
Quantitative and Molecular Genetic Analyses of Mutations Increasing Life Span
Understanding the genetic and environmental factors that affect variation in life span and senescence is of major interest for human health and evolutionary biology. Multiple mechanisms affect longevity, many of which are conserved across species, but the genetic networks underlying each mechanism and cross-talk between networks are unknown. We report the results of a screen for mutations affecting Drosophila life span. One third of the 1,332 homozygous P–element insertion lines assessed had quantitative effects on life span; mutations reducing life span were twice as common as mutations increasing life span. We confirmed 58 mutations with increased longevity, only one of which is in a gene previously associated with life span. The effects of the mutations increasing life span were highly sex-specific, with a trend towards opposite effects in males and females. Mutations in the same gene were associated with both increased and decreased life span, depending on the location and orientation of the P–element insertion, and genetic background. We observed substantial—and sex-specific—epistasis among a sample of ten mutations with increased life span. All mutations increasing life span had at least one deleterious pleiotropic effect on stress resistance or general health, with different patterns of pleiotropy for males and females. Whole-genome transcript profiles of seven of the mutant lines and the wild type revealed 4,488 differentially expressed transcripts, 553 of which were common to four or more of the mutant lines, which include genes previously associated with life span and novel genes implicated by this study. Therefore longevity has a large mutational target size; genes affecting life span have variable allelic effects; alleles affecting life span exhibit antagonistic pleiotropy and form epistatic networks; and sex-specific mutational effects are ubiquitous. Comparison of transcript profiles of long-lived mutations and the control line reveals a transcriptional signature of increased life span.
Published in the journal:
. PLoS Genet 6(7): e32767. doi:10.1371/journal.pgen.1001037
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001037
Summary
Understanding the genetic and environmental factors that affect variation in life span and senescence is of major interest for human health and evolutionary biology. Multiple mechanisms affect longevity, many of which are conserved across species, but the genetic networks underlying each mechanism and cross-talk between networks are unknown. We report the results of a screen for mutations affecting Drosophila life span. One third of the 1,332 homozygous P–element insertion lines assessed had quantitative effects on life span; mutations reducing life span were twice as common as mutations increasing life span. We confirmed 58 mutations with increased longevity, only one of which is in a gene previously associated with life span. The effects of the mutations increasing life span were highly sex-specific, with a trend towards opposite effects in males and females. Mutations in the same gene were associated with both increased and decreased life span, depending on the location and orientation of the P–element insertion, and genetic background. We observed substantial—and sex-specific—epistasis among a sample of ten mutations with increased life span. All mutations increasing life span had at least one deleterious pleiotropic effect on stress resistance or general health, with different patterns of pleiotropy for males and females. Whole-genome transcript profiles of seven of the mutant lines and the wild type revealed 4,488 differentially expressed transcripts, 553 of which were common to four or more of the mutant lines, which include genes previously associated with life span and novel genes implicated by this study. Therefore longevity has a large mutational target size; genes affecting life span have variable allelic effects; alleles affecting life span exhibit antagonistic pleiotropy and form epistatic networks; and sex-specific mutational effects are ubiquitous. Comparison of transcript profiles of long-lived mutations and the control line reveals a transcriptional signature of increased life span.
Introduction
Understanding the genetic and environmental factors affecting variation in life span and health span is of major interest for human health and evolutionary biology. As the world population ages, the incidence of age-related diseases, such as Alzheimer's disease, cancer, cardiovascular disease and Huntington's disease, is concomitantly increasing. From the evolutionary perspective, we seek to understand why aging occurs, and why there is variation in aging between and within species [1], [2].
Multiple mechanisms affecting longevity have been documented, many of which are conserved across species. Dietary restriction [3]–[6], oxidative stress [7]–[8] and insulin/IGF signaling (IIS) [9]–[17] all affect longevity. Additional processes that change with age include stress response [18], [19], telomere shortening [20] and gene silencing [21], [22]. Life span extension is often accompanied by a decline in reproduction [23]–[27], a well–known trade–off that could explain limits to life span and maintenance of genetic variation for longevity within species [28]–[30]. However, this relationship is not universal [31]–[34]. Similarly, positive correlations between life span and stress resistance [18] are not always observed [35].
Given the heterogeneity of mechanisms affecting life span and the need to understand the genetic networks underlying each mechanism as well as cross–talk between networks, there is a clear need for unbiased, genome–wide screens to identify genes and genetic networks affecting life span. Studies using microarray technology to observe changes in gene expression during normal aging or following exposure to conditions that extend or reduce life span have indeed confirmed that expression of a substantial fraction of the genome changes with age [36]–[44]. However, these analyses are correlative, and cannot distinguish between changes in gene expression that cause aging from changes in gene expression that are a consequence of aging.
Genetic screens for mutations affecting life span give unambiguous insight regarding the genes and pathways required for normal aging, as elegantly demonstrated by mutagenesis and RNAi screens in the short-lived model organism, C. elegans [45]–[49]. Genetic screens for mutations affecting life span are more laborious in longer-lived species, such as Drosophila, and consequently there have been relatively few studies reporting mutations increasing life span in this organism [50]–[54]. Here, we report the results of a screen for mutations affecting Drosophila life span, utilizing a collection of over 1,000 single, homozygous P–element insertion lines that were constructed in isogenic backgrounds [55]. We identified 58 mutations with increased longevity, only one of which is in a gene previously associated with life span. The effects of the mutations are highly sex–specific, with life span extensions ranging from 5%–33%. The mutations have pleiotropic effects on resistance to starvation stress, chill coma recovery time, and locomotion, but the pleiotropic effects are highly variable. All of the mutations associated with increased life span have at least one deleterious pleiotropic effect on stress resistance or general health, indicating the complicated mutational basis of trade–offs between putative fitness components. We performed a quantitative genetic analysis of epistasis [56]–[58] among ten of these mutations to derive genetic interaction networks, and found that epistasis is pervasive and sex–specific. Finally, we obtained whole genome transcript profiles of seven of the mutant lines and the wild type control to evaluate the biological impact of the mutant alleles [59]–[61] and derive a common transcriptional signature of increased life span.
Results
Screen for mutations affecting life span
To identify mutations affecting Drosophila life span, we quantified the life span of males and females of 1,332 homozygous P{GT1} insertion lines [55], [57], [58], [62], [63] simultaneously with their co–isogenic control lines (Table S1). Analysis of variance (ANOVA, Table 1) revealed significant variation in life span among the P–element insert lines (P<0.0001) as well as significant sex–specific effects on life span (P<0.0001). Our estimates of the broad–sense mutational heritability (HM2) and the cross–sex mutational genetic correlation (rFM±SE) of life span were HM2 = 0.557 and rFM = 0.555±0.025. Averaged over all mutations, the standardized effects (a/σP [64]) of the P–element insertions on life span were slightly negative, with a/σP = −0.41 in females and a/σP = −0.45 in males.
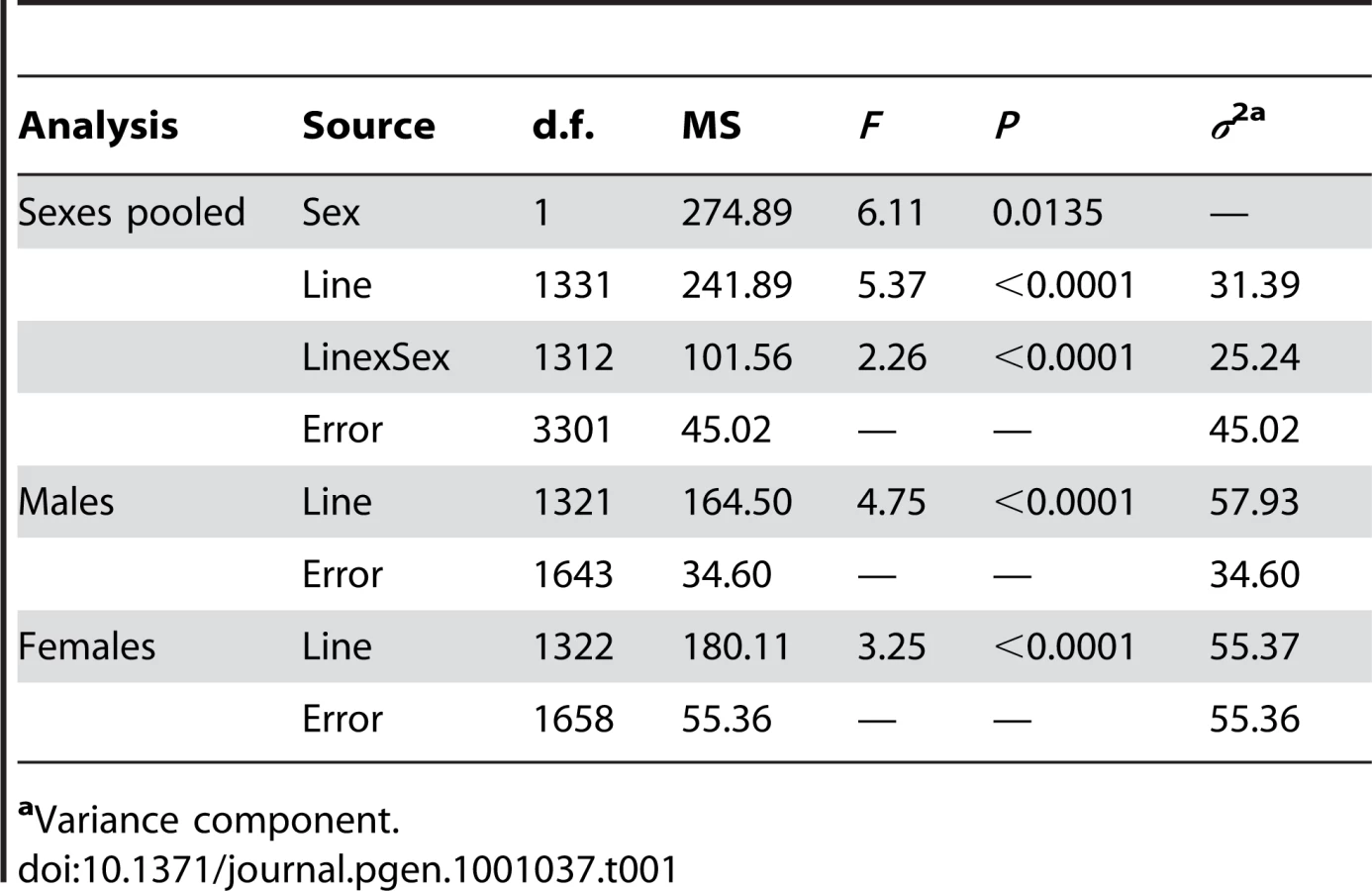
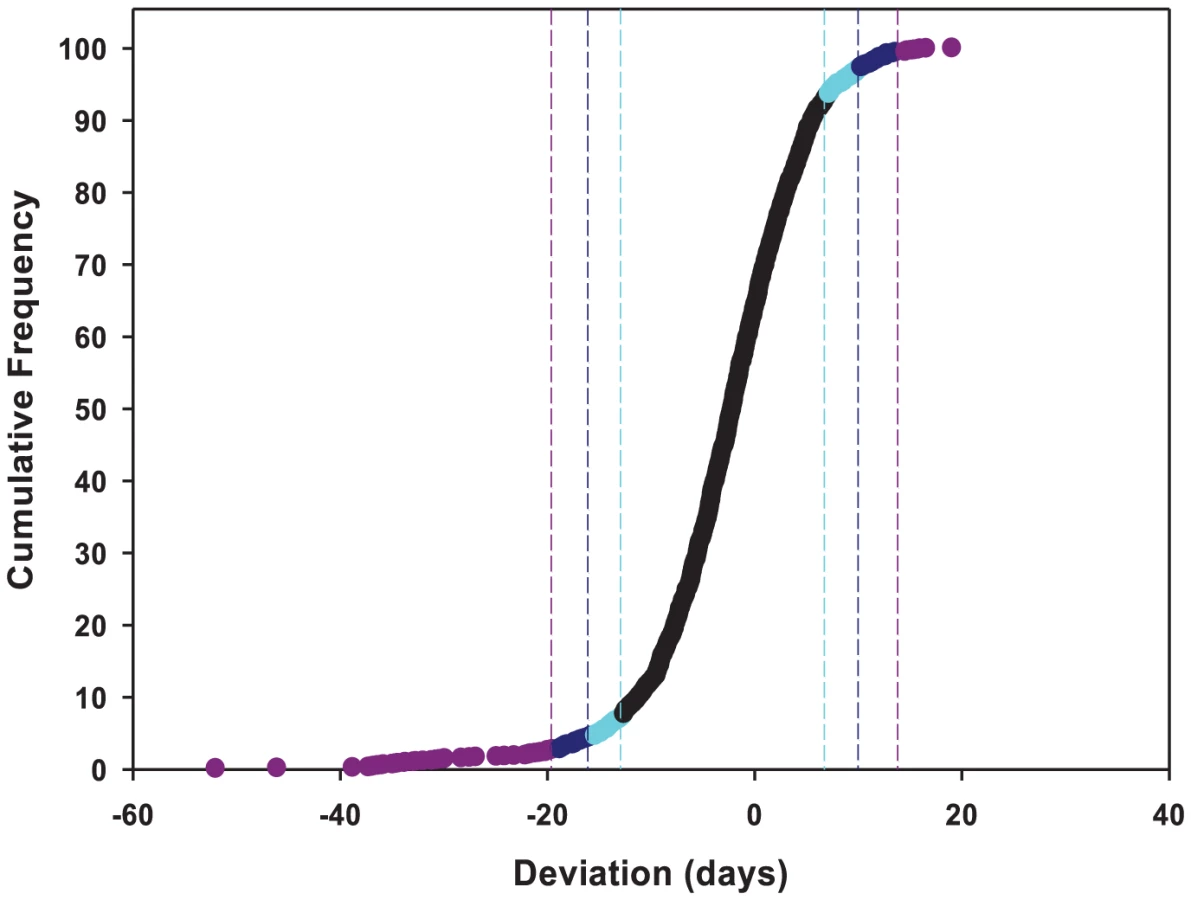
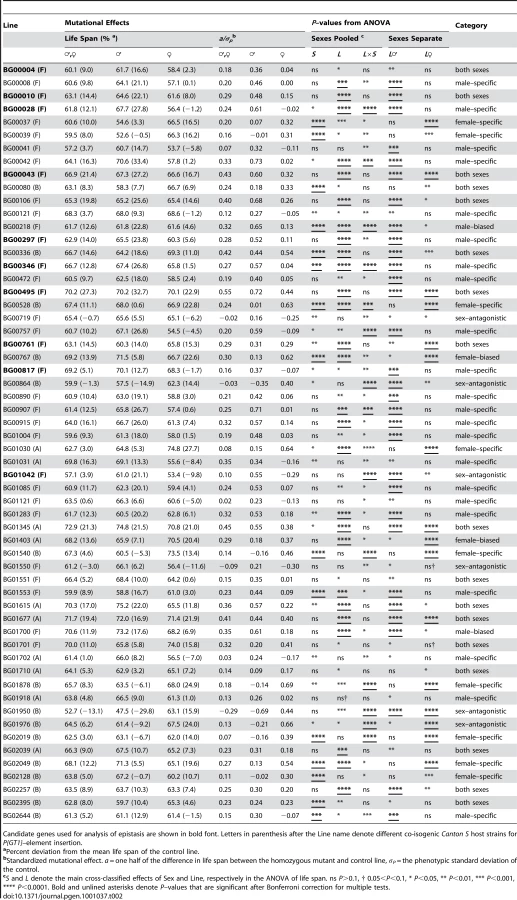
To identify the individual P–element insert lines that contributed to the significant variation in life span, we computed the confidence intervals (CIs) of deviations of line means from their corresponding controls (Figure 1), and performed Dunnett's t–tests to assess deviations of insert lines from the control line within each experimental block. Combining the results of both analyses, we identified 296 lines associated with reduced life span, 135 with increased life span, and 5 with sexually antagonistic effects on life span. At the 95%, 99% and 99.9% CIs, respectively, 139 (194), 55 (95) and 12 (49) lines had significantly increased (decreased) life span in at least one sex or averaged across sexes (Table S1). The Dunnett's tests indicated 70 (270) lines had increased (decreased) life span (P<0.05, after correction for multiple tests) (Table S1). Both analyses indicate an asymmetrical distribution of mutational effects, with more mutations decreasing than increasing longevity, as expected for components of fitness. It is generally assumed that mutations decreasing life span are less interesting than mutations increasing life span, since the former category of mutations could be generally deleterious and affect all aspects of fitness, while the latter are more likely to have specific effects on life span. Thus, we concentrated on confirming the effects of mutations associated with increased life span. We chose 83 mutations with increased life span and re-assessed their life span using larger sample sizes. We found that 58 of the 83 mutations (70%) remained formally significant for at least one sex, and 43 lines had effects that were significant following a Bonferroni correction for multiple testing (P≤0.0006) (Table 2). Thus, 4.4% of the mutations we screened are associated with increased longevity. This indicates a large mutational target for longevity and extensive pleiotropy among genes affecting life span.
Mutational effects on life span
We quantified the magnitude of the mutational effects on life span for the 58 mutations with increased life span in terms of percentage increase over the control strain, and by computing their standardized mutational effects, a/σP [64] (Table 2). The effects of the mutations on life span correspond to an average change in longevity relative to the control of 12% pooled across sexes, 17% in males and 15% in females. The average absolute value of a/σP is 0.27 pooled across sexes, 0.43 in males and 0.39 in females. Thus the average effects of P–element insertions on longevity, although statistically significant, are subtle, but effects range from 5% to as large as 33%.
We observed substantial variation in sexual dimorphism for the effects of P{GT1}–element insertions on longevity, as indicated by significant line by sex interaction terms in the ANOVAs pooled across sexes (Table 2). The cross–sex mutational genetic correlation for longevity among the 58 long-lived mutant lines was negative and significantly different from zero (rFM = −0.295±0.128, t56 = 2.308, P<0.05). Mutations associated with an increase in longevity have highly sex–specific effects, with a trend towards opposite effects in males and females.
We used the pattern of significance of the line (L) and line by sex (L×S) terms from ANOVAs comparing the life span of each long-lived mutant line to the control to infer whether the mutations affected both sexes, or were sex–specific, sex-biased, or sex antagonistic (Table 2). Mutations in 17 lines affected both sexes (the L term was significant, but the L×S term was not significant). The remaining 41 mutations (70.7%) affected males and females differently. We categorized the mutational effects as “sex–specific” if the L×S interaction from the analysis pooled across sexes was significant, and the L term from the separate sex analysis was significant only in one sex; “sex-biased” if the L and L×S terms from the analysis pooled across sexes were both significant, and the L term from the separate sex analysis was significant in both sexes; and “sex–antagonistic” if the L term from the analysis pooled across sexes was not significant, but the L×S interaction was significant, and the L term from the separate sex analysis was significant in both sexes. We found 22 male–specific, two male-biased, nine female–specific, two female-biased, and six sex–antagonistic mutations (Table 2).
Candidate genes
To identify candidate genes affecting life span, we mapped the sequences flanking the P–element insertion sites to the reference genome (Table S1, Table 3). The flanking sequences of 47 of the P–element mutations associated with increased life span mapped to unique insertion sites (Table 3). Eight of the P–element insertions were ≥2 kb from the nearest annotated gene, and either have long–range effects on the nearest neighboring gene(s) or affect an un-annotated gene in the more immediate vicinity. The remaining 39 P–element inserts were <2 kb from the nearest gene. Of these, 27 were adjacent to or within the predicted transcript of the only gene in the region, and most parsimoniously affect these genes. A total of 13 inserts were located in an intergenic region, <2 kb from each flanking gene, and could affect either or both adjacent genes.
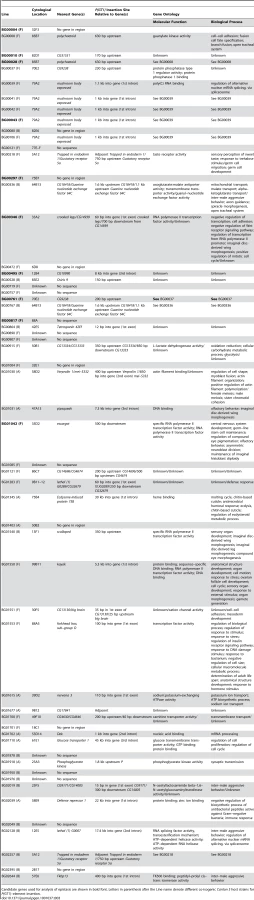
Only one of the P–element tagged candidate genes, forkhead box, sub–group O (foxo) has been previously implicated to affect adult life span [26], [34]. All others are novel candidate genes affecting longevity, and fall into a wide range of gene ontology categories, including early development, metabolism, chemosensation, immune response and transcription factors (Table 3).
Several of the P–elements inserted into identical or nearly identical positions: five inserts in the first intron of mushroom–body expressed (mub), two inserts 500 base pairs upstream of polychaetoid (pyd), two inserts adjacent to CG9238, two inserts in the Tre1/Gr5a intergenic region, and two inserts between CG8418 and Gef64C. Since this screen is far from saturation, these sites likely represent hotspots for P{GT1} element insertion [65].
The effects of multiple inserts in the same genomic region are often, but not always, heterogeneous. Two of the inserts in mub affected both sexes, two were male–specific, and one was female–specific. One insert near CG9238 was female–specific, while the other affected both sexes. One of the inserts in the CG18418/Gef64C intergenic region affected both sexes, while the other was strongly female-biased. On the other hand, both inserts in the Tre1/Gr5a intergenic region affected both sexes, and both inserts near polychaetoid were male–specific.
To add to the complexity, not all inserts in the same gene affect longevity in the same direction. The mutations in esg, pyd and mub associated with increased life span were all in the Canton S F genetic background. In addition, seven mutations in esg, two mutations in pyd, and two mutations in mub were associated with reduced life span in the initial screen. All of these mutations were in the Canton S A or B genetic backgrounds, and with one exception (mubBG02497) were in different locations from the mutations in these genes associated with increased life span (Table S1).
Analysis of revertant alleles
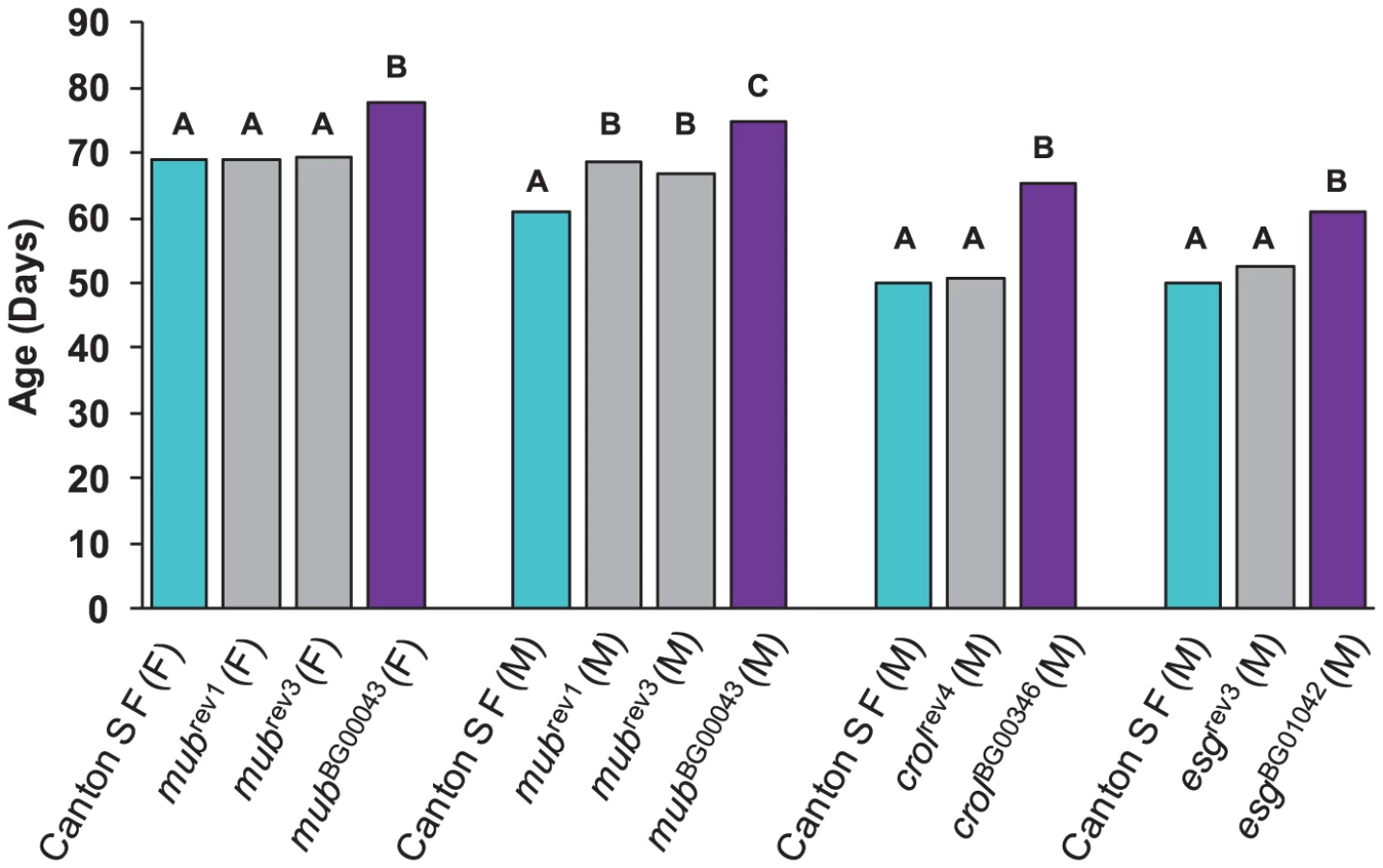
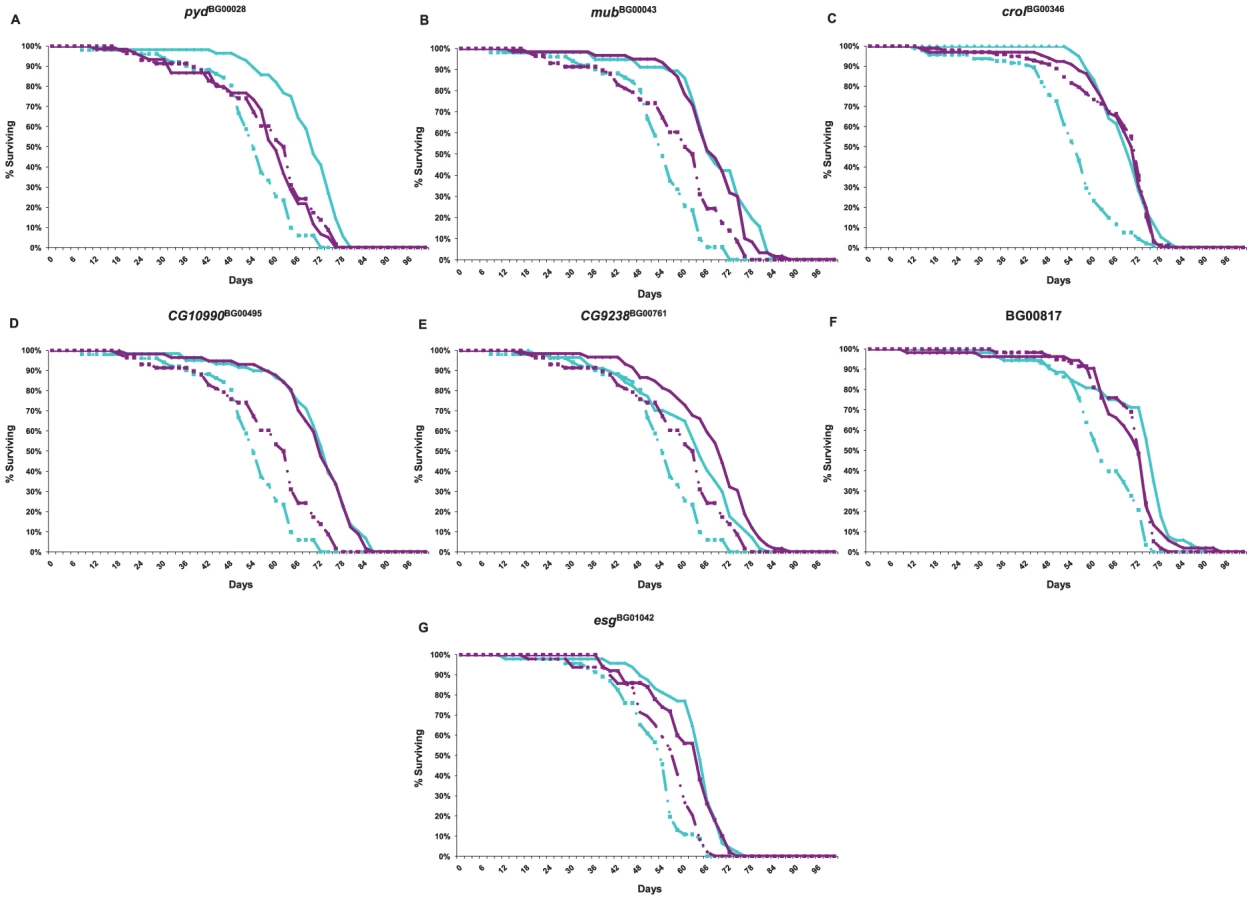
We re-mobilized the P–element insertions in three of the lines associated with increased life span (mubBG00043, crolBG00346 and esgBG01042) to create revertant alleles in which the P–element was precisely (or nearly precisely) excised, while maintaining the co–isogenic background. We measured the life span of the revertant alleles, the parental strains, and the P–element insert line simultaneously. If the disruption of the adjacent gene by the P–element insertion causes the increase in life span, we expect that the life span of the revertant alleles will not be significantly different from the control. This expectation was realized for each of the revertant alleles.
The mubBG00043 allele was associated with increased life span in both sexes. We obtained one precise revertant (mubrev1) and one imprecise revertant (mubrev3). Both revertant alleles had mean female life spans that fully reverted to the control, whereas the mean male life spans were intermediate between the control and mutant line (Figure 2). The crolBG00346 and esgBG01042 alleles were both associated with increased male life span, and the male life spans of the precise revertant alleles crolrev4 and esgrev3 were not significantly different from the control (Figure 2). These results show that the P–element mediated gene disruption is indeed responsible for the mutant phenotypes.
Epistatic interactions among mutations that increase life span
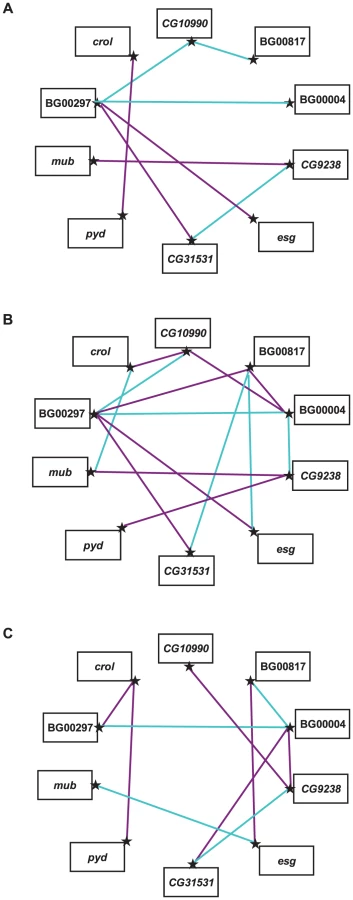
Since all independently isolated long-lived P–element insertions result in increased life span, we asked whether these genes would be part of interacting genetic networks, and, if so, to what extent such networks would differ between the two sexes. We selected 10 P–element insertion lines in the Canton S F genetic background to assess epistatic interactions affecting life span, using a half–diallel crossing scheme in which all possible double heterozygotes were constructed (without reciprocals) [56]–[58]. The mean life spans of all double heterozygote genotypes are given in Table S2. We observed significant variation in life span among the double heterozygote genotypes (P<0.0001), between males and females (P<0.0001), and the genotype by sex interaction (P<0.0001) (Table S3). The effect of double heterozygous genotype was also highly significant in the individual analyses of males and females (Table S3); however, the cross–sex genetic correlation, rFM = −0.276±0.146, is not significantly different from zero (t43 = 1.88, P>0.05). Thus, the effects of the double heterozygous genotypes on life span are independent in the two sexes.
Variation among the double heterozygote genotypes can arise from two sources: variation in mean heterozygous effects of the different mutations, and variation from epistatic interactions.
Since all mutations are in the same co–isogenic background, all genetic variation among the genotypes must be attributable to one these sources. Diallel cross analysis enables us to partition the variation among the double heterozygous genotypes into their general (GCA) and specific (SCA) combining abilities. The GCA of each mutation estimates its average dominance in combination with all other mutations. The SCA of each double heterozygous genotype is the difference in the observed life span of the genotype from that expected given the GCAs of the two parental lines. Since alleles at all other loci are fixed and homozygous, any statistically significant SCA values must be due to dominance×dominance epistasis. We found significant variation in GCA and SCA values (P<0.0001) when pooled over both sexes, as well as significant GCA×Sex and SCA×Sex interaction terms (P<0.0001), indicating sex–specific GCA and SCA effects (Table S3).
We estimated the GCA effects of each mutation and the SCA effects of all double heterozygous genotypes (Table S4). Epistatic effects can either suppress or enhance the mutant phenotype: the former occurs when the life span of a double heterozygote genotype is less than expected (closer to the wild–type, with a negative SCA), and the latter when the life span of the double heterozygote genotype is greater than expected (longer-lived, with a positive SCA). We identified eight statistically significant epistatic interactions in the analysis pooled across sexes; 10 significant interactions for females and 14 significant interactions for males (Figure 3, Table S4). The cross–sex correlation of SCA values was rFM = −0.137±0.151, which is not significantly different from zero (t43 = 0.907, P>0.05). Thus, when examined separately the two sexes displayed vastly different epistatic interactions (Figure 3, Table S4). Of the 21 significant epistatic interactions in males and/or females, only one was common to both sexes, 16 were unique to each sex, and three epistatic interactions were sexually antagonistic, with enhancing effects in one sex and suppressing effects in the other (BG00817–BG00004, BG00817–esg, BG00004–CG9238).
Pleiotropic effects of mutations increasing life span
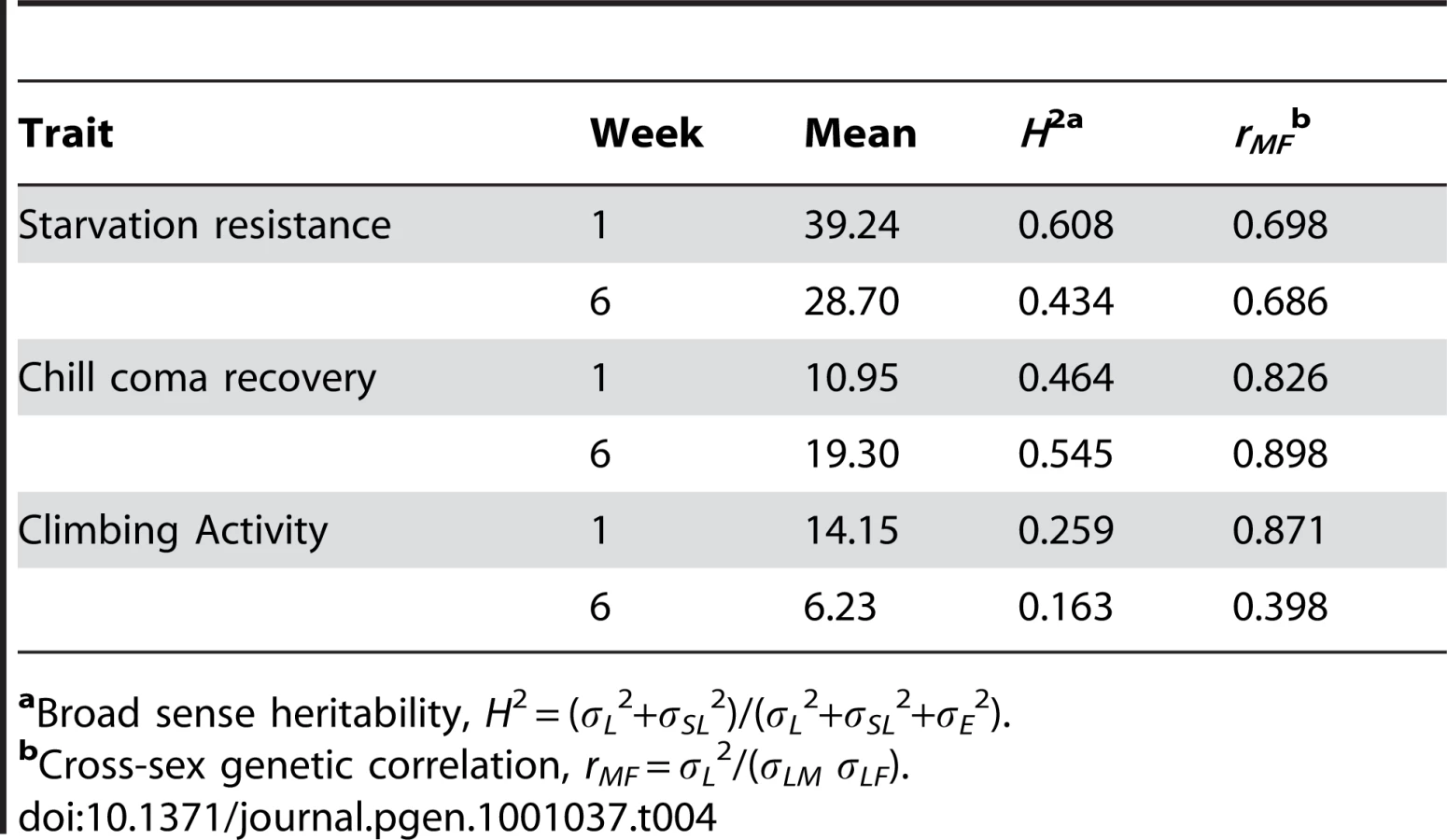
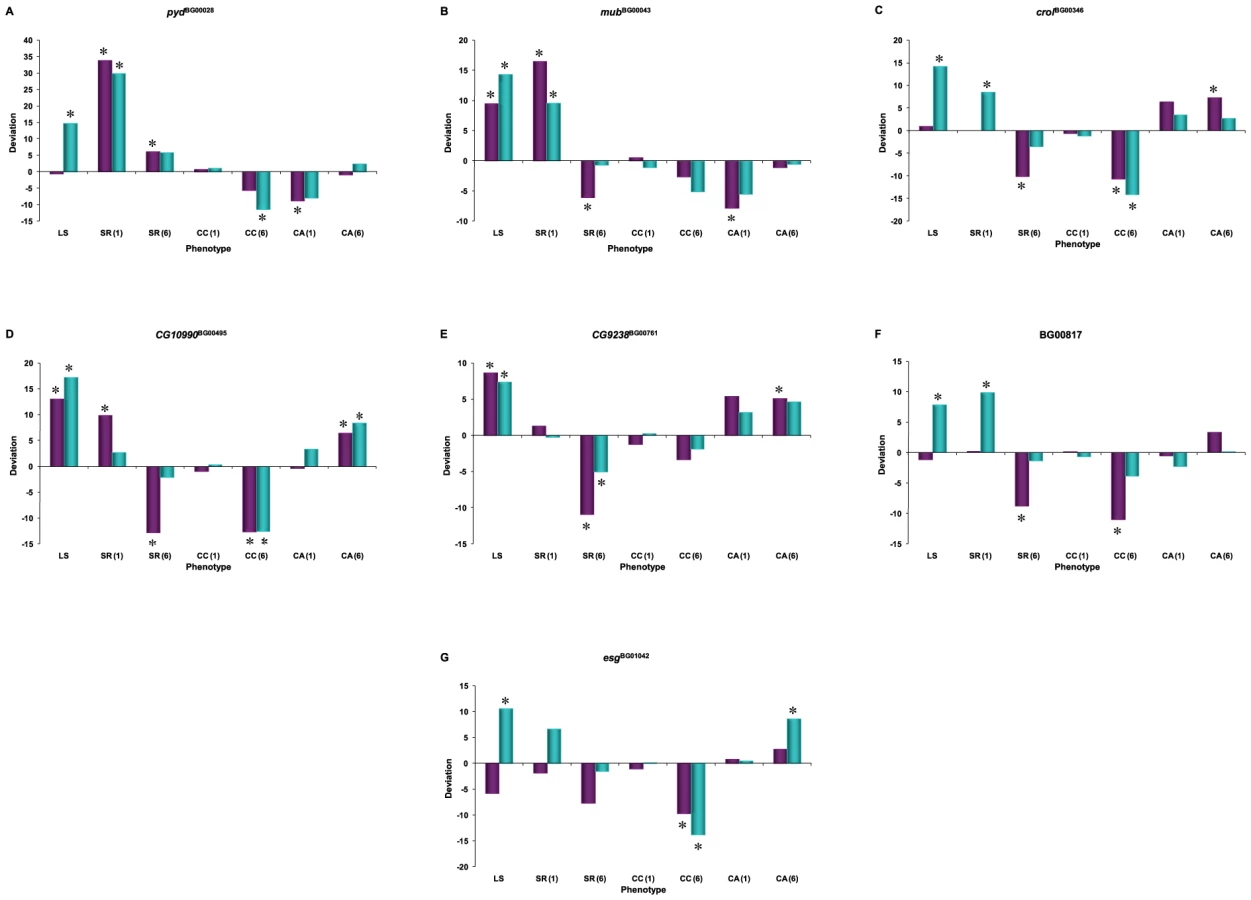
We assessed whether mutations with significantly increased life span had pleiotropic effects on stress resistance (chill coma recovery and starvation resistance) as well as a general measure of health (climbing activity) at one week and six weeks of age. We observed substantial pleiotropy. Of the 50 lines tested for starvation resistance, 44 were significantly different from the control at one week (16 with increased starvation resistance and 28 with decreased starvation resistance in one or both sexes), and 46 were significantly different from the control at six weeks (five with increased starvation resistance and 42 with decreased starvation resistance – one line had sexually antagonistic effects) (Figure 4, Table S5). Of the 50 lines tested for chill coma recovery, 32 were significantly different from the control at one week (15 with decreased chill coma recovery times and 17 with increased chill coma recovery times), and 42 were significantly different from the control at six weeks (29 with decreased chill coma recovery times and 13 with increased chill coma recovery times) (Figure 4, Table S5). We only assessed 40 of the lines for climbing ability. Of these, 23 were significantly different from the control at one week (14 with increased climbing ability and nine with decreased climbing ability), and 30 were significantly different from the control at six weeks (28 with increased climbing ability and two with decreased climbing ability) (Figure 4, Table S5). Thus, on average, by six weeks of age the lines with increased longevity have overall decreased resistance to starvation stress, but increased resistance to chill coma stress and increased general activity relative to the controls.

There was significant variation among the lines and significant sex by line interactions for all three traits (Table S6), indicating that the mutations do indeed have heterogeneous pleiotropic effects, and that the effects are sex–specific. Broad sense mutational heritabilities ranged from H2 = 0.43–0.60 for starvation resistance and chill coma recovery, but were lower for climbing ability (H2 = 0.21 averaged over week 1 and week 6 measurements) (Table 4). Although all cross–sex genetic correlations were significantly different from unity, the estimates of rMF were high for all traits except for climbing ability at six weeks (Table 4).
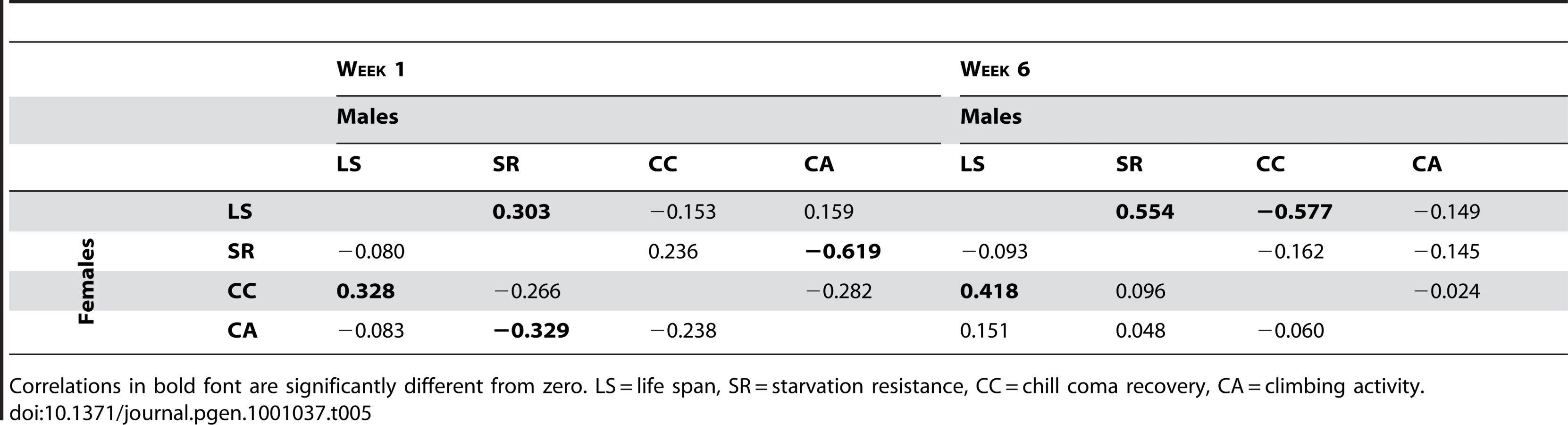
If the mutations affecting increased life span are generally more robust, we would expect positive correlations between life span and stress resistance and general health, expressed as deviations from the control. Similarly, if the mutations affecting increased life span have delayed senescence, the correlations between longevity and the other traits should be positive at six weeks of age, when the control line flies are beginning to die, but the long-lived mutant individuals are still alive. However, this was not the pattern observed. We consider the overall pleiotropic effects of the mutations separately for males and females, since the effects of the mutations on life span were not correlated between the sexes. In females, the correlation (±SE) between longevity and chill coma recovery time was positive and significant at both one week (r = 0.328±0.136, t48 = 2.41, P = 0.020) and six weeks (r = 0.418±0.131, t48 = 3.19, P = 0.0025) (Table 5). Thus, there is a tendency for mutations affecting female life span to be inversely associated with resistance to chill coma stress, at either age. The correlation between starvation resistance and climbing ability was significant and negative at one week (r = −0.329±0.153, t38 = 2.15, P = 0.038). None of the other correlations were significantly different from zero (Table 5). In males, however, the correlation (±SE) between longevity and starvation resistance was positive and significant at both one week (r = 0.303±0.138, t48 = 2.20, P = 0.038) and six weeks (r = 0.554±0.120, t48 = 4.61, P = <0.0001) (Table 5). Further, the correlation between male life span and chill coma recovery time was negative and significant at six weeks (r = −0.577±0.118, t48 = 4.90, P = <0.0001) (Table 5). Thus, mutations affecting male life span do show the expected positive associations with stress resistance and delayed senescence for stress resistance. However, the correlation between male starvation stress resistance and climbing activity was significant and negative at one week (r = −0.619±0.127, t38 = 4.86, P = <0.0001); i.e., mutations associated with increased stress resistance were less active (Table 5).
The combination of significant pleiotropy but little directional correlation between longevity and other traits indicates that the pleiotropic effects are highly variable, as illustrated in Figures S1, S2, S3, S4. The complex pattern of variation in pleiotropic effects among the P–insert lines associated with increased life span in at least one sex is depicted in Figure 4. Notably, all of the mutations are associated with at least one deleterious pleiotropic effect on stress resistance or general health, indicating the complicated mutational basis of trade–offs between putative fitness components.
Effects of mutations increasing life span on whole-genome transcript abundance
Genetic networks of mutations that affect a common phenotype can serve as focal points for the identification of additional candidate genes affecting that phenotype by transcript profiling [59]. Transcripts that are co-regulated in the genetic background of the mutant lines are themselves candidate genes affecting longevity, and the clustering of co-regulated transcripts can yield insights about the function of predicted genes tagged by the mutations. We assessed the extent to which seven of the mutations associated with increased life span (pydBG00028, mubBG00043, crolBG00346, CG10990BG00495, CG9238BG00761, BG00817 and esgBG01042) affected whole genome transcriptional regulation. We performed these analyses at six weeks of age for all mutant lines and the Canton S F co–isogenic control – the age at which the control lines are beginning to die, but at which most of the P–element insert lines remain alive, and at which we assessed differences among these lines in senescence. The survival curves for this experiment are given in Figure 5. We independently confirmed the effects of all mutations on life span, with one exception. In our initial and secondary screens, mubBG01042 females had reduced longevity, but in this assay, both males and females were long-lived.
Not all transcripts on the array are expressed in six week old adults. We eliminated all transcripts that were not considered present in both replicates of at least one line and sex, leaving 12,636 transcripts for analysis. We performed several analyses of variation of gene expression (Table S7). First, we assessed the extent to which there was variation in the main effects of sex, genotype, and the genotype by sex interaction among all lines for each transcript, using a false discovery rate criterion to account for multiple tests [66]. At a q–value of 0.001 (0.0001), we found 11,111 (10,603) sexually dimorphic transcripts. Remarkably, genotype was significant for 4,488 transcripts (35.5%) at q≤0.001, and 1,996 transcripts (15.8%) at q≤0.0001. The genotype by sex interaction was significant for 1,621 transcripts (12.8%) at q≤0.001, and 434 transcripts (3.4%) at q≤0.0001. We also ran reduced ANOVAs separately for each sex, and for each of the mutant lines compared to the control. A total of 619 and 561 transcripts were significant at q≤0.001 for females and males, respectively. The magnitude of transcriptional co–regulation varied among the mutant lines. At a significance level of q≤0.05, we observed 276 significant transcripts for CG10990BG00495; 313 for pydBG00028; 777 for CG9238BG00761; 1,815 for BG00814; 2,141 for crolBG00346; 2,193 for esgBG01042; and 3,969 for mubBG00043.
We analyzed the Biological Process Gene Ontology (GO) categories represented by the significant transcripts to determine if particular categories are over-represented. In the separate sex analyses of all genotypes, there was over–representation of significant transcripts in the DNA integration, metabolism (particularly carbohydrate metabolism) and proteolysis categories in both sexes (Table S8). Genes affecting detection of external stimuli, particularly light and abiotic stimuli, were enriched in females, while genes affecting mating and reproductive behavior and muscle development were enriched in males (Table S8).
Although all of the mutations assessed are long-lived, they have variable and sex–specific pleiotropic effects on longevity, resistance to starvation and chill coma stress, and climbing activity (Figure 4 and Figure 6). Thus, we expected to find both common and variable patterns of transcriptional co–regulation among the mutations. This is indeed what we observed.
pyd affects the biological processes of the cell–cell adhesion, fusion cell fate specification and branch fusion in the open tracheal system (Table 3). Over-represented co-regulated transcripts in the pydBG00028 mutant background fell into the categories of DNA integration; prosthetic group, pyruvate, nucleoside, lipid, chitin and glucosamine metabolism; proteolysis; and mating and reproductive behavior (Table S8).
mub is a regulator of alternative nuclear mRNA splicing via the spliceosome, and is hence likely to have far-reaching pleiotropic effects (Table 3). Categories that are over-represented among co-regulated mubBG00043 probe sets are consistent with this annotation, and include DNA replication and repair, RNA processing, the cell cycle, and chromatin modification and silencing. However, the largest over-represented categories in this mutation were in DNA, RNA, cellular and macromolecular metabolism (Table S8).
crol is an RNA polymerase II transcription factor that has pleiotropic effects on cell adhesion and proliferation, regulation of transcription, wing morphogenesis and regulation of the mitotic cell cycle and the Wnt receptor signaling pathway. Over-represented transcripts in the crolBG00346 mutation primarily affect ribosome biogenesis, histone mRNA 3′ end processing and metabolism, transcription, protein metabolism and proteolysis, sleep, and reproductive and mating behavior (Table S8).
CG10990 is a predicted gene of unknown function (Table 3). The top over-represented GO categories in CG10990BG00495 mutants are DNA integration, peptidyl–proline modification and amino acid derivative metabolism; but insulin signaling, proteolysis, and mating, reproductive and locomotor behavior are also over-represented (Table S8).
CG9238 is a predicted gene that is annotated to regulate protein phosphatase type 1 activity. Type 1 protein phosphatase is involved in the regulation of many processes so it is not that surprising that the CG9238BG00761 mutant background is over-represented in several categories, including metabolism, embryonic and larval development, as well as visual, locomotor, mating, reproductive and rhythmic (circadian) behaviors (Table S8).
We do not know the exact insertion site of the P–element in line BG00817. However, many GO categories associated with muscle development are highly over-represented among significant co-regulated transcripts in this line. Lipid catabolism, proteolysis, and lipid, carbohydrate and protein metabolism are also over-represented (Table S8).
esg is an RNA polymerase II transcription factor with pleiotropic effects on multiple biological processes: central nervous system development, germ–line stem cell maintenance, regulation of compound eye pigmentation, olfactory behavior, asymmetric neuroblast division and maintenance of imaginal histoblast diploidy. The large number of over-represented GO categories among the co-regulated transcripts in the esgBG01042 mutation is consistent with highly pleiotropic functions of esg. Genes involved in RNA processing and localization, ribosome biogenesis, RNA and DNA metabolism, primary metabolism and fertilization are over-represented. However, the most significant over-representation of co-regulated transcripts in esgBG01042 is related to vision (response to light, visual perception, phototransduction) (Table S8).
Since all seven mutant lines have increased life span relative to the control, we sought to define the transcriptional signature of increased life span from the probe sets with common patterns of co–regulation across multiple lines. A total of 553 transcripts were common to four or more of the mutant lines; of these, 187 probe sets were up-regulated relative to the control and 270 were down-regulated relative to the control (Table S9). The up-regulated probe sets are enriched for genes affecting proteolysis, whereas the down-regulated transcripts are enriched for genes affecting gene expression and RNA metabolism. However, the transcriptional signature of increased life span is most notable for the large number of computationally predicted transcripts of unknown function as well as the diversity of biological functions represented. The transcripts in common to four or more of the mutant lines are encoded by genes affecting reproduction, chemosensation, metabolism, immunity/defense response, function of the nervous system and development.
Genes that are co-regulated in the mutant backgrounds are themselves candidate genes affecting life span. Therefore, we tabulated variation in gene expression of known genes affecting life span in the mutant lines associated with increased life span (Table S10). First, five of the six focal genes for which we know the genes tagged by the P–element (pyd, mub, CG10990, CG9238 and esg) are themselves significantly differentially expressed in the analysis considering all genotypes. Three of these genes (mub, CG10990 and CG9238) are also differentially expressed relative to the control in their own mutant backgrounds. Further, mub is differentially expressed in the pydBG00028 and esgBG01042 mutant lines, and esg is differentially expressed in the pydBG00028, crolBG00346 and CG9238BG00761 mutant lines. Six additional genes in which P–element mutations were associated with increased life span in our screen were differentially regulated among the seven mutations profiled in the array analysis (CG31531, Trapped in endoderm–1, CG18418, meiotic from via Salaria 332, kayak and Dek). A further 13 genes in which P–element insertions were associated with decreased life span had differentially regulated transcripts in the mutant backgrounds (CG14478, CG31176, CG4004, CG6854, couch potato, inaF, ken and barbie, Laminin A, Lipid storage droplet–2, Malic enzyme, Protein kinase 61C, Rab23 and singed). Finally, eight genes in which mutations have been described to negatively regulate life span were also differentially co-regulated in the mutant backgrounds (I'm not dead yet, chico, Insulin–like receptor, Superoxide dismutase, Alcohol dehydrogenase, Sirt2, Vacuolar H+–ATPase SFD subunit and CTP:phosphocholine cytidylyltransferase 1).
Discussion
The mutational landscape of longevity
We performed an unbiased, forward genetic screen of 1,332 P{GT1} insertional mutations that were generated in one of six Canton S co–isogenic backgrounds for mutations affecting Drosophila longevity. In the initial screen, we identified 436 (32.7%) mutations with mean life spans that were significantly different from their co–isogenic control. Of these, 296 (67.89%) were associated with reduced life span, 135 (30.96%) were associated with increased life span, and 5 (1.15%) had sexually antagonistic effects on life span. The sample size per mutation in the initial screen was not large; therefore, many of the significant effects could be false positives. Nevertheless, if nearly one–third of mutations affect life span, the mutational target size for longevity must be large, consistent with the many mechanisms that are known to affect life span. We know the locations of the P–element inserts for 290 of the mutations associated with significant effects on longevity. Of these, 56 map to gene deserts (regions of the genome with no computationally predicted genes) and likely define novel un-annotated genes. With the exception of foxo [26], [34], none of the mutations tagged genes that have been previously associated with life span. Thus, forward genetic screens for mutations with subtle, quantitative effects on life span in a co–isogenic background is an efficient method for identifying novel genes affecting longevity and other complex traits [57], [58], [62], [63], [67].
Substantially more mutations were associated with decreased than increased life span. It is generally assumed that mutations decreasing life span are less interesting than mutations increasing life span, since the former category of mutations could be generally deleterious and affect all aspects of fitness, while the latter are more likely to have specific effects on life span. Thus, we concentrated on confirming the effects of mutations associated with increased life span with larger sample sizes in a secondary screen, and identified 58 mutations associated with increased life span. The mutations associated with significant increases in life span represent pathways known to affect life span (e.g., the insulin and metabolic pathways), as well as novel pathways involving taste, the nervous system and embryonic development.
Mutations reducing life span are typically inferred to be in genes required for normal life span; over–expression of such genes may extend longevity, as has been observed for dFOXO [26]. Conversely, mutations increasing life span are thought to be in genes that normally function to limit life span; reducing expression of these genes thus extends longevity [27], [68]. This logic presumes that all mutations in genes affecting life span have effects in the same direction. The proclivity of P–elements to insert in genomic hot–spots generated many insertions in the same genes enabled us to observe directly the distribution of mutational effects in the same genes. While many mutations in the same genes did indeed have similar effects on life span, this was not always true. Mutations in the same gene can be associated with both increased and decreased life span, often in a sex–specific manner, depending on the location and orientation of the P–element insertion, and genetic background. Examples include insertions in the Tre1/Gr5a intergenic region [63], mub, pyd and Defense repressor 1 (Dnr1) (Table 2, Table S1). These observations highlight the inaccuracy of referring to genes that are required for normal life span or that normally limit life span. Mutational analysis identifies genes that are relevant to the modulation of life span, but variable allelic effects preclude inferring directionality of wild type function.
Epistatic interactions among mutations increasing life span
Mutations in different locations in the same gene could have variable effects on longevity if they interfere with different aspects of gene regulation, or if some are in regulatory regions and others directly affect the protein. Different mutational effects could also arise due to variation in the amount of the vector inserted into the genome or by partial genomic excision during the insertion process. Variable effects of mutations in the same location and orientation but different genetic backgrounds may also be attributable to epistatic interactions with different alleles. Indeed, diallel crosses among just 10 of the mutations associated with increased life span revealed a surprisingly complex network of epistatic interactions involving all 10 mutations, suggesting pervasive epistasis between alleles affecting life span.
Mutations in the Tre1/Gr5a intergenic regions interact epistatically with mutations in genes affecting insulin signaling [63]. It will be interesting to determine to what extent the other mutations interact with components of this well-established pathway, and to what extent the effects on life span are independent of insulin signaling. Epistasis has repeatedly been observed between QTL alleles affecting variation in life span [69], [70] as well as between QTL alleles without main effects on life span [71], although the identities of the genetic loci underlying the QTLs are not known.
Further evidence for the importance of epistasis in the genetic architecture of Drosophila life span comes from observations that the effects of transgene over–expression and single mutations on longevity vary according to genetic background. The effect on increased life span of over-expressing Drosophila Superoxide dismutase was greater in the background of a relatively short-lived strain than in a long-lived strain background [72]. Similarly, the Indy mutation increased life span by 40–80% in the short-lived Shaker, Hyperkinetic and drop dead strains, but only by 15% in a strain selected for increased life span [52]. Over–expression of human SOD in Drosophila motor neurons increases life span [7], but the magnitude of the increase varies in different wild type genetic backgrounds in a sex–dependent manner [73]. Likewise, introgression of each of three morphological mutations into seven wild-derived backgrounds showed considerable background–dependent effects on life span [74]. These observations highlight the importance to assess the effects of the mutations increasing life span in a range of naturally derived genetic backgrounds, and to identify the genes with which the mutations interact.
Sex-specific effects of mutations on life span
A striking feature of our screen is that the effects of mutations increasing life span are highly sex–specific, with a low, but significant, negative cross sex–genetic correlation of rMF∼−0.3. Epistatic effects were similarly sex–specific, and in three cases the direction of the epistasis was opposite in males and females. This observation is consistent with previous studies documenting sex–specific effects on life span, beginning with Maynard Smith's [75] analysis showing that the genetic control of longevity was independent in D. subobscura males and females. More recently, QTLs affecting variation in life span between two laboratory strains, Oregon and 2b, have sex–specific effects [69], . Most studies of aging examine only one sex [80], but when both sexes are included, sex–specific mutational effects are surprisingly common. For example, the effects of mutations in the Drosophila insulin–like receptor (InR) [16], the insulin receptor substrate chico [15] and DTS–3, a gene involved in ecdysone biosynthesis [54] had female-biased or female–specific effects on life span. As noted above, over–expression of human SOD in Drosophila motor neurons in different genetic backgrounds has sex–specific effects on life span [73]. Further, the benefits of dietary restriction on increased life span of D. melanogaster are greater in females than males [81]. Conditional over–expression of both wild type and mutant p53 transgenes has sexually antagonistic effects on male and female life span that are in opposite directions depending on the developmental stage of over–expression [82].The causes of the sex–specific effects remain mysterious [80]. However, it should be noted that sex–specific effects of mutations and QTLs are a common feature of the genetic architecture of complex traits in Drosophila and other organisms [83], although such effects on life span are particularly extreme. It remains to be seen whether a common mechanism underlies sex–specificity for all traits.
Pleiotropic effects of mutations increasing life span on organismal phenotypes
The concept of trade–offs (antagonistic pleiotropy) is central to many evolutionary hypotheses for limited life span and senescence. Such trade–offs were historically envisioned to be governed by alleles with beneficial fitness effects early in life, when the force of natural selection is strong, but detrimental effects later in life, when natural selection is weak [2], [28]. Kirkwood [84] phrased this concept in terms of a physiological trade–off caused by the need to optimally allocate resources to reproduction and somatic maintenance. Support for antagonistic pleiotropy comes from quantitative genetic studies documenting negative genetic correlations between early and late fitness components [28]–[31], [85], [86]; but these negative genetic correlations are not always found [87]–[90]. Drosophila mutations affecting increased life span often exhibit antagonistic pleiotropy: mutations in chico and InR show a dwarf phenotype and have reduced fecundity [15], [16], and mutations of Indy have decreased fecundity under adult caloric restriction [91].
We have shown here that antagonistic pleiotropy is pervasive, in that all P–element insert lines associated with increased longevity were also associated with at least one deleterious pleiotropic effect on resistance to starvation stress, recovery after chill coma, and/or a general measure of health (climbing activity) at one week and/or six weeks of age (Figure 4). On average, the lines with increased longevity have overall decreased resistance to starvation stress and increased resistance to chill coma stress and increased general activity relative to the controls at six weeks of age. Mutations in genes in the insulin signaling pathway tend to have increased resistance to starvation and oxidative stress, accompanied by a trade–off in growth and fecundity [23], [25], [26], [32], [33], [92]–[94]. Thus, our observation that resistance to starvation stress actually decreases in older flies from the long-lived strains runs counter to this theme. It will be interesting in the future to assess early and late age fecundity on these mutations. However, it should be noted that the negative genetic correlation between the sexes for longevity is itself a trade–off, and that patterns of pleiotropy are different for males and females. Mutations affecting female life span have antagonistic pleiotropic effects on resistance to chill coma stress. Mutations affecting male life span have positive pleiotropic effects on resistance to starvation and chill coma stress, but there is antagonistic pleiotropy between male starvation stress resistance and climbing activity.
Pleiotropic effects of mutations increasing life span on gene expression
Whole genome expression profiling of mutations that have been derived in the same co–isogenic background is a powerful tool for identifying networks of co-regulated genes that potentially affect the trait(s) affected by the mutations. Taken at face value, our analysis of gene expression of six week old adults in seven mutant lines associated with increased life span and the control strain indicate that many genes affect life span. We identified 4,488 transcripts that were differentially expressed among all eight genotypes using a false discovery rate criterion of q≤0.001 [66]. Transcripts from many of the candidate genes identified in the P–element screen and from genes that have been previously shown to affect life span were also differentially expressed in the background of the seven focal lines. This suggests that the co-regulated genes are indeed excellent candidate genes affecting life span. The fact that transcripts of three of the focal mutations were differentially expressed in the appropriate mutant background provides independent evidence that the P–element does affect the gene in which it has inserted. Further, mutations in co-regulated genes may interact epistatically with mutations in the focal genes [59], defining genetic networks affecting longevity. The large number of co-regulated genes in each mutant background is consistent with the large number of epistatic interactions we observed among just 10 mutations associated with increased life span. The mutations in pyd, mub, crol, CG10990 and esg affected a diverse array of biological processes that were somewhat unexpected, given their functional annotations. For example, these genes were not expected a priori to affect metabolism and reproduction; yet these categories were over-represented overall. These observations suggest that these loci may interact with insulin signaling and other well-described pathways affecting life span.
Several other studies have reported whole genome changes in gene expression in aging Drosophila and C. elegans. Pletcher et al. [40] examined both aging and caloric restriction, and found considerable over–representation for biological processes involving the cell cycle, metabolism, DNA repair and replication, transcription, RNA processing, gametogenesis and perception of light. Similarly, we observed over–representation of gene ontologies for metabolism, cell cycle, mating behavior and response to light. Unfortunately, the expression data of Pletcher et al. [40] are not publicly available, precluding a direct comparison of the lists of genes that were co-regulated by mutation associated with increased life span and those implicated in the analysis of normal aging and prolonged life span through caloric restriction.
However, we were able to compare the genes that were co-regulated in the seven P–element lines associated with increased life span with the analysis of normal aging in two Drosophila strains [44]. We observe extensive overlap with the 48 candidate genes postulated by Lai et al. [44], on the basis that they exhibited significant changes in transcript abundance with age and between the two strains, and that were located in known life span QTL [74], [75], [80], [87]. Almost 23% (11) of these genes were significantly different between our genotypes at q<0.0001, 50% (24) were significant between our genotypes at q<0.001.
There was also significant overlap of the genes that were co-expressed in Drosophila mutations associated with increased life span with many of the C. elegans orthologs that were co-regulated in the long-lived daf–2 mutant background [59]. 30 of the 39 up-regulated genes and 11 of the 20 down-regulated genes identified by Murphy et al. [60] had Drosophila homologs. 30% (9) of the up-regulated genes were significant in our study at q<0.0001, and 63% (19) were significant at q<0.001. For the down-regulated genes, only 27% (3) were significant at q<0.001 (none were significant at q<0.0001). These numbers are slightly inflated as several heat shock genes in C. elegans are homologous to a single Drosophila gene, lethal (2) essential for life.
Many genes that have been previously shown to affect life span showed differential expression in the mutant lines associated with increased life span. For example, InR was down-regulated in both the CG9238 and CG10990 backgrounds, consistent with the previously observed decrease of InR expression associated with increased life span [16]. Alcohol dehydrogenase (Adh) was up-regulated in the mutant pyd, mub and esg backgrounds. Adh expression has been shown to decrease with age [37] so an increase in expression could conceivably be associated with an increase in life span. The expression of Sirt2, a member of the Drosophila Sirtuin family [95], was strongly decreased in the mub, BG00817 and esg mutant backgrounds. The mub mutant displayed an increase in Sod expression and a decrease in chico expression which mirrors previous reports of changes of the expression of these genes in association with increased life span [7], [15].
Conclusions and future prospects
We performed an unbiased, forward genetic screen for mutations affecting Drosophila longevity, and identified 58 mutations associated with increased life span. These mutations represent pathways known to affect life span (e.g., the insulin and metabolic pathways, gene silencing and immune response), as well as novel pathways involving taste and nervous system and embryonic development. Mutations in the same gene can be associated with both increased and decreased life span, which could be caused by different insertion sites or epistatic interactions with different genetic backgrounds. Pervasive epistasis for mutations affecting life span is indicated by a diallel cross analysis of ten of the mutations associated with increased life span. A striking feature of our screen is that the main and epistatic effects of mutations increasing life span are highly sex–specific. Further, antagonistic pleiotropy of mutational effects is pervasive, in that all P–element insert lines associated with increased longevity were also associated with at least one deleterious pleiotropic effect on a component of fitness. However, the patterns of pleiotropy are also sex–specific and different for males and females. The 4,488 transcripts that are differentially expressed among all eight genotypes provide a glimpse into complex genetic networks affecting longevity, which include many genes previously shown to affect life span. Further studies are required to establish that P–element disruptions of all candidate genes cause the changes in longevity and to determine interactions of these novel mutations with mutations in genes of the insulin signaling pathway and other pathways known to affect life span. The causes of the sex–specific and background–dependent epistatic effects remain to be elucidated, as do any effects on early and late reproduction, and the contribution of the novel loci to naturally occurring variation in life span – in Drosophila, and other organisms.
Materials and Methods
Drosophila stocks
The P{GT1} insertion lines [55] used in this study were constructed in six co–isogenic w1118 Canton–S backgrounds (A, B, C, D, E and F) as part of the Berkeley Drosophila Gene Disruption Project [55], and were obtained from Hugo Bellen (Baylor College of Medicine, Houston, TX). All lines were maintained at 60–80% humidity and 25°C under a 12∶12 hour light∶dark cycle.
Screen for mutations affecting life span
We screened 1,332 homozygous viable P{GT1} insertion lines for changes in life span relative to their control line. The initial screen was conducted in blocks of 50–100 insert lines and the appropriate Canton S control line. Each block was initiated with virgin males and females that had eclosed within 48 hours of each other, with two replicate vials per sex per insert line, and ten replicate vials per sex of the control line. Each replicate vial contained five flies of the same sex and 5 ml cornmeal/molasses medium. We recorded deaths every two days until all flies were dead, and transferred the flies to fresh culture medium every 1–2 days.
We evaluated mutational variation for life span using analyses of variance (ANOVAs) of the mean life span of replicate vials, expressed as deviations from the appropriate contemporaneous control means for each sex. The full two–factor mixed effects model for pooled sexes was Y = μ+L+S+L×S+ε, and the reduced model for the analysis of sexes separately was Y = μ+L+ε, where μ is the overall mean, L is the line effect (random), S is the sex effect (fixed) and ε is the environmental variance between replicate vials. We computed the mutational broad sense heritability (HM2) from the full model as HM2 = (σL2+σSL2)/(σL2+σSL2+σε2), where σL2, σSL2 and σε2 are, respectively, line, sex by line, and environmental variance components; and the cross–sex genetic correlation (rMF) as rMF = covFM/σLFσLM, where covFM is the covariance between the mean life span of males and females, and σLF and σLM are the square roots of the variance components from the separate sex analyses of females and males, respectively.
We used two methods to identify insert lines with mean life spans that were significantly different from the control. We computed the 95%, 99% and 99.9% confidence intervals of the deviations from the control mean, assuming normality, as ±zα σ/(n)½. zα is the critical value for the normal distribution (1.96, 2.575 and 3.3 respectively for the 95%, 99% and 99.9% confidence intervals). σ is the phenotypic standard deviation, estimated as (σL2+σSL2+σε2)½ for the full model and (σL2+σε2)½ for the reduced models. n is the number of replicate vials for each insert line (n = 4 in the full model and n = 2 in the reduced models). We also used the Dunnett's t–test, which corrects for multiple tests of different insert lines relative to a common control, to identify insert lines that were significantly different from the control within each block.
We re-assessed the life span of 83 lines with increased life span under the same conditions as the previous assay, but with larger sample sizes of at least 12 replicate vials per sex per line. We evaluated the significance of the difference in life span between each insert line and the control by ANOVAs pooled across sexes and for each sex separately, using the models Y = μ+L+S+L×S+R(L×S)+ε (full model) and Y = μ+L+R(L)+ε (reduced model); where μ and S are defined above; L, the fixed effect of line, is the difference between the P–element insertion line and the co–isogenic control; R is the random effect of replicate vial; and ε is the environmental variance between individuals within each replicate vial. We computed the standardized effect of each mutation as a/σP, where a is one–half the difference in mean life span between the homozygous mutant and the corresponding control line, and σP is the phenotypic variation of the control line [64].
P–element insertion sites
Bellen et al. [55] identified flanking sequences for the majority of lines using inverse PCR. We used the same technique to identify several more insertion sites. We isolated DNA from ∼25 individuals using the Puregene protocol, digested the DNA with Hinp1I and ligated it to obtain circular fragments containing both genomic and P–element DNA from both ends of the insert. We used PCR to amplify the 5′ end with oligonucleotide primers pGT1.5 (CCGCACGTAAGGGTTAATG) and pGT1.d (GAAGTTAAGCGTCTCCAGG) and the 3′ end with primers Pry1 (CCTTAGCATGTCCGTG–GGGTTTGAAT) and Pry4 (CAATCATATCGCTGTCTCACTCA), at annealing temperatures of 55°C. We sequenced the resulting products using (5′) Sp1 (ACACAACCTTTCCTCTCAA–CAA) and (3′) Spep1 (GACACTCAGAATACTATTC). We aligned the flanking sequences to the D. melanogaster genome using BLAST [68]. The inverse PCR protocol failed for lines BG00121, BG01700 and BG00817. For these lines, we determined the cytological location of the inserts by in situ hybridization to polytene chromosomes. We generated biotin-labeled probes using the BioNick Labeling System (Invitrogen) protocol, and used the Vectastain ABC kit (Vector Laboratories) for signal detection.
Revertant alleles
We generated revertant lines for two chromosome 2 insert lines (BG00346, BG01042) and one chromosome 3 insert line (BG00043), using crossing schemes that preserved the co–isogenic background of the revertant lines. To construct the chromosome 2 revertant lines, we crossed w1118; P; iso3 females to w1118; CyO/Sp; SbΔ2–3/TM6,Tb males. We mated male offspring of genotype w1118; CyO/P; SbΔ2–3/iso3 to w1118; CyO/Sp; iso3 females, and single male offspring of genotype w1118; CyO/P−; iso3 in which the P–element had excised were crossed to w1118; CyO/Sp; iso3 females. In the following generation, males and females of genotype w1118; CyO/P−; iso3 were mated inter se to make a homozygous revertant stock of genotype w1118; P−; iso3. To construct the chromosome 3 revertant lines, we crossed w1118; iso2; P females to w1118; CyO/Sp; SbΔ2–3/TM6,Tb males. We mated male offspring of genotype w1118; CyO/iso2; SbΔ2–3/P to w1118; iso2; TM3, Sb/H females, and single male offspring of genotype w1118; iso2; H/P− were crossed to w1118; iso2; TM3, Sb/H females. In the following generation, males and females of genotype w1118; iso2; TM3, Sb/P− were mated inter se to make a homozygous revertant stock of genotype w1118; iso2; P−. Here w1118, iso2 and iso3 are the three isogenic chromosomes of the Canton S F strain; P refers to the chromosome from the Canton S F strain with the P–element insertion associated with increased life span; and P− indicates a P–element excision allele.
We assessed the life span of the revertant lines simultaneously with the corresponding insert and control lines, with 12 replicates for each line and sex. The analysis of the BG00043 revertants was done in Raleigh under the same conditions described for the previous tests. The analysis of the BG00346 and BG01042 revertants was done in Moscow, Russia. We used the same ANOVA models described above for the second analysis of life span to assess differences in life span among the lines, and Tukey tests to identify significant differences between mutant, revertant and control lines.
Epistasic interactions
We evaluated epistatic interactions among 10 mutations, generated in different genes in the F background, that had increased life span relative to the Canton S F strain (BG00004, BG00010, BG00028, BG00043, BG00297, BG00346, BG00495, BG00761, BG00817, BG01042). We crossed these lines in a half–diallel crossing scheme (excluding homozygous insert lines and reciprocal crosses) to create all 45 possible double heterozygote F1 genotypes following Griffing's [96] Method 4 and Model 1. We measured the life span of each F1 genotype as described above, with eight replicate vials per genotype per sex. The general combining ability (GCA) for each mutation is the difference between the mean life span of all genotypes containing that mutation and the overall mean [97]. We estimated GCA values as GCAi = [Ti/(n−2)]−ΣT/n(n−2), where Ti is the sum of mean life spans for all genotypes with the ith mutation, ΣT is twice the sum of mean life spans of all double–heterozygote genotypes, and n is the total number of mutant lines [64]. The specific combining ability (SCA) for any particular genotype is the difference between the mean life span of the genotype and the life span expected from the sum of the GCAs of the mutants involved in the cross [97]. We estimated SCA values as SCAij = Xij−(Ti+Tj)/(n−2)+ΣT/(n−1)(n−2), where Xij is the mean life span of the offspring resulting from the cross of the ith and jth mutant lines. We also estimated GCAs and SCAs separately for each sex. We used Diallel–SAS05 [98] to estimate individual GCA and SCA effects and their standard errors; to perform ANOVAs to assess the significance of variation among the double heterozygous genotypes (G) for the full model pooled across sexes (Y = μ+G+S+G×S+R(G×S)+ε) and for the analyses of each sex separately (Y = μ+G+R(G)+ε); and to partition the G term into its GCA and SCA components, pooled across sexes (Y = GCA+SCA+S+GCA×S+SCA×S+R(S)+ε) and separately by sex (Y = GCA+SCA+R(S)+ε).
Pleiotropic effects on organismal phenotypes
We assessed pleiotropic effects of mutations with significantly increased life span on stress resistance (chill coma recovery and starvation resistance) as well as a general measure of health (climbing ability) for virgin flies at one week and six weeks of age. We tested the F lines in three blocks, the A lines in two blocks, and all B lines simultaneously. Each block included the appropriate control line. We measured chill coma recovery time and climbing ability for individuals within each block within 48 hours, and scored all individuals within a block for starvation resistance at the same time.
Chill coma recovery
We transferred 30 same–sex individuals without anesthesia into empty vials and placed the vials in chambers containing melting ice. After three hours, we transferred the vials to room temperature, and placed the flies from each vial on their backs. We scored the time it took for each individual to stand on its legs in one minute intervals, for a maximum of 30 minutes for one week old flies and 45 minutes for six week old flies. The total sample size was 30 males and 30 females per line per age. We evaluated the significance of the difference in chill coma recovery time between each insert line and the control using ANOVAs pooled across sexes (Y = μ+L+S+L×S+ε) and for each sex separately(Y = μ+L+ε).
Starvation resistance
We assessed survival time of flies in vials containing non–nutritive medium (1.5% agar in 5 ml water). The sample size was 30 males and 30 females per line per age, with 10 same–sex flies in each of three replicate vials per sex per line. We recorded survival every eight hours until all flies were dead. We evaluated the significance of the difference in survival between each insert line and the control using ANOVAs pooled across sexes (Y = μ+L+S+L×S+R(L×S)+ε) and for each sex separately (Y = μ+L+R(L)+ε).
Climbing assay
We assessed the climbing ability of 30 flies per sex per line. We placed single flies in empty glass vials (15cm high×2.5cm diameter) without medium for one hour. We then tapped the flies to the bottom of the vial to stimulate a negative geotactic climbing response, and scored the height of the fly in the vial after 10 seconds (1 height unit = 5 mm). All climbing assays were conducted between 11:00 am and 12:30 pm. We evaluated the significance of the difference in climbing activity between each insert line and the control using ANOVAs pooled across sexes (Y = μ+L+S+L×S+ε) and for each sex separately(Y = μ+L+ε).
We also assessed variation for these three traits among all the long-lived mutant lines, expressed as deviations from controls, by similar two–factor mixed effects ANOVA pooled across sexes and for sexes separately, but treating the L term as a random effect. We computed cross–sex genetic correlations as described above. We estimated Pearson's product–moment correlations among line means, expressed as deviations from the control, to quantify directional pleiotropy among the traits.
Whole-genome expression analysis
We chose seven P–element mutations in the Canton S F genetic background that were associated with increased life span in at least one sex, and for which we knew the exact P–element insertion sites (with the exception of BG00817) for whole genome expression analysis. These seven lines were a subset of the lines used for the epistasis analysis: BG00028, BG00043, BG00346, BG00495, BG00761, BG00817 and BG01042. We collected over 100 virgin flies of each sex over a 4–day interval from each of the P–element insert lines and the co–isogenic Canton S F control line, and maintained them as described for the previous life span assays. We froze 42 day–old flies, and created two replicate pools of 25 flies per sex per line for RNA extraction. We used a TRIZOL (Gibco BRL)/chloroform protocol to extract RNA from whole flies, and prepared cRNA from 5 µg RNA following the recommended protocol for eukaryotic one–cycle target labeling. We hybridized fragmented cRNA to Affymetrix Drosophila Genome 2.0 GeneChip arrays.
Analysis
For each of the 18,800 probe sets on the array there are 14 25mer perfect match (PM) oligonucleotides and 14 25mer single nucleotide mis–match (MM) pairs, with the mis–match at the 13th base. We used the weighted log(PM–MM) intensity of each probe set as the quantitative measure of expression, and scaled the expression scores to a median intensity of 500. Each probe set is identified as being present, marginal or absent. After excluding probe sets that did not have a present signal for both replicates for at least one sex and line, we retained 12,635 probe sets for analysis. We performed ANOVAs of gene expression on each probe set using the model Y = μ+L+S+L×S+ε. We considered probe sets for which the P–value for the L term exceeded a q–value threshold of q<0.0001 [66] to be significant after correction for multiple tests. We also performed reduced ANOVAs for males and females separately for the probe sets with a significant L×S term. To identify in which mutant lines gene expression was different from the control line, we used Tukey tests on probe sets for which the L or L×S terms were significant, and full model ANOVAs comparing each individual line with the control. We used χ2 tests to evaluate over– and under–representation of Gene Ontology (GO) biological process categories for probe sets with a significant L effect for all mutant lines together (q<0.0001) as well as individual mutant lines (q<0.05) and individual sexes (q<0.001). We based the expected values on the ratio of the biological process probe sets in the significant lists to the total number of biological process probe sets on the array.
Data from all arrays used in the study are located at the Gene Expression Omnibus (GEO) public data repository (GSM216501–GSM216513, GSM216515–GSM216533).
Statistics
We performed all statistical tests using SAS V8.2, V9.1 and Microsoft Office Excel. We used QVALUE software [66] to compute q–values.
Supporting Information
Zdroje
1. MedawarPB
1952 An unsolved problem of biology London HK Lewis and Co 1 24
2. WilliamsGC
1957 Pleiotropy, natural selection and the evolution of senescence. Evolution 11 398 411
3. HarrisonDE
ArcherJR
1987 Genetic differences in effects of food restriction on aging in mice. J Nutr 117 376 382
4. LakowskiB
HekimiS
1998 The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A 95 13091 13096
5. LinSJ
DefossezPA
GuarenteL
2000 Requirement of NAD and SIR2 for life–span extension by caloric restriction in Saccharomyces cerevisiae. Science 289 2126 2128
6. LinSJ
KaeberleinM
AndalisAA
SturtzLA
DefossezPA
2002 Caloric restriction extends Saccharomyces cerevisiae lifespan by increasing respiration. Nature 418 344 348
7. ParkesTL
EliaAJ
DickinsonD
HillikerAJ
PhillipsJP
1998 Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet 19 171 174
8. KharadeSV
MittalN
DasSP
SinhaP
RoyN
2005 Mrg19 depletion increases S. cerevisiae lifespan by augmenting ROS defence. FEBS Letters 579 6809 6813
9. KenyonC
ChangJ
GenschE
RudnerA
TabtiangR
1993 A C. elegans mutant that lives twice as long as wild type. Nature 366 461 464
10. Brown–BorgHM
BorgKE
MeliskaCJ
BartkeA
1996 Dwarf mice and the ageing process. Nature 384 33
11. MorrisJZ
TissenbaumHA
RuvkunG
1996 A phosphatidylinositol–3–OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382 536 539
12. KimuraKD
TissenbaumHA
LiuY
Ruvkun
1997 daf–2, an insulin receptor–like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277 942 946
13. ParadisS
RuvkunG
1998 Caenorhabditis elegans Akt/PKB transduces insulin receptor–like signals from AGE–1 PI3 kinase to the DAF–16 transcription factor. Genes Dev 12 2488 2498
14. GilEB
LinkEM
LiuLX
JohnsonCD
LeesJA
1999 Regulation of the insulin–like developmental pathway of Caenorhabditis elegans by a homolog of the PTEN tumor suppressor gene. Proc Natl Acad Sci U S A 96 2925 2930
15. ClancyDJ
GemsD
HarshmanLG
OldhamS
StockerH
2001 Extension of life–span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292 104 106
16. TatarM
KopelmanA
EpsteinD
TuMP
YinCM
2001 A mutant Drosophila insulin receptor homolog that extends life–span and impairs neuroendocrine function. Science 292 107 110
17. TatarM
BartkeA
AntebiA
2003 The endocrine regulation of aging by insulin–like signals. Science 299 1346 1351
18. RoseMR
VuLN
ParkSU
GravesJL
1992 Selection on stress resistance increases longevity in Drosophila melanogaster. Exp Gerontol 27 241 250
19. KhazaeliAA
TatarM
PletcherSD
CurtsingerJW
1997 Heat–induced longevity extension in Drosophila. I. Heat treatment, mortality, and thermotolerance. J Gerontol A Bio Sci Med Sci 52A B48 B52
20. HaussmannMF
WinklerDW
O'ReillyKM
HuntingtonCE
NisbetICT
2003 Telomeres shorten more slowly in long–lived birds and mammals than in short lived ones. Proc R Soc Lond B 270 1387 1392
21. KimS
BenguriaA
LaiCY
Jazwinski
1999 Modulation of life–span by histone deacetylase genes in Saccharomyces cerevisiae. Mol Biol Cell 10 3125 3136
22. TissenbaumHA
GuarenteL
2001 Increased dosage of a sir–2 gene extends lifespan in Caenorhabditis elegans. Nature 227 230
23. FriedmanDB
JohnsonTE
1987 A mutation in the age–1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics 118 75 86
24. FowlerK
PartridgeL
1989 A cost of mating in female fruitflies. Nature 338 760 761
25. TissenbaumHA
RuvkunG
1998 An Insulin–like signaling pathway affects both longevity and reproduction in Caenorhabditis elegans. Genetics 148 703 717
26. GiannakouME
GossM
JungerMA
HafenE
LeeversSJ
2004 Long–lived Drosophila with overexpressed dFOXO in adult fat body. Science 305 361
27. MockettRJ
SohalRS
2006 Temperature–dependent trade–offs between longevity and fertility in the Drosophila mutant, methuselah. Exp Gerontol 41 566 573
28. RoseMR
CharlesworthB
1980 A test of evolutionary theories of senescence. Nature 287 141 142
29. RoseMR
CharlesworthB
1981 Genetics of life history in Drosophila melanogaster. I. Sib analysis of adult females. Genetics 97 173 186
30. RoseMR
1984 Laboratory evolution of postponed senescence in Drosophila melanogaster. Evolution 38 1004 1010
31. PartridgeL
FowlerK
1992 Direct and correlated responses to selection on age at reproduction in Drosophila melanogaster. Evolution 46 76 91
32. GemsD
SuttonAJ
SundermeyerML
AlbertPS
KingKV
1998 Two pleiotropic classes of daf–2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150 129 155
33. HolzenbergerM
DupontJ
DucosB
LeneuveP
GeloenA
2003 IGF–1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421 182 187
34. HwangboDS
GershmanB
TuMP
PalmerM
TatarM
2004 Drosophila dFOXO controls lifespan and regulates insulin signaling in brain and fat body. Nature 429 562 566
35. HarshmanLG
MooreKM
StyMA
MagwireMM
1999 Stress resistance and longevity in selected lines of Drosophila melanogaster. Neurobiol Aging 20 521 529
36. LeeCK
KloppRG
WeindruchR
ProllaTA
1999 Gene expression profile of aging and its retardation by caloric restriction. Science 285 1390 1393
37. ZouS
MeadowsS
SharpL
JanLY
JanYN
2000 Genome–wide study of aging and oxidative stress response in Drosophila melanogaster. Proc Natl Acad Sci U S A 97 13726 13731
38. WeindruchR
KayoT
LeeCK
ProllaTA
2001 Microarray profiling of gene expression in aging and its alteration by caloric restriction in mice. J Nutr 131 918S 923S
39. LeeCK
AllisonDB
BrandJ
WeindruchR
ProllaTA
2002 Transcriptional profiles associated with aging and middle age–onset of caloric restriction in mouse hearts. Proc Natl Acad Sci U S A 99 14988 14993
40. PletcherSD
MacdonaldSJ
MarguerieR
CertaU
StearnsSC
2002 Genome–wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr Biol 12 712 723
41. LandisGN
AbduevaD
SkvortsovD
YangJ
RabinBE
2004 Similar gene expression patterns characterize aging and oxidative stress in Drosophila melanogaster. Proc Natl Acad Sci U S A 101 7663 7668
42. McCarrollSA
MurphyCT
ZouS
PletcherSD
ChinCS
2004 Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet 36 197 204
43. KimSN
RheeJH
SongYH
ParkDY
HwangM
2005 Age–dependent changes of gene expression in the Drosophila head. Neurobiol Aging 26 1083 1091
44. LaiCQ
ParnellLD
LymanRF
OrdovasJM
MackayTFC
2007 Candidate genes affecting Drosophila life span identified by integrating microarray gene expression analysis and QTL mapping. Mech Ageing Dev 128 237 249
45. DuhonSA
MurakamiS
JohnsonTE
1996 Direct isolation of longevity mutants in the nematode Caenorhabditis elegans. Dev Genet 18 144 153
46. LeeSS
LeeRYN
FraserAG
KamathRS
AhringerJ
2003 A systematic RNAi screen identifies a critical role in C. elegans longevity. Nat Genet 33 40 48
47. HamiltonB
DongY
ShindoM
LiuW
OdellI
2005 A systematic RNAi screen for longevity genes in C. elegans. Genes Dev 19 1544 1555
48. HansenM
HsuAL
DillinA
KenyonC
2005 New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet 1 e17 doi:10.1371/journal.pgen.0010017
49. CurranSP
RuvkunG
2007 Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS 3 e56 doi:10.1371/journal.pgen.0030056
50. ClarkAG
GuadalupeRN
1995 Probing the evolution of senescence in Drosophila melanogaster with P–element tagging. Genetica 96 225 234
51. LinYJ
SeroudeL
BenzerS
1998 Extended life–span and stress resistance in the Drosophila mutant methuselah. Science 282 943 946
52. RoginaB
ReenanRA
NilsenSP
HelfandSL
2000 Extended life–span conferred by cotransporter gene mutations in Drosophila. Science 290 2137 2140
53. LandisGN
BholeD
TowerJ
2003 A search for doxycycline–dependent mutations that increase Drosophila melanogaster life span identifies the VhaSFD, Sugar baby, filamin, fwd and Cct1 genes. Genome Biology 4 R28
54. SimonAF
ShihC
MackA
BenzerS
2003 Steroid control of longevity in Drosophila melanogaster. Science 229 1407 1410
55. BellenHJ
LevisRW
LiaoG
HeY
CarlsonJW
2004 The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics 167 761 781
56. FedorowiczGM
FryJD
AnholtRR
MackayTF
1998 Epistatic interactions between smell-impaired loci in Drosophila melanogaster. Genetics 148 1885 1891
57. SambandanD
YamamotoAH
FanaraJJ
MackayTF
AnholtRR
2006 Dynamic genetic interactions determine odor-guided behavior in Drosophila melanogaster. Genetics 174 1349 1363
58. YamamotoA
ZwartsL
CallaertsP
NorgaK
MackayTF
2008 Neurogenetic networks for startle-induced locamotion in Drosophila melanogaster. Proc Natl Acad Sci U S A 105(34) 12393 8
59. AnholtRRH
DildaCL
ChangS
FanaraJJ
KulkarniNH
2003 The genetic architecture of odor-guided behavior in Drosophila: Epistasis and the transcriptome. Nat Genet 35 180 184
60. McElweeJ
BubbK
ThomasJH
2003 Transcriptional outputs of the Caenorhabditis elegans forkhead protein DAF–16. Aging Cell 2 111 121
61. MurphyCT
McCarrollSA
BargmannCI
FraserA
KamathRS
2003 Genes that act downstream of DAF–16 to influence the lifespan of Caenorhabditis elegans. Nature 424 277 284
62. HarbisonST
YamamotoAH
FanaraJJ
NorgaKK
MackayTF
2004 Quantitative trait loci affecting starvation resistance in. Genetics 166 1807 1823
63. RollmannSM
MagwireMM
MorganTJ
OzsoyED
YamamotoA
2006 Pleiotropic fitness effects of the Tre1–Gr5a region in Drosophila melanogaster. Nat Genet 38 824 829
64. FalconerDS
MackayTFC
1996 Introduction to Quantitative Genetics, 4th ed Essex, England Pearson Education Limited
65. LiaoG
RehmEJ
RubinGM
2000 Insertion site preferences of the P transposable element in Drosophila melanogaster. Proc Natl Acad Sci U S A 97 3347 3351
66. StoreyJD
TibshiraniR
2003 Statistical significance for genome–wide experiments. Proc Natl Acad Sci U S A 100 9440 9445
67. NorgaKK
GurganusMC
DildaCL
YamamotoA
LymanRF
2003 Quantitative analysis of bristle number in Drosophila mutants identifies genes involved in neural development. Curr Biol 13 1388 1396
68. LibertS
ZwienerJ
ChuX
VanvoorhiesW
RomanG
2007 Regulation of Drosophila life span by olfaction and food-derived odors. Science 315 1133 1137
69. LeipsJ
MackayTFC
2000 Quantitative trait loci for life span in Drosophila melanogaster: interactions with genetic background and larval density. Genetics 155 1773 1788
70. LeipsJ
MackayTFC
2002 The complex genetic architecture of life span. Exp Aging Res 28 361 390
71. MackayTFC
RoshinaNV
LeipsJW
PasyukovaEG
2006 Complex genetic architecture of Drosophila longevity.
MasaroEJ
AustadSN
Handbook of the biology of aging, sixth edition Boston Elsevier Academic Press 181 216
72. SunJ
TowerJ
1999 FLP recombinase-mediated induction of Cu/Zn–superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Mol Cell Biol 19 216 228
73. SpencerCC
HowellCE
WrightAR
PromislowDE
2003 Testing an ‘aging gene’ in long-lived Drosophila strains: increased longevity depends on sex and genetic background. Aging Cell 2 123 130
74. SpencerCC
PromislowDE
2005 Age–specific changes in epistatic effects on mortality rate in Drosophila melanogaster. J Hered 96 513 521
75. SmithJM
1958 Prolongation of the life of Drosophila subobscura by a brief exposure of adults to a high temperature. Nature 181 496 497
76. VieiraC
PasyukovaEG
ZengZB
HackettJB
LymanRF
2000 Genotype–environment interaction for quantitative trait loci affecting life span in Drosophila melanogaster. Genetics 154 213 227
77. NuzhdinSV
PasyukovaEG
DildaCL
ZengZB
MackayTFC
1997 Sex–specific quantitative trait loci affecting longevity in Drosophila melanogaster. Proc Natl Acad Sci U S A 94 9734 9739
78. PasyukovaEG
VieiraC
MackayTFC
2000 Deficiency mapping of quantitative trait loci affecting longevity in Drosophila melanogaster. Genetics 156 1129 1146
79. PasyukovaEG
RoshinaNV
MackayTFC
2004 Shuttle craft: a candidate quantitative trait gene for Drosophila lifespan. Aging Cell 3 297 307
80. BurgerJM
PromislowDE
2004 Sex–specific effects of interventions that extend fly life span. Sci Aging Knowledge Environ 28 pe30
81. MagwereT
ChapmanT
PartridgeL
2004 Sex differences in the effect of dietary restriction on life span and mortality rates in female and male Drosophila melanogaster. J Gerontol A Biol 59 3 9
82. WaskarM
LandisGN
ShenJ
CurtisC
TozerK
2009 Drosophila melanogaster p53 has developmental stage–specific and sex–specific effects on adult life span indicative of sexual antagonistic pleiotropy. Aging 1 903 936
83. MackayTF
StoneEA
AyrolesJF
2009 The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10(8) 565 77
84. KirkwoodTBL
1977 Evolution of ageing. Nature 270 301 304
85. LuckinbillLS
ArkingR
ClareMJ
CiroccoWC
BuckS
1984 Selection for delayed senescence in Drosophila melanogaster. Evolution 38 996 1004
86. ZwaanB
BijlsmaR
HoekstraRF
1995 Direct selection on life span in Drosophila melanogaster. Evolution 49 649 659
87. RoperC
PignatelliPM
PartridgeL
1993 Evolutionary effects of selection on age at reproduction in larval and adult Drososphila melanogaster. Evolution 47 445 455
88. HouleD
HughesKA
HoffmasterDK
IharaJ
AssimacopoulosS
1994 The effects of spontaneous mutation on quantitative traits. I. Variances and covariances of life history traits. Genetics 138 773 785
89. HughesKA
1995 The evolutionary genetics of male life–history characters in Drosophila melanogaster. Evolution 49 521 537
90. TatarM
PromislowDEL
KhazaeliAA
CurtsingerJW
1996 Age–specific patterns of genetic variance in Drosophila melanogaster. II. Fecundity and its genetic covariance with age–specific mortality. Genetics 143 849 858
91. MardenJH
RoginaB
MontoothKL
HelfandSL
2003 Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc Natl Acad Sci USA 100 3369 3373
92. KramerJM
DavidgeJT
LockyerJM
StaveleyBE
2003 Expression of FOXO regulates growth and can phenocopy starvation. BMC Dev Biol 3 5
93. FantinVR
WangQ
LienhardGE
KellerSR
2000 Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am J Endocrinol Metab E127 E133
94. BroughtonSJ
PiperMDW
IkeyaT
BassTM
JacobsonJ
2005 Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin–like ligands. Proc Natl Acad Sci U S A 102 3105 3110
95. FryeRA
2000 Phylogenetic classification of prokaryotic and eukaryotic Sir2–like proteins. Biochem Biophys Res Commun 273 793 798
96. GriffingB
1956 Concept of general and specific combining ability in relation to diallel crossing systems. Aust J Biol Sci 9 463 493
97. SpragueGF
TatumLA
1942 General vs. specific combining ability in single crosses of corn. J Amer Soc Agron 34 923 932
98. ZhangY
KangMS
LamkeyKR
2005 DIALLEL–SAS05: A comprehensive program for Griffing's and Gardner–Eberhar analysis. Agron J 97 1097 1106
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 7
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- Extensive DNA End Processing by Exo1 and Sgs1 Inhibits Break-Induced Replication
- Question and Answer: An Anniversary Interview with Jane Gitschier
- Multi-Variant Pathway Association Analysis Reveals the Importance of Genetic Determinants of Estrogen Metabolism in Breast and Endometrial Cancer Susceptibility
- Lysosomal Dysfunction Promotes Cleavage and Neurotoxicity of Tau