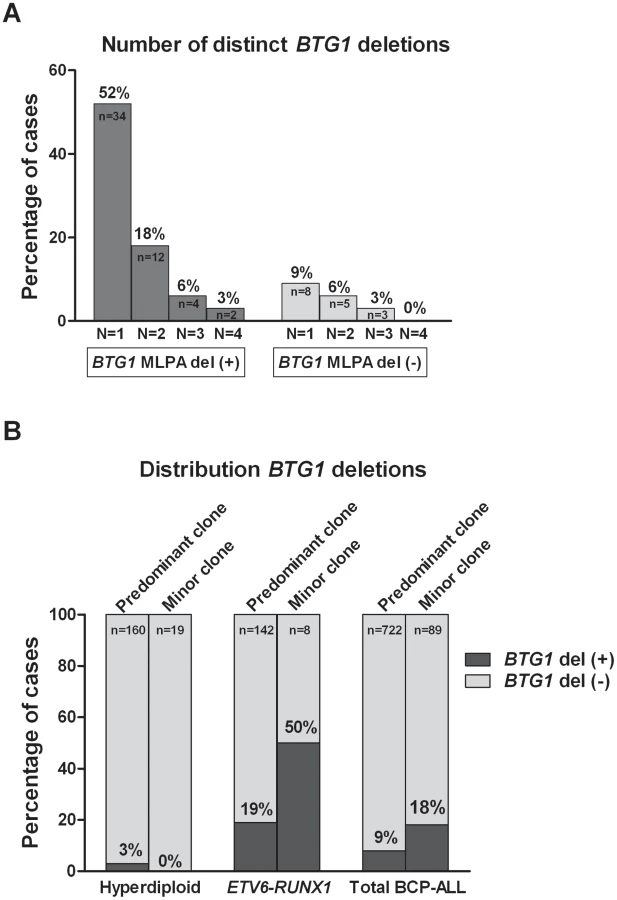
BTG1 Deletion (Sub)Clones May Reoccur in Relapse
To examine whether minor subclones or progenitors thereof that harbor a BTG1 deletion can evolve into relapse clones, we analyzed 62 matched relapse samples using MLPA for focal BTG1 deletions. All deletion-positive cases were subjected to breakpoint-spanning PCR for each of the eight deletion variants (deletions I–VIII). Again, we found unique BTG1 deletions both in the predominant clone and/or in (multiple) subclones (Table 2). An additional deletion was sequenced of which the telomeric breakpoint is located 2 kb downstream in the 3′UTR of BTG1. Backtracking and sequencing of the deletion breakpoints in matched diagnosis samples revealed that these BTG1 deletion clones originated from a (sub)clone at diagnosis in at least four cases. These data confirm that BTG1 deletions arise repeatedly and independently during disease progression and that these leukemic (sub)clones frequently reoccur at relapse.
Truncated BTG1 Transcripts Are Expressed in BCP-ALLs with BTG1 Deletions
Virtually all deletions affecting the BTG1 locus target the second exon of BTG1, leaving the possibility of expression of a truncated protein product. To determine whether a truncated BTG1 mRNA was expressed from the rearranged allele, RT-PCR using breakpoint-flanking primers was performed on three BCP-ALL cell lines with a BTG1 deletion (380, REH and SUP-B15 with deletions II, III and IV, respectively) and two cell lines that are BTG1 deletion negative (Nalm6 and RS4;11) (Figure 3). Wild-type BTG1 mRNA was found to be present in all cell lines, whereas truncated read-through transcripts specific for each type of deletion could be detected only in the three deletion positive BCP-ALL cell lines (Figure 3B). Similarly, we detected BTG1 read-through transcripts in primary BCP-ALL samples with BTG1 deletions III, V and VIII, respectively, several of which could be confirmed on genomic DNA of these patients (Figure 3C). Notably, the presence of multiple BTG1 deletion subclones as observed at the genomic level was also detectable at the transcript level and subsequent sequencing confirmed the unique identity of each clone (Table S5). Quantitative RT-PCR revealed that in REH, SUP-B15 and 380 cells the rearranged BTG1 allele was expressed at significantly higher levels compared to the wild-type BTG1 allele, which may reflect differences in mRNA stability (Figure 3D). These results indicate that monoallelic BTG1 deletions result in the expression of truncated BTG1 transcripts, encoding more than half of the BTG1 protein.
To assess the function of the truncated BTG1 protein, a C-terminal deletion variant of BTG1, which terminates at the common breakpoint after amino acid residue 100, was cloned in vector pcDNA3.1 and tested for protein expression. For comparison, wild-type BTG1 was included in the analyses (Figure S1A). Full-length HA-tagged BTG1 protein is subject to proteosomal degradation [8], but in the presence of proteosome inhibitor MG132, expression could be readily detected (Figure S1B). In contrast, truncated BTG1 (HA-BTG1-Trunc) protein levels appeared to be highly unstable and hardly detectable even in the presence of MG132 (Figure S1B), while mRNA expression levels were equal to those of full-length HA-BTG1 (Figure S1C). Based on these findings, we conclude that the read-through transcripts that are expressed upon focal loss of BTG1, are unlikely to give rise to functionally active protein, which favors a BTG1 haploinsufficiency scenario.
BTG1 Deletions May Result from Illegitimate RAG-Mediated Recombination
Because illegitimate RAG-mediated recombination has been implicated in the origin of several translocations and deletions in leukemia [18]–[20], we examined the sequences flanking each of the BTG1 breakpoints for the presence of recombination signal sequences (RSS). RSS motifs represent moderately conserved heptamer (CACAGTG) and nonamer (ACAAAAACC) sequences separated by either a spacer of 12±1 bp (12-RSS) or a spacer of 23±1 bp (23-RSS). The RAG complex only joins gene segments containing RSSs with different spacer lengths, complying to the 12/23 rule for efficient V(D)J recombination (Figure S2). Notably, we found that a 23-RSS was present at the recombination hotspot in the second exon of BTG1, while all but one of the centromeric breakpoints were found to harbor a 12-RSS (Table 3; Table S2). Furthermore, all deletion breakpoints contained a random addition of single nucleotides between the joining ends, which is most likely due to the action of terminal deoxynucleotidyl transferase (TdT). Together, these findings strongly support a role for illegitimate RAG-mediated recombination in the occurrence of BTG1 deletions.
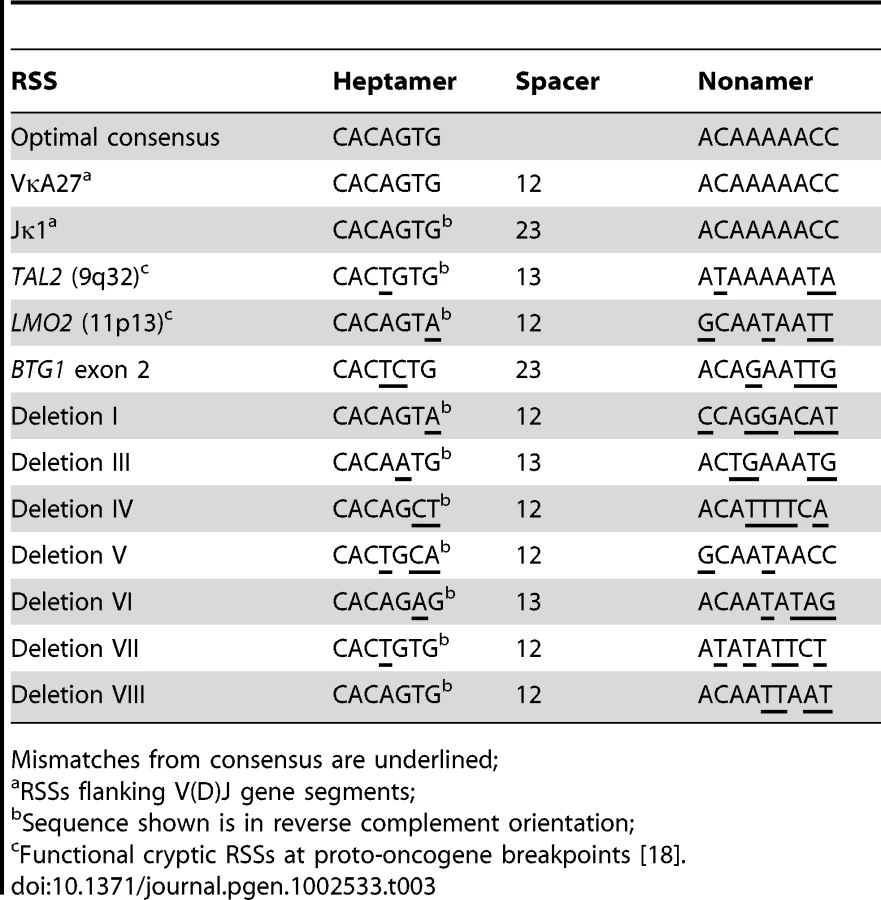
The BTG1 Gene Exhibits Elevated H3K4me3 Levels in B-Lineage Cells
Recent studies have shown that RAG-mediated V(D)J recombination is controlled by histone modifications, including histone H3 and H4 acetylation and H3 trimethylation, that are normally present at promoter regions of actively transcribed genes [26], [27]. More specifically, it has been demonstrated that the RAG2 plant homeodomain (PHD) finger binds directly to H3K4me3, thereby stimulating the catalytic activity of the RAG enzyme complex [28]. To reveal whether differences in levels of H3K4 trimethylation and/or H3K9/14 acetylation could explain the lineage-specific occurrence of BTG1 deletions, chromatin immunoprecipitation (ChIP) experiments were performed targeting different positions within the BTG1 gene in several B-lineage and T-lineage cell lines exhibiting abundant BTG1 expression (Figure 4A). As expected, prominent levels of H3K4me3 were present at the proximal promoter region close to the transcription start site in both B-lineage and T-lineage cell lines (Figure 4B). In contrast, only BCP-ALL cell lines displayed significantly higher levels of H3K4me3 near the breakpoint hotspot within the second exon of BTG1. H3K9/14 acetylation levels were found to significantly differ at the proximal promoter region between B- and T-lineage cells, but not at the body of the BTG1 gene (Figure 4C). In conclusion, our data suggest that illegitimate RAG-mediated recombination at the deletion breakpoint hotspot within the second exon of BTG1 in B-lineage cells may be facilitated by increased levels of H3K4me3.

Discussion
In this study, we demonstrate that BTG1, a recurrent target in pediatric ALL, is affected by gene truncating deletions, which occur predominantly in two cytogenetic subgroups of pediatric BCP-ALL, i.e., the ETV6-RUNX1 positive (19%) and BCR-ABL1 positive (26%) subgroups. Within these subgroups, BTG1 deletions frequently arise independently in different subclones, which is in full conformity with the recently reported complex multi-clonal evolution model of ALL [24], [25]. In addition, our results suggest a role for BTG1 deletions in the clonal selection and outgrowth in these BCP-ALL subgroups.
Through high-resolution genomic profiling, BTG1 was only recently identified as a recurrent target in pediatric ALL [2]–[4]. Here we demonstrate, using a cohort of 831 pediatric ALL cases, that BTG1 is targeted by a restricted number of well-demarcated genomic deletions. These microdeletions were found predominantly as monoallelic events at the clonal level in about 9% of the BCP-ALL cases, and were found to be completely absent in the T-ALL cases studied. Promoter hypermethylation events or sequence mutations were not detected. In contrast, BTG1 mutations have frequently been found in diffuse large B-cell lymphoma, arguing that other mutation mechanisms may target BTG1 in more maturated B-lineage malignancies [10]. Eight different deletions in 52 cases were molecularly cloned and sequenced. Together, the three most prevalent deletions (III, V and VIII) were found in 81% of the total. Of note, the vast majority of telomeric BTG1 deletion breakpoints were found to be tightly clustered within a stretch of 10 bp in the last exon of BTG1.
This remarkable finding has both structural and functional implications. Structurally, this breakpoint clustering implies the presence of sequence features underlying the origin of BTG1 deletions. Virtually all breakpoints that were mapped, including those within exon 2, were flanked by non-canonical RSS sequences, which strongly suggests that they have arisen from illegitimate RAG-mediated recombination. In line with these observations, BCP-ALL derived cell lines showed local accumulation of H3K4me3 within the coding region of the BTG1 gene including the second exon, which harbors the breakpoint hotspot. This epigenetic mark is normally associated with the proximal promoter of actively transcribed genes and, in addition, has been shown to act as a docking site for RAG2 binding, thereby facilitating V(D)J recombination [28]. In contrast, in T-ALL-derived cell lines these H3K4me3 marks were not enriched at the site of the BTG1 gene body, which indicates that in these cells the locus is less accessible for the RAG enzyme complex. Illegitimate recombination events at cryptic RSS motifs have been implicated in several recurrent chromosomal abnormalities in lymphoid malignancies, such as translocations and deletions. For example, both ETV6-RUNX1 and BCR-ABL1 translocations have been suggested to arise from mistargeting of RAG proteins that facilitate RAG-mediated transposition or at least create one of the initiating lesions in the form of nicks [29]–[31]. This mistargeting of RAG proteins may occur as a rare spontaneous event during lymphocyte differentiation, but may also result from increased or prolonged RAG activity in leukemic lymphoid blasts. This latter phenomenon may explain the subtype specificity of BTG1 deletions and/or the co-occurrence of these deletions with ETV6-RUNX1 and BCR-ABL1 translocations.
The recurrence of truncating BTG1 deletions in BCP-ALL may also point towards a functional role in leukemogenesis. BTG1 belongs to the BTG/TOB family of anti-proliferative genes (BTG1-BTG4, TOB1 and TOB2) implicated in several types of cancer [9]. We recently reported that in BCP-ALL, BTG1 regulates the glucocorticoid receptor (GR)-dependent transcriptional response in leukemia cells, while loss of BTG1 expression leads to glucocorticoid resistance in cell line models [8]. In addition, we have shown that leukemia clones carrying BTG1 deletions can survive therapy and rearise as a predominant clone in relapse or, as shown also by others, can occur as new lesions in relapse [21], [32]. Many BTG1 deletions result in almost identical truncations of the open reading frame and the consequent loss of two conserved C-terminal protein interaction domains. Nevertheless, dominant-negative or gain-of-function effects of the truncated BTG1 protein are less likely since the truncated BTG1 protein appears to be highly unstable. Furthermore, we also encountered deletions encompassing the entire open reading frame or only part of the 3′-UTR, indicating that truncation of the BTG1 open reading frame is highly frequent but not essential.
The fact that BTG1 deletion breakpoints are tightly clustered provides an opportunity to perform rapid and sensitive deletion screening through breakpoint-spanning PCRs. By applying this screening approach to our pediatric ALL cohort we identified, besides the most prominent BTG1 deletions, additional deletions in a substantial fraction (18%) of BCP-ALL cases at the subclonal level. These deletions remained undetected using standard procedures like MLPA or SNP arrays. In several cases, multiple deletions co-occurred in a single patient, each carrying unique breakpoint-spanning sequences that were detectable both at the genomic and at the transcript level. Analogous to the most prominent deletions, these subclonal lesions displayed a very similar distribution over different cytogenetic subgroups in BCP-ALL and were not present in T-ALL or normal bone marrow samples. Moreover, in one patient, this subclonal BTG1 deletion-positive clone at diagnosis reappeared as the major clone at relapse, whereas in relapse samples of five other patients new (sub)clonal BTG1 deletions appeared. These findings are completely in line with recent studies describing the clonal evolution of ETV6-RUNX1 and BCR-ABL1 positive ALLs with a clonal evolution during leukemia development showing that, upon treatment, recurrent lesions occur repeatedly and independently within a single patient, giving rise to a complex variety of slightly different subclones [24], [25]. Our current data provide additional evidence for this model at the molecular level by showing that BTG1 deletions arise independently in multiple ALL subclones in a context-dependent manner.
In addition, our results provide insight into the genetic lesions that act in concert with BTG1 deletions in BCP-ALL development. We found, for example, that deletions in ETV6, EBF1 and RB1 co-occur with BTG1 deletions, indicating that loss of normal BTG1 function may add to defects in pRb/E2F-mediated cell cycle and EBF1-mediated B-cell differentiation pathways. Illegitimate RAG-mediated recombination has been suggested as the responsible mechanism for IKZF1 [19], [20], CDKN2A/B [33], [34] and LMO2 [35] deletions, as well as ETV6-RUNX1 translocations [30]. It remains to be established whether RAG-mediated recombination, or other mechanisms like CSR, are implicated in the occurrence of RB1 and EBF1 deletions. However, similar to BTG1, these recurrent gene deletions may repeatedly arise in different subclones due to local subtype-specific accessibility of these loci to specific recombination machineries, followed by clonal selection and outgrowth.
In conclusion, our comprehensive analysis of BTG1 aberrations revealed that this gene is recurrently and exclusively affected by deletions in specific BCP-ALL subtypes. BTG1 deletions can arise independently in different ALL subclones, which either develop into a predominant clone at diagnosis, remain present as minor subclones during the course of the disease, or develop into the major clone at relapse. As such, this phenomenon may provide a molecular explanation for the model of multiclonal evolution during leukemogenesis.
Materials and Methods
Clinical Samples
A total of 831 patients diagnosed with BCP-ALL (n = 722) and T-ALL (n = 109) were included in this study. Diagnosis samples (n = 831) and matched relapse samples (n = 62) were collected by the Dutch Childhood Oncology Group (DCOG) and the Radboud University Nijmegen Medical Centre, the Netherlands. Mononuclear cells were harvested through Ficoll gradient separation and DNA was isolated using a QiaAmp purification kit (Qiagen, Venlo, The Netherlands). DNA from non-leukemic bone marrow samples was provided by Dr. Joop Jansen (Department of Hematology, Radboud University Nijmegen Medical Centre, the Netherlands). Written informed consent was obtained for all patient and control samples.
Cell Lines
The human BCP-ALL (CCRF-SB, RS4;11, Nalm6, REH, SUP-B15, 380, 697, SEM, TOM1, MUTZ5) and T-ALL cell lines (MOLT4, Jurkat) were purchased either from ATCC or DSMZ. T-ALL cell lines CML-T1, HSB2, and KARPAS45 were obtained from Dr. Adolfo Ferrando (Columbia University, NY, USA) and T-ALL cell line TK6 was obtained from Dr. Albert Fornace (NCI, Bethesda, USA). Leukemia cell lines were maintained in RPMI-1640 medium (Invitrogen) supplemented with 10% fetal calf serum, 100 U/mL penicillin sodium, and 100 µg/mL of streptomycin sulfate at 37°C in a humidified air atmosphere containing 5% carbon dioxide.
Multiplex Ligation-Dependent Probe Amplification
Leukemic patient samples and cell lines (380, 697, CCRF-SB, RS4;11, SEM, TOM1, REH, MUTZ5, SUP-B15, Nalm6, Jurkat, KARPAS45, MOLT4 and TK6) were analyzed for copy number changes in BTG1 using multiplex ligation-dependent probe amplification (MLPA). DNA was isolated using a QiaAmp purification kit (Qiagen). A total of 9 probes was developed using MeltIngeny software and guidelines provided by MRC-Holland (Amsterdam, The Netherlands; Table S6). MLPA analyses were performed using SALSA MLPA reaction mixture and P200-A1 reference probe-mix (MRC Holland) as previously described [5]. In addition, we determined the copy number status of several leukemia-associated genes (PAX5, IKZF1, EBF1, CDKN2A, CDKN2B, RB1, ETV6, BTG1) with the SALSA MLPA kit P335-A2 ALL-IKZF1 (MRC Holland) according to manufacturer's instructions.
Bisulfite Sequencing
In 25 BCP-ALL patient diagnosis samples, we determined the methylation status of the BTG1 promoter using bisulfite sequencing. A total of 500 ng DNA per sample was bisulphite converted using the EZ DNA Methylation™ Kit (Zymo research corporations, Leiden, The Netherlands) according to manufacturer's protocol. Subsequently, the CpG island was PCR amplified (for primers see Table S7), cloned in a pGEM-T vector (Promega Benelux BV, Leiden, The Netherlands), transformed in competent E. Coli DH5α cells and plated on an LB agar plate containing 7 µg/ml ampicillin, 200 nM Isopropyl β-D-1-thiogalactopyranoside (IPTG) and 200 µg/ml X-Gal. After overnight incubation, individual white clones were selected for colony PCR and sequenced using a 3730 Sequence Analyzer (Applied Biosystems, Foster City, CA, USA). Sequences were analyzed using Vector NTI software (Advance TM 11.0, release December 15 2008, Invitrogen, Breda, the Netherlands). In vitro methylated DNA was used as a positive control. Briefly, 1 µg DNA was incubated with 4 U M.SssI CpG Methyltransferase (New England Biolabs, Ipswich, United Kingdom), 160 µM S-adenosylmethionine, 50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2 and 1 mM Dithiothreitol (pH7.9) for 4 hours at 37°C. As a negative control, HEK293 DNA was amplified by using the GenomePlex Complete Whole Genome Amplification Kit (Sigma-Aldrich, Zwijndrecht, The Netherlands), according to manufacturer's protocol.
BTG1 Mutation Detection
Sequencing of the first (n = 135) and second (n = 337) exon of the BTG1 gene was performed on PCR-amplified exons using flanking intron-based primers (listed in Table S7). PCR amplifications were performed with 10 ng DNA, 200 nM of each primer, 2 (ex1) or 2.5 (ex2) mM MgCl2, 200 µM dNTPs, 1 M GC melt (Clontech, Saint-Germain-en-Laye, France), 10× PCR buffer II, 2.5 U AmpliTaq Gold (Applied Biosystems) using 5 minutes preheating at 96°C, followed by 35 cycles of 30 seconds at 96°C, 30 seconds at 55.3°C and 1 min at 72°C, and termination for 3 min at 72°C. The samples were sequenced using a 3730 Sequence Analyzer (Applied Biosystems) and the results were analyzed using Vector NTI software (Invitrogen).
Genomic SNP Arrays
To characterize the BTG1 deletions in further detail, we performed high-resolution copy number and genotyping analyses using Affymetrix SNP6.0 arrays according to standard protocols provided by the manufacturer. Genotypes were generated using Affymetrix Genotyping Console v2.1 and Nexus 5.0 software (BioDiscovery, Inc 2010, build version 4621). Normal copy number variation was filtered out using data from our in-house database (containing over 600 healthy control samples), Hapmap, and the Database of Genomic Variants (http://projects.tcag.ca/variation/).
Breakpoint Mapping and PCR-Based Validation
Starting from the SNP array data, we used a stepwise approach to zoom in on eight individual breakpoints of different BTG1 deletions. We determined the exact genomic breakpoints and interstitial sequences using Q-PCR, long-range PCR and finally, standard PCR and sequencing. Q-PCR was performed on a 7900HT FAST Real-Time PCR system (Applied Biosystems) with SYBR Green to detect PCR product. Long range PCR was performed using TaKaRa La Taq (Takara Bio Inc, Otsu, Shiga, Japan). PCR amplicons were sequenced using a 3730 Sequence Analyzer (Applied Biosystems) and analyzed with VectorNTI software (Invitrogen). The primer pairs used to detect and sequence the eight different breakpoints are listed in Table S7. For PCR-based validation we used 20 ng DNA, 200 nM of each primer, 2 or 2.5 mM MgCl2, 200 µM dNTPs, 10× PCR buffer II, 2.5 U AmpliTaq Gold (Applied Biosystems) using 5 minutes preheating at 95°C, followed by 10 cycles of 30 seconds at 95°C, 2 min at 60°C or 57.8°C and 2 min at 72°C, and a further 30 cycles of 30 seconds at 95°C, 30 seconds at 60°C or 57.8°C and 2 min at 72°C, and termination for 10 min at 72°C.
RT–PCR
Total RNA from the BCP-ALL and T-ALL cell lines was extracted with Trizol (Invitrogen), while total RNA from diagnosis patient samples was isolated using the RNeasy kit (Qiagen), both according to the manufacturer's instructions. Total RNA was converted to cDNA via RT-PCR using random 6-mers and Superscript III Reverse Transcriptase (Invitrogen). Thereafter, cDNA was resolved in 50 µl ddH2O, and PCR amplifications were performed in 30 µl reactions at standard concentrations (1.5 mM MgCl2, 0.2 mM dNTP, 1× PCR buffer (Invitrogen), 2 U Platinum Taq (Invitrogen), 0.3 µM of each primer, and 2 µl cDNA template) (see Table S7 for primer sequences). PCR reactions were performed for 35 cycles at an annealing temperature of 58°C using an Eppendorf Mastercycler. The PCR fragments obtained were resolved by 1% agarose gel electrophoresis, isolated using Qiagen gel extraction kit and cloned into pGEM-T easy for sequencing with M13 primers. Quantitative real-time PCR was performed to determine the expression of BTG1 mRNA sequences 5′ to the breakpoint hotspot (exon primers flanking intron 1), BTG1 exon 2 (primers flanking breakpoint hotspot) and HPRT with Power SYBR Green (Applied Biosystems) (Table S7). Reactions were performed in triplicate on two independent cDNA templates using an Applied Biosystems 7500 Real-Time PCR thermocycler (ABI). Data were represented as fold differences relative to BTG1 mRNA expression in Nalm6 cells normalized to HPRT expression based on calculation of 2−ΔΔCt.
Cloning and Protein Expression Analyses BTG1 Constructs
HA-BTG1-Trunc was generated by PCR based on full-length HA-BTG1 [8], using primers indicated in Table S7. PCR products were purified, cloned into the EcoRI and XhoI sites of pcDNA3.1 (Invitrogen) and verified by Sanger sequencing. To analyze protein expression of the different BTG1 constructs, HEK293 cells were transfected with 10 µg of plasmid DNA using linear Polyethyleneimine (PEI, Polysciences, Warrington, PA). The next day cells were splitted over two dishes and, 24 hours after transfection, treated with 5 µM MG132 (Peptides International, Louisville, KY) in DMSO or vehicle for 16 hours. For detection of HA-BTG1 protein expression, 5×106 cells were lysed in Laemmli sample buffer containing 2% SDS and 100 mM DTT, boiled for 5 minutes, loaded on 15% SDS-PAGE, blotted on PVDF membrane, and stained with HA antibody, clone 3F10 (Roche Diagnostics). Protein expression was visualized using ECL Plus Western Blot Detection System (GE Healthcare) and FluorchemE digital imaging device (Cell Biosciences).
Chromatin Immunoprecipitations
After crosslinking for 15 min with 1% formaldehyde, the leukemia cells were re-suspended in ice-cold lysis buffer (50 mM HEPES [pH7.6], 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate) supplemented with Complete protease inhibitors (Roche) at a density of 35×106 cells/mL and sonicated for 15 min, 30 seconds on and 30 seconds off (Bioruptor Diagenode). Chromatin was incubated overnight in the presence 0.1% BSA, 20 mM HEPES (pH 7.6), 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.15% SDS, 1% Triton X-100, protein A/G beads (Santa Cruz), Complete protease inhibitors and 2 µg H3K4me3 or H3K9/14Ac antibody (both from Diagenode). Beads were washed two times in buffer 1 (0.1% SDS, 0.1% Sodium Deoxycholate, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH7.6]), one time in buffer 2 (0.1% SDS, 0.1% Sodium Deoxycholate, 1% Triton X-100, 500 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH7.6]), one time in buffer 3 (0.5% Sodium Deoxycholate, 0.5% NP40, 250 mM LiCl , 1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH7.6]), and two times in buffer 4 (1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES [pH 7.6]). Chromatin was eluted from the beads with 1% SDS/100 mM NaHCO3 and de-crosslinked together with input material for 4 hrs at 65°C with 200 mM NaCl. After phenol∶chloroform∶isoamylalcohol extraction, DNA was precipitated in the presence of 10 µg glycogen. Subsequently, diluted DNA was subjected to quantitative PCR using Power SYBR Green (Applied Biosystems) and an Applied Biosystems 7500 Real-Time PCR thermocycler (primers are listed in Table S7). The percentage recovery was equal to dilution factor of input DNA×2(Ct Input-Ct ChIP)×100.
Statistical Analyses
Co-occurrence of copy number alterations and cytogenetic subgroups was compared using crosstabs and a standard chi-square test or Fisher's exact test when sample groups were small. The differences between H3K4me3 and H3K9/14Ac levels in T-ALL versus BCP-ALL samples was assessed using a Student's t-test. Statistical analyses were carried out using the SPSS statistical package (IBM, Chicago, IL, USA; release 16.0.2, April 2008) and two-sided P-values below 0.05 were considered to be statistically significant.
Supporting Information
Zdroje
1. PuiCHRellingMVDowningJR 2004 Acute lymphoblastic leukemia. N Engl J Med 350 1535 1548
2. MullighanCGGoorhaSRadtkeIMillerCBCoustan-SmithE 2007 Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446 758 764
3. KuiperRPSchoenmakersEFReijmersdal vanSVHehir-KwaJYGeurts van KesselA 2007 High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia 21 1258 1266
4. TsuzukiSKarnanSHoribeKMatsumotoKKatoK 2007 Genetic abnormalities involved in t(12;21) TEL-AML1 acute lymphoblastic leukemia: analysis by means of array-based comparative genomic hybridization. Cancer Sci 98 698 706
5. KuiperRPWaandersEvan der VeldenVHJvan ReijmersdalSVVenkatachalamR 2010 IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 24 1258 1264
6. WaandersEvan der VeldenVHJvan der SchootCEvan LeeuwenFNvan ReijmersdalSV 2011 Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia 25 254 258
7. HarveyRCMullighanCGWangXDobbinKKDavidsonGS 2010 Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 116 4874 4884
8. Van GalenJCKuiperRPvanELLeversMTijchonE 2010 BTG1 regulates glucocorticoid receptor autoinduction in acute lymphoblastic leukemia. Blood 115 4810 4819
9. WinklerGS 2010 The mammalian anti-proliferative BTG/Tob protein family. J Cell Physiol 222 66 72
10. MorinRDMendez-LagoMMungallAJGoyaRMungallKL 2011 Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476 298 303
11. LinWJGaryJDYangMCClarkeSHerschmanHR 1996 The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem 271 15034 15044
12. RouaultJPPrevotDBerthetCBirotAMBillaudM 1998 Interaction of BTG1 and p53-regulated BTG2 gene products with mCaf1, the murine homolog of a component of the yeast CCR4 transcriptional regulatory complex. J Biol Chem 273 22563 22569
13. BussonMCarazoASeyerPGrandemangeSCasasF 2005 Coactivation of nuclear receptors and myogenic factors induces the major BTG1 influence on muscle differentiation. Oncogene 24 1698 1710
14. PrevotDVoeltzelTBirotAMMorelAPRostanMC 2000 The leukemia-associated protein Btg1 and the p53-regulated protein Btg2 interact with the homeoprotein Hoxb9 and enhance its transcriptional activation. J Biol Chem 275 147 153
15. LieberMRMaYPannickeUSchwarzK 2003 Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol 4 712 720
16. JungDGiallourakisCMostoslavskyRAltFW 2006 Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 24:541–70 541 570
17. StavnezerJGuikemaJESchraderCE 2008 Mechanism and regulation of class switch recombination. Annu Rev Immunol 26:261–92 261 292
18. MarculescuRLeTSimonPJaegerUNadelB 2002 V(D)J-mediated translocations in lymphoid neoplasms: a functional assessment of genomic instability by cryptic sites. J Exp Med 195 85 98
19. MullighanCGMillerCBRadtkeIPhillipsLADaltonJ 2008 BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453 110 114
20. IacobucciIStorlazziCTCilloniDLonettiAOttavianiE 2009 Identification and molecular characterization of recurrent genomic deletions on 7p12 in the IKZF1 gene in a large cohort of BCR-ABL1-positive acute lymphoblastic leukemia patients: on behalf of Gruppo Italiano Malattie Ematologiche dell'Adulto Acute Leukemia Working Party (GIMEMA AL WP). Blood 114 2159 2167
21. MullighanCGPhillipsLASuXMaJMillerCB 2008 Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322 1377 1380
22. ZhangJMullighanCGHarveyRCWuGChenX 2011 Key pathways are frequently mutated in high risk childhood acute lymphoblastic leukemia: a report from the Children's Oncology Group. Blood 118 3080 3087
23. GreavesM 2010 Cancer stem cells: back to Darwin? Semin Cancer Biol 20 65 70
24. AndersonKLutzCvan DelftFWBatemanCMGuoY 2011 Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature 469 356 361
25. NottaFMullighanCGWangJCPoepplADoulatovS 2011 Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 469 362 367
26. MatthewsAGKuoAJRamon-MaiquesSHanSChampagneKS 2007 RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature 450 1106 1110
27. McMurryMTKrangelMS 2000 A role for histone acetylation in the developmental regulation of VDJ recombination. Science 287 495 498
28. ShimazakiNTsaiAGLieberMR 2009 H3K4me3 stimulates the V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in cis: implications for translocations. Mol Cell 34 535 544
29. ThandlaSPPloskiJERaza-EgilmezSZChhalliyilPPBlockAW 1999 ETV6-AML1 translocation breakpoints cluster near a purine/pyrimidine repeat region in the ETV6 gene. Blood 93 293 299
30. NumataMSaitoSNagataK 2010 RAG-dependent recombination at cryptic RSSs within TEL-AML1 t(12;21)(p13;q22) chromosomal translocation region. Biochem Biophys Res Commun 402 718 724
31. ScoreJCalasanzMJOttmanOPaneFYehRF 2010 Analysis of genomic breakpoints in p190 and p210 BCR-ABL indicate distinct mechanisms of formation. Leukemia 24 1742 1750
32. Van DelftFWHorsleySColmanSAndersonKBatemanC 2011 Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 117 6247 6254
33. NovaraFBeriSBernardoMEBellazziRMaloviniA 2009 Different molecular mechanisms causing 9p21 deletions in acute lymphoblastic leukemia of childhood. Hum Genet 126 511 520
34. RaschkeSBalzVEfferthTSchulzWAFlorlAR 2005 Homozygous deletions of CDKN2A caused by alternative mechanisms in various human cancer cell lines. Genes Chromosomes Cancer 42 58 67
35. Van VlierberghePvan GrotelMBeverlooHBLeeCHelgasonT 2006 The cryptic chromosomal deletion del(11)(p12p13) as a new activation mechanism of LMO2 in pediatric T-cell acute lymphoblastic leukemia. Blood 108 3520 3529
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 2
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- Gene Expression and Stress Response Mediated by the Epigenetic Regulation of a Transposable Element Small RNA
- Contrasting Properties of Gene-Specific Regulatory, Coding, and Copy Number Mutations in : Frequency, Effects, and Dominance
- Homeobox Genes Critically Regulate Embryo Implantation by Controlling Paracrine Signaling between Uterine Stroma and Epithelium
- Nondisjunction of a Single Chromosome Leads to Breakage and Activation of DNA Damage Checkpoint in G2