
An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype
Epithelial-mesenchymal transition (EMT), a mechanism important for embryonic development, plays a critical role during malignant transformation. While much is known about transcriptional regulation of EMT, alternative splicing of several genes has also been correlated with EMT progression, but the extent of splicing changes and their contributions to the morphological conversion accompanying EMT have not been investigated comprehensively. Using an established cell culture model and RNA–Seq analyses, we determined an alternative splicing signature for EMT. Genes encoding key drivers of EMT–dependent changes in cell phenotype, such as actin cytoskeleton remodeling, regulation of cell–cell junction formation, and regulation of cell migration, were enriched among EMT–associated alternatively splicing events. Our analysis suggested that most EMT–associated alternative splicing events are regulated by one or more members of the RBFOX, MBNL, CELF, hnRNP, or ESRP classes of splicing factors. The EMT alternative splicing signature was confirmed in human breast cancer cell lines, which could be classified into basal and luminal subtypes based exclusively on their EMT–associated splicing pattern. Expression of EMT–associated alternative mRNA transcripts was also observed in primary breast cancer samples, indicating that EMT–dependent splicing changes occur commonly in human tumors. The functional significance of EMT–associated alternative splicing was tested by expression of the epithelial-specific splicing factor ESRP1 or by depletion of RBFOX2 in mesenchymal cells, both of which elicited significant changes in cell morphology and motility towards an epithelial phenotype, suggesting that splicing regulation alone can drive critical aspects of EMT–associated phenotypic changes. The molecular description obtained here may aid in the development of new diagnostic and prognostic markers for analysis of breast cancer progression.
Published in the journal:
. PLoS Genet 7(8): e32767. doi:10.1371/journal.pgen.1002218
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002218
Summary
Epithelial-mesenchymal transition (EMT), a mechanism important for embryonic development, plays a critical role during malignant transformation. While much is known about transcriptional regulation of EMT, alternative splicing of several genes has also been correlated with EMT progression, but the extent of splicing changes and their contributions to the morphological conversion accompanying EMT have not been investigated comprehensively. Using an established cell culture model and RNA–Seq analyses, we determined an alternative splicing signature for EMT. Genes encoding key drivers of EMT–dependent changes in cell phenotype, such as actin cytoskeleton remodeling, regulation of cell–cell junction formation, and regulation of cell migration, were enriched among EMT–associated alternatively splicing events. Our analysis suggested that most EMT–associated alternative splicing events are regulated by one or more members of the RBFOX, MBNL, CELF, hnRNP, or ESRP classes of splicing factors. The EMT alternative splicing signature was confirmed in human breast cancer cell lines, which could be classified into basal and luminal subtypes based exclusively on their EMT–associated splicing pattern. Expression of EMT–associated alternative mRNA transcripts was also observed in primary breast cancer samples, indicating that EMT–dependent splicing changes occur commonly in human tumors. The functional significance of EMT–associated alternative splicing was tested by expression of the epithelial-specific splicing factor ESRP1 or by depletion of RBFOX2 in mesenchymal cells, both of which elicited significant changes in cell morphology and motility towards an epithelial phenotype, suggesting that splicing regulation alone can drive critical aspects of EMT–associated phenotypic changes. The molecular description obtained here may aid in the development of new diagnostic and prognostic markers for analysis of breast cancer progression.
Introduction
About 90% of human malignancies are carcinomas, tumors of epithelial origin [1]. The early steps in carcinoma metastasis often bear a striking resemblance to developmental programs involving Epithelial-to-Mesenchymal Transition (EMT), a process that converts organized epithelial cells into isolated, migratory cells with a mesenchymal morphology [2]. A growing body of work implicates EMT-like mechanisms in tumor cell invasion and dissemination in experimental systems and, recently, in human cancer [3], [4]. Normal epithelia are comprised of cells with aligned apical-basal polarity that are interconnected laterally by several types of junctions, including adherens junctions (AJs), which play important roles in establishing and regulating cell-cell adhesion [5]. During EMT, apico-basolateral polarity is lost, cell-cell junctions dissolve and the actin cytoskeleton is remodeled to endow cells with mesenchymal characteristics, including an elongated, migratory and invasive phenotype. Importantly, as a consequence of EMT cells may escape tumors, invade the surrounding tissue and migrate towards blood- or lymphatic vessels guided by the cells and extracellular matrix present in their microenvironment [6].
While EMT is thought to promote carcinoma invasion and metastasis, it is clear that other mechanisms for carcinoma progression exist [3], [7], and direct in vivo evidence linking EMT to metastasis in clinical subjects has been challenging to obtain. Some studies have shown that a poor clinical outcome correlates with markers of EMT progression [8]–[11]. Conversely, some reports have identified carcinoma cells in primary and metastatic lesions with well-differentiated epithelial morphology [7], [12]. Detection of EMT in vivo during metastasis is complicated further by a reverse process, Mesenchymal-to-Epithelial transition (MET), that is also important during embryonic development and is thought to occur during metastatic colonization at secondary sites [13]. New approaches are needed to detect EMT and MET during metastatic progression and to clarify their clinical significance [14], [15].
The molecular mechanisms underlying EMT have been studied extensively in the last decade. EMT-inducing growth factors can trigger signaling cascades that activate a network of transcription factors, including Snail, ZEB-1, Goosecoid, FOXC2, Twist and others [16], that orchestrate the EMT program. Ectopic expression of a number of the EMT-associated transcription factors can initiate the program as well. Twist, a potent EMT driver, was identified originally as an inducer of mesoderm formation in Drosophila [17]. Ectopic Twist expression in epithelial cells results in loss of E-cadherin-mediated cell-cell adhesion, acquisition of mesenchymal markers and increased motility of isolated cells [18], a hallmark of the mesenchymal phenotype.
EMT is also likely regulated by post-transcriptional mechanisms including alternative pre-mRNA splicing. Alternative splicing expands the diversity of the proteome by producing multiple mRNA and protein isoforms per gene [19]. More than 90% of human genes are estimated to undergo alternative splicing, with a majority of alternative splicing events exhibiting tissue-specific splicing differences [20]. A variety of cancer-associated genes express alternatively spliced isoforms [21], indicating that regulation at the level of splicing may play important roles in cancer onset and progression. Alternative splicing of FGFR2 correlates with EMT in rat bladder carcinoma cells, where mutually exclusive inclusion of one of two exons defines the ligand binding specificity of the receptor during EMT [22]. ENAH (also known as Mena), an actin cytoskeleton regulatory protein, contains a small coding exon 11a that is included only in epithelial cells and excluded in mesenchymal cell lines and during EMT [23], [24]. Alternative splicing of p120catenin (CTNND1) generates protein isoforms that display opposite effects on cell motility in epithelial and mesenchymal cells [25]. Recently, two epithelial-specific RNA binding proteins, ESRP1 and ESRP2, homologs of the nematode splicing factor Sym-2, were identified in a screen for regulators of FGFR2 splicing [24]. The RBFOX2 splicing factor has recently been demonstrated to regulate subtype-specific splicing in a panel of breast cancer cell lines [26]. The ESRPs and RBFOX2 promote epithelial splicing of a number of transcripts (including FGFR2 and ENAH), some of which play important roles in EMT [24], [27]. Loss of ESRPs in epithelial cells induces some EMT-like changes in cell morphology [28]. However, the full extent of alternative splicing during EMT and its functional consequences to cell phenotype has yet to be elucidated.
We used an established in vitro model of EMT to evaluate the amount of gene expression and alternative splicing changes during EMT. Using deep sequencing analysis of the transcriptomes of epithelial and mesenchymal cells, we discovered a global alternative splicing program that alters splicing of key regulators of cell phenotype, including proteins that control cell adhesion and cytoskeletal dynamics. Our analysis indicates that EMT-associated splicing is likely regulated by several splicing factors, including the ESRPs and members of the RBFOX, CELF, MBNL, and hnRNP classes of splicing factors. We found that partial induction of the epithelial splicing program in mesenchymal cells via ectopic expression of ESRP1 or by depletion of RBFOX2 conferred epithelial properties to mesenchymal cells, supporting a key role for alternative splicing during MET. Multiple EMT-associated alternative splicing events were identified in breast cancer cell lines and in primary human breast cancer samples where epithelial and mesenchymal splicing patterns were negatively correlated. This EMT-associated splicing signature likely represents a broadly conserved program involved in the acquisition of mesenchymal-like phenotypes in vivo that could be used to detect EMT in primary human cancers with a potentially significant prognostic value.
Results
Large-scale changes in gene expression accompany EMT
To assess gene and alternative mRNA isoform expression during EMT, we utilized an in vitro model in which mammary epithelial cells (HMLE) expressing Twist fused to a modified estrogen receptor (ER) undergo EMT when the fusion protein is activated by addition of the ER ligand 4-hydroxytamoxifen (4-OHT; tamoxifen) [29]. Untreated HMLE/Twist-ER epithelial cells maintained highly organized cell-cell adhesions and cell polarity (Figure 1A). Following tamoxifen treatment, the cobblestone-like appearance of HMLE/Twist-ER cells was replaced by a spindle-like, fibroblastic morphology, consistent with previously published results (Figure 1A; [29]). This morphological transformation represents one of the hallmarks of EMT. As expected, phenotypic changes coincided with a change in expression of canonical EMT markers, including loss of E-cadherin and induction of N-cadherin, Fibronectin and Vimentin expression (Figure 1B). Tamoxifen competes with estrogen for binding to ER to form a complex that translocates into the nucleus where it recruits co-repressors of transcription, thus preventing activation of ER downstream targets [30]. Since HMLE cells do not express endogenous ER (Figure S1B), EMT induction in HMLE/Twist-ER cells is likely initiated exclusively by downstream targets of Twist, making HMLE/Twist-ER cells a useful in vitro model of EMT.

To obtain an in-depth analysis of gene expression and splicing changes during EMT, we collected mRNA from untreated (epithelial) and from tamoxifen-treated (mesenchymal) HMLER/Twist-ER cells. Deep sequencing of fragments of polyA-selected mRNAs (RNA-Seq) was used to obtain a digital inventory of gene and mRNA isoform expression (Figure 1A). Between 27 million and 30 million 39-base-pair (bp) cDNA fragments were sequenced from each sample (Figure S1A). Sequenced cDNA fragments (reads) were mapped to the human genome (hg18 version) and to a splice junction database derived from AceView annotation [31]. In total, ∼75% of reads mapped uniquely to the genome or to splice junctions, allowing up to 2 mismatches. Less than 1% of total reads mapped uniquely to rRNA sequences (data not shown). Read density (coverage) was over 400-fold higher in exons than in introns or intergenic regions (Figure S1C), indicating that most reads derived from mature mRNA.
We first estimated gene expression changes during EMT using ‘Reads Per Kilobase of Exon Model per Million Mapped Reads’ (RPKM), a measure of expression that reflects the molar concentration of a transcript in the sample by normalizing read counts for mRNA length and for the total read number in the sample [32]. Applying both a statistical cut-off based on Audic-Claverie statistics for read-based expression profiling [33] and requiring a minimum 3- fold change, we observed that ∼2,060 genes were downregulated, while ∼950 were upregulated in EMT (Figure S2A), indicating a large-scale reorganization of the transcriptome during this process in agreement with recently published data [34]. As expected, E-cadherin was downregulated, while N-cadherin was upregulated during EMT [18]; actin transcript levels remained unchanged (Figure S2A). These observations revealed that Twist-induced EMT is accompanied by massive changes in gene expression similar to those observed in developmental EMT [35].
Gene ontology (GO) enrichment analysis of up- and down-regulated genes was used to gain insight into functional significance of the EMT-driven expression changes. Genes involved in epithelial cell differentiation, encoding components of cell cycle machinery and cell-cell junction components, were downregulated during EMT (Figure S2B). Concomitantly, genes associated with cell-matrix adhesion, extracellular matrix organization and cell motility, were upregulated (Figure S2C). Thus, the most significant EMT-driven changes in gene expression are associated with gene categories involved in the phenotypic conversion that occurs during EMT, in agreement with previously published data [36].
Alternative isoform expression is grossly affected in EMT
To explore the extent of regulated RNA processing during EMT, we examined eight common types of alternative isoform expression events, each capable of producing multiple mRNA isoforms from a gene through alternative splicing, alternative cleavage and polyadenylation (APA) and/or alternative promoter usage (Figure 1D). These eight types of events included: skipped exons (SE), retained introns (RI), mutually exclusive exons (MXEs), alternative 5′ and 3′ splice sites (A5SS and A3SS), alternative first exons (AFE), alternative last exons (ALE) and tandem 3′ untranslated regions (tandem 3′ UTRs). A comprehensive set of ∼136,000 events of these eight types was derived from the AceView gene annotations [31]. The fraction of mRNAs that contained an alternative exon – the ‘percent spliced in’ (PSI or Ψ) value – was estimated by the ratio of the density of inclusion reads to the sum of the densities of inclusion reads and exclusion reads, with a variant of this method used for tandem 3′ UTRs, as described previously [20]. Thus, Ψ values range from ∼0, indicating predominant exclusion of an alternative exon from mRNAs, to ∼1, indicating predominant inclusion of the exon.
The extent of EMT-specific regulation of these events was assessed by comparison of the mesenchymal (post-EMT) to the epithelial (pre-EMT) RNA-Seq data (Figure 1D). In all, for ∼40% of genes with documented alternative isoforms, both isoforms were detected by RNA-Seq reads. Of the events where both isoforms were detected, about 1 in 10 skipped exons and 1 in 20 mutually exclusive exons exhibited a significant change in Ψ value >10%, with hundreds of alternative splicing events of other types also regulated at this level (Figure 1D). At the gene level, 4.5% of genes contained an event(s) with an absolute change in Ψ value greater than 10% during EMT, and 2% of genes contained an event(s) with a Ψ value change greater than 30% (Table S1). These data indicate that a substantial change in splicing accompanies EMT.
To confirm the accuracy of RNA-Seq analysis of alternative splicing during EMT, a subset of SE and MXE events was chosen from the set with False Discovery Rate (FDR) below <0.05 and |ΔΨ|>0.1 for semi-quantitative RT-PCR (sqRT-PCR) analysis using cDNA from cells before and after EMT induction. Alternative splicing events with |ΔΨ|>0.1 between human tissues are enriched for evolutionarily conserved sequences surrounding the alternative exons as compared to constitutive exons, suggesting that use of this cutoff enriches for functional events [20]. The tested subset included 37 alternative exons that showed relatively large changes in splicing based on the analysis of the RNA-seq data, or whose host genes encoded functionally interesting molecules with respect to EMT (e.g., adhesion molecules). This subset also included a few events that showed relatively small changes in isoform expression in order to assess the robustness of our statistical test. In all cases, the change in splicing ΔΨ ( = ΨM−ΨE) detected by RT-PCR was in the same direction as that determined by RNA-Seq (Figure S1D), and in 78% of cases, the change in Ψ observed by sqRT-PCR was 20% or higher. Altogether, a strong concordance (R2 = 0.86; Figure S1D) was observed between splicing changes detected by RNA-Seq and measurements by sqRT-PCR. The high validation rate and quantitative concordance by an independent method (sqRT-PCR) support the reliability of the alternative splicing events identified by the RNA-seq analysis.
Genes with altered splicing during EMT showed strong enrichment for involvement in biological processes related to the regulation of the actin cytoskeleton, cell-cell junctions, regulation of cell migration and wound healing. Pathway analysis using KEGG and GO detected enrichment of EMT-associated alternative splicing events in the Wnt, Ras and Insulin pathways (Figure 1C). These enriched terms suggested that alternative splicing plays a role in pathways that direct morphological and motility-related changes associated with EMT. Interestingly, although the types of gene functions most affected at the splicing and expression levels were largely similar, the actual sets of genes undergoing splicing-level and expression-level changes did not overlap more than expected by chance (Figure S3). This observation suggests that the EMT splicing program functions in a manner that is parallel to the transcriptional program and that gene expression and alternative splicing, may coordinately drive changes to specific aspects of cell morphology related to the cytoskeleton, cell adhesion and cell motility.
Regulatory motifs and factors associated with the EMT splicing program
A substantial shift in the levels or activity of the major splicing factors likely underlies the large-scale program of splicing changes that occur during EMT. To explore the nature of this shift, we first analyzed the incidence of oligonucleotide motifs occurring in regulated alternative transcripts. As most splicing factors bind short RNA oligomers a few bases long, we identified pentanucleotides (5mers) that were enriched in regions adjacent to the splice sites involved in the splicing of exons induced or repressed upon EMT (Figure 2A). This analysis identified a few dozen 5mers enriched in each region relative to control alternative introns, including motifs corresponding to the RBFOX, CELF, ESRP and MBNL families of tissue-specific factors, as well as motifs for several heterogeneous nuclear ribonucleoprotein (hnRNP) factors, including hnRNPs F and H, PTB/hnRNP I, and hnRNP L (Table S2, S3). A subset of these motifs was specifically enriched adjacent to exons whose Ψ values increased following EMT relative to exons whose splicing did not change (Figure 2A). These included motifs associated with RBFOX and ESRP splicing factors and with hnRNPs F/H and L. An overlapping subset of motifs were enriched adjacent to exons whose Ψ values decreased following EMT, again including motifs associated with the RBFOX and hnRNP F/H families and also motifs associated with PTB and MBNL family proteins (Figure 2A). Several 5-mers, without clear RNA binding protein partners, that may represent binding sites of uncharacterized splicing regulators in EMT were also identified.

We also examined changes in the expression of RNA binding protein (RBP) genes. The most striking changes in RBP expression occurred for the related epithelial specific splicing factors ESRP1 (RBM35A) and ESRP2 (RBM35B) [24]. During EMT, the expression of these factors decreased by ∼90-fold and ∼35-fold, respectively, from relatively high initial levels (Figure 2B). Motif enrichment for ESRP splicing factors was observed in the upstream sequence of cassette exons upregulated during EMT (Figure 2A) consistent with the recent observation that ESRP binding sites are present at greater numbers upstream of silenced exons, than included exons [28]. As ESRPs are downregulated during EMT, these silenced exons are relieved from ESRP inhibition and thus appear upregulated.
Splicing factor activity often switches between positive and negative regulation depending on the location of binding relative to the regulated exon. RBFOX family splicing factors tend to enhance splicing when bound downstream and to repress splicing when bound upstream of alternative exons [27]. The observed pattern of enrichment of RBFOX motifs downstream of exons whose inclusion increased during EMT and upstream of exons whose inclusion decreased (Figure 2A) is therefore consistent with an increase in the activity of RBFOX family factors during EMT. Expression of the RBFOX2 gene increased moderately but significantly by about 15% following EMT (Table S4) while at the same time splicing of a MXE encoding the RNA-binding domain of the RBFOX2 protein increased by about 20%. Thus, these changes together should increase the levels of splicing-active RBFOX2 mRNA by at least a third. Recently, it has been suggested that RBFOX2 activity plays a role in regulating a set of breast cancer subtype–specific alternative splicing events [26]. The expression levels of many other RBPs associated with motifs enriched near EMT-regulated exons changed during EMT (Figure 2B, Table S4), including downregulation of the splicing repressor PTBP1 (PTB/hnRNP I) by ∼2.5-fold, downregulation of the PTB-associated splicing co-repressor RAVER1 by ∼4-fold, and downregulation of the myotonic dystrophy-associated splicing factors MBNL2 and MBNL3 and hnRNP F by ∼1.6- to 2.5-fold. These observations suggested that changes in the levels and activity of several different splicing factors may contribute to the splicing changes observed in EMT.
To explore the potential contributions of splicing factors to EMT-regulated alternative splicing, we analyzed published cross-linking/immunoprecipitation-sequencing (CLIP-Seq) data from human cell lines. Dozens of EMT-regulated skipped exons were associated with RBFOX2 CLIP-Seq clusters, and hundreds were associated with PTB CLIP-Seq clusters (Figure 2C; [27], [37]). In addition, a fraction of the observed EMT-regulated splicing events overlapped with a set of ESRP1-regulated exons recently identified by Carstens and coworkers using RNAi and a splicing-sensitive microarray analysis (Figure 3C; [28]). Together, the RNAi and CLIP-Seq data demonstrate the potential for regulation of a substantial portion – perhaps a majority of EMT-regulated exons – by these three factors. Thus, our data are consistent with a model in which several splicing factors collaborate in the regulation of splicing during EMT, adding a layer of post-transcriptional regulation to the EMT program.

EMT–associated alternative transcripts correlate with the phenotype of breast cancer cell lines
Alternatively spliced mRNA isoforms that exhibit EMT-associated changes in exon inclusion might serve as valuable prognostic markers for metastatic disease, since EMT is considered an early event in metastatic progression. As an initial step towards eventual analysis of primary human samples, we assessed alternative isoform expression in a panel of human breast cancer cell lines of luminal (generally poorly metastatic) and basal-like origin (generally aggressive and metastatic). Luminal cell lines, like MCF7 and T47D, express high levels of epithelial markers including E-cadherin, while basal-like cell lines express mesenchymal markers including N-cadherin, vimentin and fibronectin [14]. In addition, in our analysis we included two cell lines – derivatives of MDA-MB-231 cell metastases to the brain and bone – that exhibited a more aggressive phenotype compared to the parental MDA-MB-231 cells [38]. We hypothesized that splicing events with high epithelial inclusion (i.e. high inclusion in the pre-EMT/epithelial sample) would be expressed in luminal breast cancer cell lines, and conversely that splicing events with high mesenchymal inclusion (defined analogously) would be expressed in basal-like cell lines. A quantitative RT-PCR (qRT-PCR) analysis of nine skipped exons demonstrating the largest ΔΨ in the validated set of 37 alternative splicing events, using cDNA from the panel of luminal and basal-like cell lines, indicated that four epithelial inclusion events, in the SLC37A2, KIF13A, FLNB, and MBNL1 genes, were included at high frequency in luminal cell lines, whereas inclusion of these events was low in basal-like cells compared to T47D epithelial cells (Figure 3A). Conversely, five mesenchymal-enriched inclusion events in the PLEKHA1, MLPH, ARHGEF11, CLSTN1 and PLOD2 genes were enriched in basal-like cell lines with only low inclusion levels in luminal cells relative to BT549 mesenchymal cells (Figure 3B), consistent with recently published results [28]. Thus, taken together, epithelial inclusion events were identified in corresponding mRNA transcripts in luminal cells and were detected at very low levels in basal-like cells, while mesenchymal inclusion events were detected at low levels in luminal cells but showed a high inclusion ratio in basal-like cells (Figure 3C, 3D). Therefore, the qRT-PCR analysis of skipped exons using cDNA from a panel of luminal and basal-like breast cancer cell lines detected EMT-associated splicing events, as predicted by the RNA-seq analysis of Twist-induced EMT.
To explore the expression of EMT-associated alternative splicing events in breast cancer cell lines further and to determine whether EMT-associated alternative exons could classify breast cancer cell line subtypes, we compared the expression of skipped exons (SEs) from our EMT RNA-seq analysis to available exon array data from luminal and basal B breast cancer cell lines in the NCI-60 panel [39]. Unsupervised hierarchical clustering of exon array data relating to 307 EMT-associated SE events (|ΔΨ|>0.1, FDR<0.05; foreground set) detected by the array, segregated basal B cell lines from luminal cell lines with only two basal cell lines (MDA-MB-436 and SUM149) misclassified in the luminal cluster (Figure 4A). In contrast, clustering of the exon array data using the background set of 8839 events resulted in cell line subtype classification with nine misclassifications, indicating lack of intrinsic bias in the whole set of analyzed events and that the SE events identified by our EMT RNA-Seq better classify the luminal and basal B cell lines. Furthermore, a randomized-clustering procedure demonstrated that the clustering classification using our set of SE events was statistically significant (p-value = 0.0014). Therefore, the EMT-associated splicing program identified by our RNA-seq analysis is conserved in breast cancer cell lines and correlates with their invasive and metastatic properties.
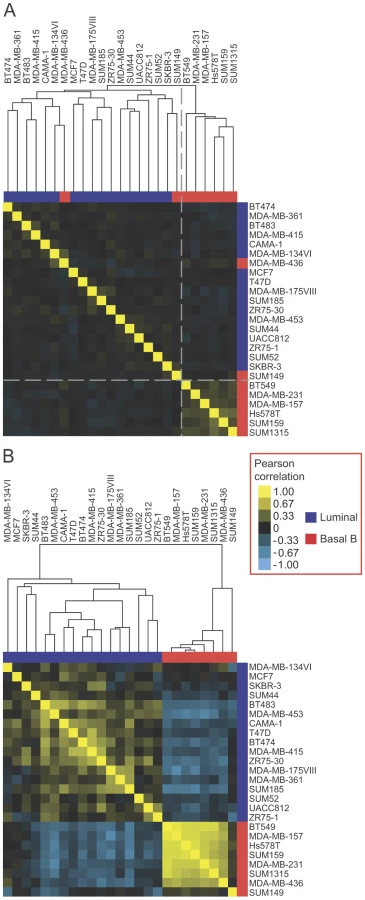
We hypothesized that some of the heterogeneity in splicing observed across the cell lines stemmed from cell type specific splicing events that may not be linked directly to EMT regulation (Figure 4A). To find the “core” EMT alternative splicing signature that can unambiguously distinguish between breast cancer cell line subtypes, we compared EMT-driven SE events to the SE events that were differentially regulated between the luminal and basal B cell lines. Of the SE events represented on the array that changed significantly in our EMT RNA-Seq dataset (|ΔΨ|>0.1, FDR<0.05), a total of 24 events changed significantly between luminal and basal B cell lines at an FDR<0.25. Of these, 19 (79%) changed in a “coherent” manner in the sense that the change in exon inclusion was in the same direction between mesenchymal and epithelial samples in the EMT RNA-seq dataset as between basal B and luminal cell lines in the exon array dataset (Figure S4). Interestingly, coherence increased for events that changed more dramatically in the EMT RNA-Seq dataset, with 11 (100%) of SE events (RNA-seq |ΔΨ|>0.3) exhibiting coherence between the two datasets (Figure S4). Notably, clustering analysis of luminal and basal B breast cancer cell lines using 19 coherent SE events demonstrated that luminal cell lines could be unambiguously distinguished from basal B cell lines based exclusively on these splicing events alone (Figure 4B). These “core” EMT-associated alternative splicing events may comprise a common program that contributes to the phenotypic changes that endow cancer cells with invasive and metastatic capabilities.
Alternative isoforms detected in the in vitro EMT model are expressed in primary human breast cancer samples
To determine whether the alternative mRNA isoforms confirmed in human breast cancer cell lines are relevant to human disease, we assessed expression of these events in fine needle aspiration (FNA) biopsies from breast cancer patients. FNA is the least invasive available method of collecting diagnostic material from patients with breast mass. This procedure is performed using a small gauge needle that gently disrupts the tissue and allows loose tumor cells to travel up the needle via capillary action. The FNA sample is usually enriched in tumor cells and can be analyzed by qRT-PCR [40]. However, due to the small volume of the sample, RNA recovery is low – tens of nanograms of total RNA at most. As expected, a subset of patient FNA spreads contained tumor cells that appeared cohesive and tightly attached to each other, typical of benign ductal lesions, while another subset of FNA smears from invasive ductal carcinomas (IDCs) contained discohesive populations of enlarged tumor cells (Figure 5A), typical for a highly invasive phenotype. Analysis of 15 random FNA smears from IDCs used in this study for the percentage of tumor, inflammatory and stromal cells demonstrated an almost a complete absence of adipocytes, macrophages and inflammatory cells (Figure S5), indicating that all of the cells present in FNA samples were ductal cancer cells. Therefore, the phenotypic characteristics of FNA collected samples indicated that they represent an appropriate human sample for assessment of alternative mRNA transcript expression found in our in vitro screen for EMT-associated splicing.
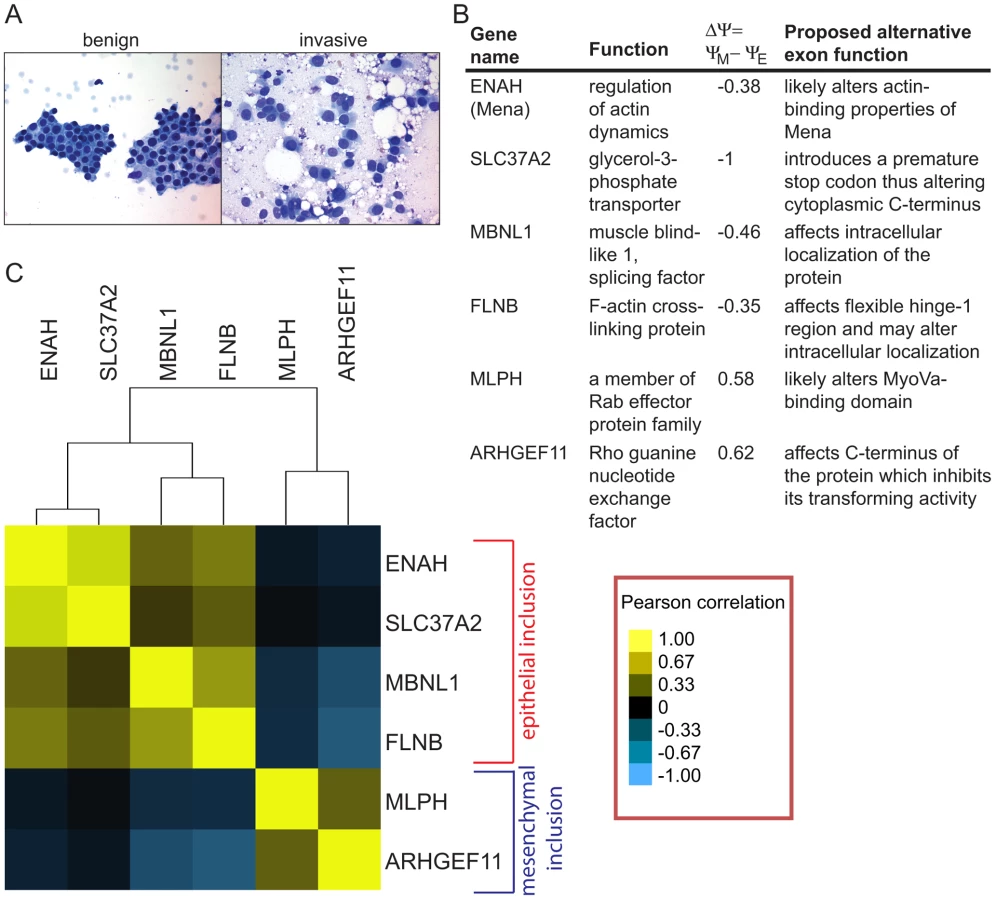
To check expression of alternative mRNA isoforms, we obtained FNA samples from 40 patients with IDCs of various grades and growth hormone receptor status. IDCs in patients were classified as well, moderately or poorly differentiated according to the modified Bloom Richardson scale. The clinical and demographic data including patients' age, tumor size, lymph node status, estrogen, progesterone and Her2/neu receptor status were also collected (Table S5). Using the cDNA from 40 IDC samples, we determined inclusion ratios for six SE events that exhibited the largest change in exon inclusion levels based on the analysis of breast cancer cell lines. These included epithelial inclusion events in ENAH, MBNL1, FLNB and SLC37A2, and mesenchymal inclusion events in MLPH and ARHGEF11 (Figure 5B). The small amount of RNA isolated from FNA samples permitted analysis of only six alternative splicing events per sample. Inclusion ratios of splicing events in IDC samples were normalized to the average inclusion ratio of the same splicing event measured in six fibroadenoma (FA) samples. For each pair of splicing events, the Pearson correlation between normalized inclusion ratios of the two splicing events across 40 IDC samples was calculated and used for clustering analysis to assess the relationships between events (Figure 5C). Interestingly, ENAH and SLC37A2 as well as MLPH and ARHGEF11 inclusion events were highly correlated. Some epithelial and mesenchymal inclusion events were inversely correlated, e.g., increases in FLNB inclusion tended to be associated with decreases in inclusion of the ARHGEF11 alternative exon. Little or no correlation was observed between SLC37A2 and MLPH inclusion events. Overall, many IDCs expressed the mesenchymal mRNA isoforms, indicating that EMT-associated splicing occurs in human tumors in vivo.
Unsupervised clustering of splicing ratios of six alternative exons in 34 FNA samples demonstrated a significant correlation between the two mesenchymal markers, MLPH and ARHGEF11, and between the four epithelial markers, ENAH, SLC37A2, FLNB and MBNL1, while epithelial and mesenchymal marker groups exhibited anti-correlation (Figure S6A). Approximately Unbiased (AU) p-values obtained from the Pvclust analysis (http://www.is.titech.ac.jp/~shimo/prog/pvclust/) were >99%, thus supporting reliability of the clustering tree (Figure S6B). This result suggests that the IDC samples tended to have either epithelial or mesenchymal splicing patterns but rarely exhibited mixed inclusion patterns, indicating that IDCs could be unambiguously classified into two groups on this basis.
The ESRP1 splicing factor confers epithelial-like properties to mesenchymal cells
By far the most strongly downregulated RBPs in EMT were the related factors ESRP1 and ESRP2 (Figure 2B; Table S4). These factors have been proposed to promote an epithelial phenotype by facilitating epithelial-specific splicing of a number of genes, some of which have well documented and essential roles in EMT [24], [41]. Silencing of ESRP1/2 in epithelial cells induced N-cadherin expression without affecting E-cadherin levels and led to a slight, but significant, increase in the rate of monolayer wound healing [28]. We hypothesized that expression of ESRP1 in mesenchymal cells would convert a portion of the mesenchymal splicing program to an epithelial state and allow us to examine the role of alternative splicing in the context of Mesenchymal-to-Epithelial Transition (MET). We introduced ESRP1-EGFP into HMLE/pBP-Twist cells, immortalized human mammary epithelial cells that ectopically express Twist [18], and analyzed expression of canonical EMT markers. As expected, control HMLE/pBP epithelial cells expressed high levels of E-cadherin while HMLE/pBP-Twist mesenchymal cells expressed high levels of N-cadherin (Figure 6A; [18]). Expression of ESRP1 in HMLE/pBP-Twist cells was sufficient to switch ENAH splicing to an epithelial pattern, as evident by the inclusion of epithelial-specific 11a exon of ENAH (Figure 6A). However, ESRP1-expressing cells still had high levels of N-cadherin and low levels of E-cadherin. Thus, ESRP1 expression is sufficient to alter splicing of some targets but is not sufficient to alter expression of EMT markers in mesenchymal cells.
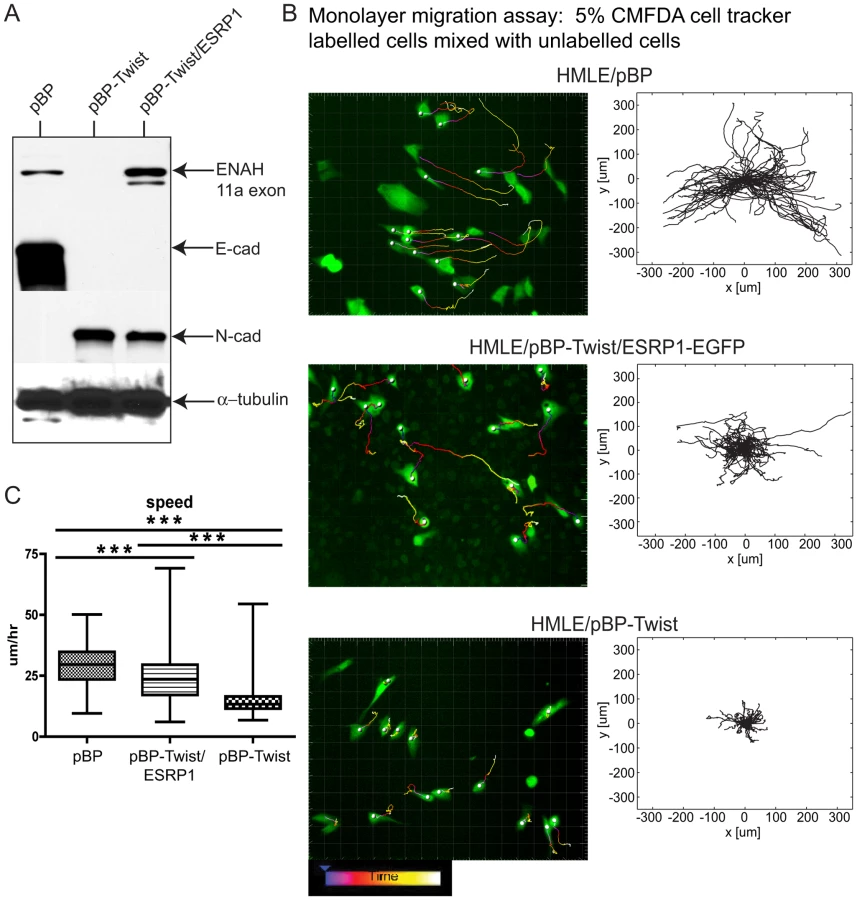
One important consequence of EMT is altered cell migration. To assess the effect of ESRP1 expression on cell migration qualitatively, we analyzed cell the movement of cells migrating out of a matrigel drop by time-lapse microscopy. This assay is similar to a standard ex vivo EMT assay used in the studies of developmental EMT to assess cell migration of endocardial cushion explants [42]. Cells were reconstituted in a small volume of matrigel and allowed to migrate out of the cell-matrigel drop for 24 hrs (Figure S7). Almost no difference in migration was observed in 8 hrs between control epithelial cells, mesenchymal cells and the same cells expressing ESRP1. However, by 19 hrs the epithelial HMLE/pBP cells continued to migrate as an epithelial sheet, keeping in tight contact with each other, while HMLE/pBP- Twist mesenchymal cells acquired a spindle-shaped morphology, migrated as individual cells and for a longer distance than epithelial cells during the same time period (Figure S7). Interestingly, HMLE/pBP-Twist cells expressing ESRP1 became elongated but continued to move in contact with each other. These differences in migration were further manifested at 24 hrs, suggesting that ESRP1 expression conferred epithelial-like properties to the migration of mesenchymal HMLE/pBP-Twist cells (Figure S7). To analyze the migration characteristics of mesenchymal cells upon ESRP1 expression quantitatively, we utilized an “in monolayer” migration assay [43] that evaluates the movement of individual cells within a monolayer in contrast to a “sheet monolayer” motility assay which assesses collective cell migration towards an open wound [44]. Epithelial HMLE/pBP cells migrate efficiently only when plated in a monolayer in contact with other cells, while mesenchymal HMLE/pBP-Twist cell movement is attenuated by cell-cell contact (H.D. Kim, FBG and D.Lauffenburger, unpublished observations). Control HMLE/pBP epithelial cells, HMLE/pBP-Twist mesenchymal cells and HMLE/pBP-Twist cells expressing ESRP1-EGFP were labeled with whole-cell tracking dye and plated along with the equivalent unlabeled cell types such that the labeled cells represented 5% of cells within a confluent monolayer to assess migration in the presence of cell-cell contact (Figure S8A). As expected, epithelial cells exhibited significant movement in a 17 hr cell tracking experiment [43], [45], while confluent mesenchymal cells moved a little, if at all (Figure 6B; Videos S1, S2). Surprisingly, upon expression of ESRP1, mesenchymal cells demonstrated significant locomotion, bypassing their typical contact inhibition of motility and instead resembling the movement of epithelial cells in a monolayer (Figure 6B; Video S3). Windrose plots of cell movement, where all cell tracks are placed at the same starting point, clearly demonstrated the extent of motion for each cell type (Figure 6B). While many epithelial HMLE/pBP cells traversed paths of up to 300 µm in length, mesenchymal HMLE/pBP-Twist cells moved less than 100 µm. Interestingly, many ESRP1 expressing mesenchymal cells exhibited intermediate range of motion of about 200 µm (Figure 6B). Analysis of the cell movement parameters revealed that the speed of ESRP1- expressing cells was significantly increased compared to the speed demonstrated by mesenchymal cells without ectopic ESRP1 expression (Figure 6C). The total path and overall displacement of HMLE/pBP-Twist/ESRP1 cells were also increased significantly (Figure S8B). The most likely explanation of this data is that splicing changes resulting from ESRP1 expression are sufficient to shift the migration properties of mesenchymal cells towards an epithelial-like phenotype. However, we cannot rule out indirect effects or possible uncharacterized functions of ESRP1.
The actin organization and structure of cell-cell contacts have a substantial effect on the migration of cells within monolayers. To characterize phenotypic changes underlying differences in cell migration behavior of epithelial HMLE/pBP cells, mesenchymal HMLE/pBP-Twist cells, and HMLE/pBP-Twist cells expressing ESRP1, immunofluorescence analysis was used to visualize actin organization and cell-cell junctions (Figure 7A; Figure S9). As expected, three-dimensional structured illumination microscopy revealed the presence of circumferential actin belt in epithelial cells, while actin stress fibers prevailed in mesenchymal cells (Figure 7B). Interestingly, actin organization was altered in mesenchymal cells upon expression of ESRP1. While some stress fibers were present in the central part of the cell, prominent accumulation of peripheral circumferential actin, characteristic of epithelial cell morphology, was also observed. p120catenin, a marker for cell-cell adhesions, decorated areas of cell-cell contact in HMLE/pBP cells, while in HMLE/pBP-Twist cells p120catenin localization was barely visible at cell contact points and could be observed only in areas where adjacent cells overlapped without forming obvious junctions (Figure 7B). Expression of ESRP1 led to increased recruitment of p120catenin to the sites of cell-cell adhesion (Figure 7B). The tight junction marker ZO-1 as well as alpha-catenin localized to actin filaments that perpendicularly terminated at cell-cell borders in immature cell-cell junctions of epithelial cells. In contrast, ZO-1 and alpha-catenin localized to the sites of focal cell-cell contact at the ends of stress fibers in mesenchymal cells (Figure 7A; Figure S9A). Interestingly, expression of ESRP1 in mesenchymal cells led to patterns of ZO-1 and alpha-catenin resembling their localization in epithelial cells. Thus, ESRP1 expression in mesenchymal cells partially reverted actin organization and cell-cell junction morphology towards the epithelial phenotype.

A defining feature of epithelia and endothelia is to separate compositionally distinct fluid phase compartments by providing a barrier to ion and solute passage, a prerequisite for the development of most organ systems in vertebrates [46], [47]. To assess whether the change in actin organization and cell-cell junction morphology in mesenchymal cells upon expression of ESRP1 would have functional consequences, we compared the ability of fluorescently tagged dextran to cross a confluent monolayer of epithelial HMLE/pBP cells, mesenchymal HMLE/pBP-Twist cells and the same cells expressing ESRP1. As expected, permeability of the HMLE/pBP-Twist cell monolayer was almost two-fold higher than permeability of the HMLE/pBP cell monolayer (Figure 7C). Strikingly, expression of ESRP1 in HMLE/pBP-Twist cells increased their barrier function significantly, resulting in permeability that was less then 1.5 fold higher than control epithelial cells (Figure 7C). Thus, expression of ESRP1 lead to a substantial increase in barrier function of mesenchymal cells, caused by the epithelial-specific splicing induced in mesenchymal HMLE/pBP-Twist cells. These results suggest that ESRP1-mediated splicing changes may drive epithelial-like re-organization of peripheral actin and cell-cell junctions that underlie barrier function.
Depletion of RBFOX2 in mesenchymal cells leads to a partial reversion towards epithelial phenotype
As noted above, our analysis along with published data [26] suggest that the RBFOX2 splicing factor likely controls a substantial subset of EMT-dependent alternative splicing (Figure 2A, 2C). To assess the effect of RBFOX2 depletion on cell phenotype, we treated HMLE/pBP-Twist mesenchymal cells with scrambled shRNA or with shRNA targeting RBFOX2. qRT-PCR analysis demonstrated ∼80% depletion of RBFOX2 mRNA (Figure S10A), while RBFOX2 protein levels became virtually undetectable (Figure S10C). RT-PCR analysis of the known RBFOX2 targets FAT and PLOD2 [26] confirmed changes consistent with depletion of RBFOX2 activity. In mesenchymal cells treated with RBFOX2 shRNA, FAT alternative exon inclusion was reduced from 40% to 5%. A less dramatic but significant effect on exon inclusion was also observed for the PLOD2 alternative exon (Figure S10B).
Expression of many EMT markers was unaffected by RBFOX2 depletion. No difference in expression was observed for N-cadherin and fibronectin compared to scrambled shRNA-treated control cells (Figure S10C). However, vimentin levels were reduced, indicating a partial loss of the mesenchymal expression program in HMLE/pBP-Twist cells upon RBFOX2 knockdown. Immunofluorescence analysis revealed that RBFOX2 depletion in mesenchymal HMLE/pBP-Twist cells shifted their morphology from spindle-shaped to cobblestone-like, resembling epithelial cell morphology (Figure S10D). Stress fibers, prominent in HMLE/pBP-Twist cells, were not readily observed after RBFOX2 depletion. Junctional markers like ZO-1, p120catenin and alpha-catenin brightly decorated cell-cell contacts, suggesting that cell junctions were formed in these cells in contrast to HMLE/pBP-Twist mesenchymal cells, where these markers were barely visible at sites of cell-cell contact (Figure S10D). Qualitative assessment of cell migration properties using a matrigel drop assay described above demonstrated that HMLE/pBP-Twist cells expressing a scrambled shRNA exhibited an individual cell migration pattern and scattered in 24 hrs of plating characteristic of mesenchymal cells. In contrast, cells expressing RBFOX2 shRNA migrated as a sheet, staying in contact with each other (Figure S10E). Together, these data suggests that, similar to ectopic ESRP1 expression, knockdown of RBFOX2 conferred a number of epithelial features to mesenchymal cells, presumably by shifting their splicing program from mesenchymal to partially epithelial.
Discussion
In the present study, we profiled the transcriptome of human mammary epithelial cells induced to undergo EMT by activation of Twist, a transcription factor important for EMT induction during embryonic development and metastasis. Using this system, we observed an EMT-associated global change in alternative splicing of a number of genes that are involved in functions crucial for EMT progression, such as cell adhesion, cell motility, and cytoskeletal remodeling. Several of the splicing changes discovered in vitro were also found to occur in a panel of breast cancer cell lines and in vivo in primary human breast cancer samples. We also demonstrated that expression of an epithelial specific splicing factor, ESRP1, was sufficient to cause a substantial shift in the actin organization, migration properties and barrier function of mesenchymal cells towards the epithelial phenotype, while depletion of the splicing factor RBFOX2 also conferred some epithelial properties to mesenchymal cells. Altogether, the present evidence leads us to propose that alternative splicing plays a major role in EMT and tumor progression by changing alternative isoform expression of genes important for epithelial and mesenchymal cell morphology and motility.
Changes in alternative splicing contribute to pathological EMT
Transcriptional regulation of EMT has been a focus of numerous studies in cancer cell lines and primary tumor samples in the last decade [15]. A number of transcription factors have been identified that repress key regulators of EMT such as E-cadherin, and induce transcription of the drivers of mesenchymal phenotype, including N-cadherin and vimentin [16], [18], [48], [49]. Changes in alternative isoform expression during EMT have been observed previously only for a handful of genes including FGFR2, p120catenin, ENAH and CD44 [22]–[25], [50]. Recently, epithelial-specific splicing factors ESRP1 and ESRP2 have been shown to regulate splicing of a subset of genes that contribute to the epithelial phenotype [28]. However, the extent to which coordinated changes in splicing might contribute to phenotypic and morphological changes during EMT has not been investigated systematically. Our results demonstrate that thousands of genes undergo changes in alternative isoform expression during EMT, establishing the existence of a program of alternative RNA processing accompanying EMT.
Many of the alternative splicing events we observed may have a major effect on protein functions important for EMT, including regulation of cell migration, cell adhesion and actin cytoskeleton remodeling (See Figure 5B; Table 1). For example, inclusion of alternative exon in the C-terminus of ARHGEF11, a Rho guanine nucleotide exchange factor (GEF) 11, also known as PDZ-RhoGEF, is increased in mesenchymal cells. Interestingly, removal of the C-terminus of ARHGEF11 results in a remarkable increase in its ability to induce RhoA activation in vivo and promotes neoplastic transformation [51]. Furthermore, components of key pathways that control cell motility, invasion and EMT itself are affected by alternative splicing (Table 1), including components of the Wnt and TGF-β signaling pathways. Some RNA regulatory proteins were also affected. For example, increased inclusion of exon 5 of the splicing factor MBNL1 was detected in epithelial cells, a change that occurs in models of myotonic dystrophy and alters the intracellular localization of the protein from cytoplasmic to nuclear [52]–[54]. Interestingly, several previously uncharacterized mRNA isoforms of genes that control important aspects of EMT have been found in this analysis. For example, a 40% increase in inclusion of a 26 aa region in SCRIB (a homolog of Drosophila scribble), involved in regulation of apical-basal polarity and directional migration of epithelial cells [55], [56], was observed in mesenchymal cells that might alter a PKC phosphorylation site. This suggests that a cDNA containing this 26 aa exon may not encode the appropriate isoform to use when studying the function of SCRIB in epithelial cells. Altogether, our analysis demonstrates that alternative splicing in EMT leads to changes in protein functions in ways that contribute to the establishment of mesenchymal phenotype, and identifies many widely studied molecules with the potential for significant isoform-dependent functions during EMT.

Could key aspects of EMT and/or MET be driven by splicing changes alone, independent of the transcriptional machinery? Recent data suggests that systemic dissemination of tumor cells occurs at early stages of tumor development [57], therefore, targeting MET therapeutically might prove more effective since at the time of diagnosis it may already be too late to successfully target EMT-inducing events. Thus we chose to assess contribution of changes in alternative splicing to MET. Our experiments with ESRP1 and RBFOX2 splicing factors suggest that epithelial splicing induced in mesenchymal cells by expression of ESRP1 or the loss of mesenchymal splicing resulting from depletion of RBFOX2 are not sufficient to convert gene expression into an epithelial pattern. However, mesenchymal cells expressing ectopic ESRP1 or depleted of RBFOX2 exhibited actin organization, barrier function and migration characteristics shifted significantly towards an epithelial phenotype, indicative of a partial MET. Our data along with other reports [58], [59] suggest that although transcriptional control is extremely important to drive EMT, alternative splicing is required to execute the complex changes needed for cells to undergo the dramatic phenotypic change from epithelial to mesenchymal states.
Comparison of EMT-dependent skipped exon events identified in the current study to ESRP-regulated ones [28], [41] revealed that out of ∼1500 EMT-dependent events (FDR<0.05), only 116 seem to be regulated by ESRP1,2 (Figure S11A). Interestingly, ESRP1 expression in clinical samples correlated with the inclusion of an alternative exon of ENAH but no correlation was observed with the presence of lymph node metastasis (Figure S11B). PTB and RBFOX2 may control a number of EMT-driven splicing events, as evident by a significant overlap between exons associated with CLIP-Seq tags and exons that undergo splicing changes during EMT. However, ESRP1, RBFOX2 and PTB together may regulate only a fraction of all EMT-associated alternative splicing (Figure 2A, 2C; Figure S11A), so it is likely that other splicing factors also play important roles in executing the EMT splicing program. Our RBP motif enrichment analysis suggests involvement of the MBNL family of splicing factors and several hnRNP proteins, including hnRNPs F/H, L and PTB. Potentially, alteration of a combination of ESRP1, RBFOX2 and/or other specific splicing factors could be sufficient to drive many phenotypic aspects of EMT. In other words, epithelial cells might potentially bypass the traditional EMT-inducing transcriptional networks to acquire mesenchymal-like phenotypes, when triggered by global changes in splicing programs that enable an EMT-like transformation. This raises the intriguing possibility that instances where invasion and metastasis occurs without changes in canonical EMT expression markers may arise from splicing-driven phenotypic changes.
EMT in primary breast cancers
Evidence for EMT in clinical carcinomas has been difficult to obtain, leading to a controversy regarding the role of EMT as a prerequisite for metastasis. Although EMT and MET have been observed in the animal model of prostate cancer [60], the presence of regions of well-differentiated epithelial morphology within some invasive primary tumors and metastatic lesions appears to conflict with a role for EMT in metastatic progression [7]. A number of factors that may account for this discrepancy have been suggested, including: 1) incomplete EMT may be sufficient for cells to metastasize; 2) EMT might only occur in a small number of cells within the tumor mass that would quickly disappear by intravasating into blood or lymphatic vessels; and, 3) after colonization, tumor cells revert to an epithelial morphology at metastatic sites through a reciprocal process of mesenchymal to epithelial transformation (MET) [13], [15]. Thus, clinical samples of primary tumor and metastatic nodules often do not show evidence of EMT because the relevant cells display a mesenchymal phenotype only when they are in transit from the primary tumor to the site of mestastasis. Moreover, if indeed only a few cells in the primary tumor undergo EMT prior to migration, RNA from these cells would be diluted by RNA from the luminal parts of the tumor in qRT-PCR analyses. FNA samples seem to be an attractive alternative to assess EMT. FNA of some IDCs, where many cells are loosely attached to the tumor mass, collect motile cells that may already be ‘in transit’ from the primary tumor to secondary sites, some of which might presumably have undergone EMT.
In our analysis of EMT-associated splicing changes in IDCs from breast cancer patients collected by FNA, we identified two groups of IDCs. In one group, inclusion of a set of epithelial splicing events was observed; while in a second group inclusion of mesenchymal splicing events was detected, suggesting a post-EMT phenotype. These data indicate that in some of the IDCs, tumor cells underwent EMT, consistent with the idea that EMT is associated with, and can contribute to cancer progression. We hypothesize that IDCs where mesenchymal splicing events were identified are more likely to metastasize than tumors exhibiting the epithelial splicing pattern, since recent studies suggest that expression of EMT program is associated with poor clinical outcome in some tumor types [61], [62].
EMT–associated alternative splicing events as potential prognostic and diagnostic markers for breast cancer metastasis
Splicing aberrations have been associated with several diseases, including cancer, where altered splicing can lead to production of protein isoforms with oncogenic properties [63]. A large-scale analysis of alternative splicing in ductal breast tumors of 600 cancer-associated genes identified 41 breast cancer-specific markers that discriminate between normal breast tissue and ductal breast tumors [64]. A number of shared splicing events have been recently demonstrated in a panel of breast and ovarian cancers using a high throughput RT-PCR approach [65]. Exon array analysis was recently used to identify subtype-specific alternative splicing events in a panel of breast cancer cell lines [26]. Therefore, it appears likely that alternative splicing analysis will dramatically increase the pool of potential biomarkers for cancer diagnostics.
Since EMT is considered an early event in the metastatic process, splicing changes associated with EMT in particular have the potential to become useful prognostic and diagnostic markers for breast cancer metastasis. Analysis of the EMT-driven splicing events in the NCI-60 panel of breast cancer cell lines [39] demonstrated that many of the EMT-associated alternative isoforms are expressed. Furthermore, luminal and basal B cell lines could be distinguished based solely on their splicing patterns, suggesting that EMT-associated alternative splicing events may serve as useful markers for classification of breast cancer cell lines and potentially of human cancers. Moreover, we identified splicing events that might be considered novel markers of EMT in vivo. Alternative splicing of ENAH, MLPH, ARHGEF11, MBNL1, FLNB and SLC37A2 transcripts have been confirmed in a number of IDC FNAs, suggesting that our EMT-associated splicing signature may have a prognostic or diagnostic potential. However, a mesenchymal splicing pattern did not correlate with the presence of lymph node metastasis. This finding is not surprising since FNA samples were obtained from recently diagnosed cancer patients and no follow up information is available regarding a possible relapse or metastatic status of the tumor. Therefore, we cannot draw a meaningful conclusion about the overall relevance of the splicing pattern in relation to outcome or presence of lymph node metastasis. Although lymph node status is considered an independent prognostic factor for relative survival of breast cancer patients (National Cancer Institute website), the presence of regional lymph node metastases does not always correlate with subsequent distant spread possibly because the mechanisms of hematogenous spread are different from those for lymphatic spread [66]. Within the clinical groups created using tumor size and lymph node involvement, there is a spectrum of disease behavior. Even patients with stage I lymph node negative breast cancer have 15–25% chance of developing distant metastasis, so breast cancers of early stage must be composed of mixed phenotypes that cannot be stratified using standard approaches such as lymph node status, or tumor size [67]. Thus, an EMT splicing signature may help to stratify early stage breast cancers, however additional studies will be required to determine the prognostic potential of EMT-associated splicing events that we have validated in FNA samples.
A growing body of evidence suggests that EMT is responsible for acquisition of therapeutic resistance by cancer cells [13]. EMT has been implicated in the generation of cancer cells with stem-like characteristics that have a high tumor-initiating potential [68]. Cancer stem cells have been found enriched in residual breast tumors after chemo- or endocrine therapy [68], in colorectal cancer cells after oxaliplatin treatment [69], or in ovarian carcinoma cells after exposure to paclitaxel [70]. Clinical evidence suggests that expression of mesenchymal markers is increased in breast tumors after letrozole or docetaxcel treatment [68]. This indicates that EMT-associated alternative splicing events that we confirmed in FNA samples from patients with IDCs may potentially become predictive biomarkers that can be used for patient selection and/or provide information early during therapy. Further studies specifically designed to identify alternative splicing markers that reflect distinct breast cancer biology in relation to clinical outcomes and prognoses show promise to improving our understanding of EMT and breast cancer at the molecular level.
Materials and Methods
Ethics statement
MIT Committee on the Use of Humans as Experimental Subjects
To: Frank Gertler
From: Leigh Firn, Chair
COUHES
date: 04/16/2009
Committee action: Exemption granted
COUHES protocol#: 0904003185
Study title: Gene Expression and splicing analysis during cancer progression.
The above referenced protocol is considered exempt after review by MIT Committee on the use of Humans as experimental subjects pursuant to Federal Regulations 45 CFR Part 46, 101(b)(4).
Cell culture
Immortalized human mammary epithelial cells (HMLEs) expressing either the empty pBabe puro vector (pBP), pBP-Twist or pWZL-Twist-ER were obtained from Robert Weinberg's laboratory at the Whitehead Institute for Biomedical Research (Cambridge, MA) and cultured as described previously [71]. 4-hydroxy tamoxifen (4-OHT) treatment was performed as described previously [29]. Other plasmids used in this study, procedures used to produce virus, the procedure for the infection of target cells, and the derivation of different cell lines are provided as Text S1.
Antibodies, Western blotting, and immunofluorescence
Cells were lysed in the presence of 50 mM Tris, pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% Na-Deoxycholate and 1.0% NP-40 on ice. Twenty micrograms of total protein from each sample were resolved on an 8%–10% SDS-PAGE Gel with Laemmli Running Buffer and transferred to PVDF membranes. The blots were then probed with various antibodies, such as anti-Mena, and anti-Mena-11a, anti-E-cadherin (BD Transduction), anti-Fibronectin (BD Transduction), anti-vimentin V9 (NeoMarkers), or anti-N-cadherin (BD Transduction). Detailed immunofluorescence methods are provided in the Text S1.
cDNA library preparation for Illumina sequencing
Total RNA was extracted from untreated HMLE/Twist-ER cells (epithelial sample) and after prolonged 4-OHT treatment (mesenchymal sample) using RNeasy Plus Mini kit (Qiagen). Poly-T capture beads were used to isolate mRNA from 10 mg of total RNA. mRNA was fragmented and used for a first-strand cDNA synthesis by random hexamer-primed reverse transcription and subsequent second-strand cDNA synthesis. Sequencing adaptors were ligated using the Illumina Genomic DNA sample prep kit. Fragments 200 bp long were isolated by gel electrophoresis, amplified by 16 cycles of PCR, and sequenced on the Illumina Genome Analyser, as described previously [20].
Computational analyses of RNA–Seq, exon array data, motif analysis, and clustering
Computational and statistical methods are described in the Text S1. Briefly, for analysis of RNA-seq data, reads were mapped to the union of the genome and a database of junctional sequences derived from AceView/Acembly annotation. Expression analysis was based on reads that were mapped to constitutive exons among annotated RefGene transcripts of each gene. Splicing analysis was based on read density supporting either isoforms of an alternative splicing event from a database of alternative isoform events. For more details see the Text S1. Alignment and raw sequencing reads were deposited in Gene Expression Omnibus with accession number GSE30290.
Reverse transcriptase PCR analysis
Total RNA for validation of splicing events in HMLE/Twist-ER cells was extracted using RNeasy Plus Mini kit (Qiagen) and reverse transcribed with Superscript II (Invitrogen). The resulting cDNA was used for 25 cycles of PCR with primers listed in the Text S1. Then samples were subjected to 10%TBE gel electrophoresis (Bio-Rad), stained with SYBR Safe DNA Gel Stain (Invitrogen), scanned (Typhoon, GE Healthcare) and quantified (ImageQuant 5.2). Total RNA from FNA samples was extracted using RNeasy Plus Micro kit (Qiagen). The resulting cDNAs were used for qPCR analysis using iQ Syber-Green Supermix (BioRad) in triplicates. qPCR and data collection were performed on iCycler (BioRad). Primer sequences used to amplify cDNAs and the detailed description of quantification analysis are listed in the Text S1.
Human tissue selection and FNA biopsy procedure
Lumpectomy and mastectomy specimens that arrive to grossing rooms at AECOM hospitals Montefiore and Weiler for pathological examination were used for tissue collection. The specimens were sectioned as usual at 0.5 or 1.0 cm intervals to locate and visualize the lesion of interest. Four to 5 FNA aspiration biopsies (passes) were performed on grossly visible lesions using 25 gauge needles. When an FNA needle is inserted into a malignant tumor it preferentially collects loose tumor cells, as can be noted on FNA obtained smears in Figure 5 and Figure S5. A small number of other cell types may also be present, most commonly inflammatory cells and macrophages. The aspirated material was collected in the cryo-vials, and to assess the adequacy of the sample, a small portion of the aspirated material was taken out of the vial, smeared on a glass slide, air-dried and stained by standard Diff-Quick protocol. The adequacy of the sample was determined by cytopathologic microscopic examination of the smears. Only samples composed of 95% of either benign or malignant epithelial cells were used in the study. Standard cytopathologic criteria such as cell size, nuclear/cytoplasmic ratio, nuclear contours, cell crowding and cohesiveness of the cells were the major criteria for classification into benign or malignant category. Samples containing a mixture of malignant and benign cells, necrotic cell debris, or more than 5% of inflammatory or stromal cells as determined by cytopathologic microscopic examination were discarded. FNA biopsy samples were immediately snap frozen in liquid nitrogen and stored frozen for RNA isolation followed by a qPCR analysis. Specimens were collected without patient identifiers following protocols approved by the Montefiore Medical Center Institutional Review Board.
Cell migration assays
Matrigel overlay assay was performed as previously described [42]. 105 cells were mixed with 3.5 mg/ml matrigel and polymerized in a drop on top of the matrigel-covered coverslip. Images of migrating cells at 0, 8 hr, 19 hrs, 24 hrs time points were obtained on a Nikon Eclipse TE200 using a 10× DIC objective. Cell migration assay was performed as previously described [43], [72]. Cells were incubated with CMFDA (Invitrogen) for 10 minutes and seeded overnight. Labeled and unlabeled cells were seeded at a 1∶20 ratio. In 24 hrs, cells were placed on an environment-controlled Nikon TE2000 microscope (Nikon Instruments; Melville, NY) and were imaged every 10-minutes for 12 hrs. Image sequences were analyzed with Bitplane Imaris software (Zurich, Switzerland) using the built-in ‘Spots’ function. 12-hour tracks were generated using the ‘Brownian Motion’ algorithm.
Permeability assay
HMLE/pBP-EGFP, HMLE/pBP-Twist-EGFP and HMLE/pBP-Twist/ESRP1-EGFP cells were seeded at confluence on polycarbonate transwell membrane inserts (3.0 µm pore size; Falcon 353492) and cultured for 3 d. 70 kD of Texas red–dextran (Invitrogen) was added to the top chamber at 2 mg/ml, and its movement into the bottom chamber was monitored over 4 hrs by spectrophotometer.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. ChristoforiG 2006 New signals from the invasive front. Nature 441 444 450
2. Vincent-SalomonAThieryJP 2003 Host microenvironment in breast cancer development: epithelial-mesenchymal transition in breast cancer development. Breast Cancer Res 5 101 106
3. YangJWeinbergRA 2008 Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell 14 818 829
4. YilmazMChristoforiG 2009 EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev 28 15 33
5. NelsonWJ 2008 Regulation of cell-cell adhesion by the cadherin-catenin complex. Biochem Soc Trans 36 149 155
6. CondeelisJPollardJW 2006 Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124 263 266
7. ChristiansenJJRajasekaranAK 2006 Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 66 8319 8326
8. AbbaMCDrakeJAHawkinsKAHuYSunH 2004 Transcriptomic changes in human breast cancer progression as determined by serial analysis of gene expression. Breast Cancer Res 6 R499 513
9. SorlieTPerouCMTibshiraniRAasTGeislerS 2001 Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 98 10869 10874
10. ThompsonEWNewgreenDFTarinD 2005 Carcinoma invasion and metastasis: a role for epithelial-mesenchymal transition? Cancer Res 65 5991 5995; discussion 5995
11. SabbahMEmamiSRedeuilhGJulienSPrevostG 2008 Molecular signature and therapeutic perspective of the epithelial-to-mesenchymal transitions in epithelial cancers. Drug Resist Updat 11 123 151
12. RubinMAPutziMMucciNSmithDCWojnoK 2000 Rapid (“warm”) autopsy study for procurement of metastatic prostate cancer. Clin Cancer Res 6 1038 1045
13. PolyakKWeinbergRA 2009 Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9 265 273
14. BlickTWidodoEHugoHWalthamMLenburgME 2008 Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis 25 629 642
15. HugoHAcklandMLBlickTLawrenceMGClementsJA 2007 Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol 213 374 383
16. ManiSAYangJBrooksMSchwaningerGZhouA 2007 Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A 104 10069 10074
17. ThisseBel MessalMPerrin-SchmittF 1987 The twist gene: isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Res 15 3439 3453
18. YangJManiSADonaherJLRamaswamySItzyksonRA 2004 Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117 927 939
19. BlencoweBJ 2006 Alternative splicing: new insights from global analyses. Cell 126 37 47
20. WangETSandbergRLuoSKhrebtukovaIZhangL 2008 Alternative isoform regulation in human tissue transcriptomes. Nature 456 470 476
21. SrebrowAKornblihttAR 2006 The connection between splicing and cancer. J Cell Sci 119 2635 2641
22. SavagnerPVallesAMJouanneauJYamadaKMThieryJP 1994 Alternative splicing in fibroblast growth factor receptor 2 is associated with induced epithelial-mesenchymal transition in rat bladder carcinoma cells. Mol Biol Cell 5 851 862
23. PinoMSBalsamoMDi ModugnoFMottoleseMAlessioM 2008 Human Mena+11a isoform serves as a marker of epithelial phenotype and sensitivity to epidermal growth factor receptor inhibition in human pancreatic cancer cell lines. Clin Cancer Res 14 4943 4950
24. WarzechaCCSatoTKNabetBHogeneschJBCarstensRP 2009 ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33 591 601
25. KeirsebilckABonneSStaesKvan HengelJNolletF 1998 Molecular cloning of the human p120ctn catenin gene (CTNND1): expression of multiple alternatively spliced isoforms. Genomics 50 129 146
26. LapukAMarrHJakkulaLPedroHBhattacharyaS Exon-level microarray analyses identify alternative splicing programs in breast cancer. Mol Cancer Res 8 961 974
27. YeoGWCoufalNGLiangTYPengGEFuXD 2009 An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat Struct Mol Biol 16 130 137
28. WarzechaCCJiangPAmirikianKDittmarKALuH An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. Embo J 29 3286 3300
29. ManiSAGuoWLiaoMJEatonENAyyananA 2008 The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133 704 715
30. ShangYHuXDiRenzoJLazarMABrownM 2000 Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103 843 852
31. Thierry-MiegDThierry-MiegJ 2006 AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol 7 Suppl 1 S12 11
32. MortazaviAWilliamsBAMcCueKSchaefferLWoldB 2008 Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 621 628
33. AudicSClaverieJM 1997 The significance of digital gene expression profiles. Genome Res 7 986 995
34. TaubeJHHerschkowitzJIKomurovKZhouAYGuptaS Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A 107 15449 15454
35. LaGambaDNawshadAHayED 2005 Microarray analysis of gene expression during epithelial-mesenchymal transformation. Dev Dyn 234 132 142
36. ThomsonSPettiFSujka-KwokIMercadoPBeanJ A systems view of epithelial-mesenchymal transition signaling states. Clin Exp Metastasis 28 137 155
37. XueYZhouYWuTZhuTJiX 2009 Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 36 996 1006
38. YonedaTWilliamsPJHiragaTNiewolnaMNishimuraR 2001 A bone-seeking clone exhibits different biological properties from the MDA-MB-231 parental human breast cancer cells and a brain-seeking clone in vivo and in vitro. J Bone Miner Res 16 1486 1495
39. RiazMElstrodtFHollestelleADehghanAKlijnJG 2009 Low-risk susceptibility alleles in 40 human breast cancer cell lines. BMC Cancer 9 236
40. MaasRABruningPFBreedijkAJTopBPeterseHL 1995 Immunomagnetic purification of human breast carcinoma cells allows tumor-specific detection of multidrug resistance gene 1-mRNA by reverse transcriptase polymerase chain reaction in fine-needle aspirates. Lab Invest 72 760 764
41. WarzechaCCShenSXingYCarstensRP 2009 The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 6 546 562
42. MoriMNakagamiHKoibuchiNMiuraKTakamiY 2009 Zyxin mediates actin fiber reorganization in epithelial-mesenchymal transition and contributes to endocardial morphogenesis. Mol Biol Cell 20 3115 3124
43. JoslinEJOpreskoLKWellsAWileyHSLauffenburgerDA 2007 EGF-receptor-mediated mammary epithelial cell migration is driven by sustained ERK signaling from autocrine stimulation. J Cell Sci 120 3688 3699
44. VitorinoPMeyerT 2008 Modular control of endothelial sheet migration. Genes Dev 22 3268 3281
45. EwaldAJBrenotADuongMChanBSWerbZ 2008 Collective epithelial migration and cell rearrangements drive mammary branching morphogenesis. Dev Cell 14 570 581
46. BaldaMSWhitneyJAFloresCGonzalezSCereijidoM 1996 Functional dissociation of paracellular permeability and transepithelial electrical resistance and disruption of the apical-basolateral intramembrane diffusion barrier by expression of a mutant tight junction membrane protein. J Cell Biol 134 1031 1049
47. TroxellMLGopalakrishnanSMcCormackJPoteatBAPenningtonJ 2000 Inhibiting cadherin function by dominant mutant E-cadherin expression increases the extent of tight junction assembly. J Cell Sci 113 Pt 6 985 996
48. BolosVPeinadoHPerez-MorenoMAFragaMFEstellerM 2003 The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: a comparison with Snail and E47 repressors. J Cell Sci 116 499 511
49. MediciDHayEDOlsenBR 2008 Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol Biol Cell 19 4875 4887
50. BrownRLReinkeLMDamerowMSPerezDChodoshLA CD44 splice isoform switching in human and mouse epithelium is essential for epithelial-mesenchymal transition and breast cancer progression. J Clin Invest
51. ChikumiHBaracABehbahaniBGaoYTeramotoH 2004 Homo- and hetero-oligomerization of PDZ-RhoGEF, LARG and p115RhoGEF by their C-terminal region regulates their in vivo Rho GEF activity and transforming potential. Oncogene 23 233 240
52. TerenziFLaddAN Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol 7
53. LinXMillerJWMankodiAKanadiaRNYuanY 2006 Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15 2087 2097
54. TerenziFLaddAN Conserved developmental alternative splicing of muscleblind-like (MBNL) transcripts regulates MBNL localization and activity. RNA Biol 7 43 55
55. PhuaDCHumbertPOHunzikerW 2009 Vimentin regulates scribble activity by protecting it from proteasomal degradation. Mol Biol Cell 20 2841 2855
56. QinYCapaldoCGumbinerBMMacaraIG 2005 The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J Cell Biol 171 1061 1071
57. HusemannYGeiglJBSchubertFMusianiPMeyerM 2008 Systemic spread is an early step in breast cancer. Cancer Cell 13 58 68
58. GhignaCGiordanoSShenHBenvenutoFCastiglioniF 2005 Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 20 881 890
59. ValaccaCBonomiSBurattiEPedrottiSBaralleFE Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J Cell Biol 191 87 99
60. OlteanSFebboPGGarcia-BlancoMA 2008 Dunning rat prostate adenocarcinomas and alternative splicing reporters: powerful tools to study epithelial plasticity in prostate tumors in vivo. Clin Exp Metastasis 25 611 619
61. LiuRWangXChenGYDalerbaPGurneyA 2007 The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med 356 217 226
62. SheridanCKishimotoHFuchsRKMehrotraSBhat-NakshatriP 2006 CD44+/CD24− breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res 8 R59
63. PajaresMJEzpondaTCatenaRCalvoAPioR 2007 Alternative splicing: an emerging topic in molecular and clinical oncology. Lancet Oncol 8 349 357
64. VenablesJPKlinckRBramardAInkelLDufresne-MartinG 2008 Identification of alternative splicing markers for breast cancer. Cancer Res 68 9525 9531
65. VenablesJPKlinckRKohCGervais-BirdJBramardA 2009 Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol 16 670 676
66. RobinsonBDSicaGLLiuYFRohanTEGertlerFB 2009 Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin Cancer Res 15 2433 2441
67. HeimannRHellmanS 2000 Clinical progression of breast cancer malignant behavior: what to expect and when to expect it. J Clin Oncol 18 591 599
68. CreightonCJLiXLandisMDixonJMNeumeisterVM 2009 Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A 106 13820 13825
69. YangADFanFCampERvan BurenGLiuW 2006 Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 12 4147 4153
70. KajiyamaHShibataKTerauchiMYamashitaMInoK 2007 Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 31 277 283
71. ElenbaasBSpirioLKoernerFFlemingMDZimonjicDB 2001 Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev 15 50 65
72. KimHDGuoTWWuAPWellsAGertlerFB 2008 Epidermal growth factor-induced enhancement of glioblastoma cell migration in 3D arises from an intrinsic increase in speed but an extrinsic matrix- and proteolysis-dependent increase in persistence. Mol Biol Cell 19 4249 4259
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2011 Číslo 8
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- An EMT–Driven Alternative Splicing Program Occurs in Human Breast Cancer and Modulates Cellular Phenotype
- Chromosome Painting Reveals Asynaptic Full Alignment of Homologs and HIM-8–Dependent Remodeling of Chromosome Territories during Meiosis
- Discovery of Sexual Dimorphisms in Metabolic and Genetic Biomarkers
- Regulation of p53/CEP-1–Dependent Germ Cell Apoptosis by Ras/MAPK Signaling