
Extensive DNA End Processing by Exo1 and Sgs1 Inhibits Break-Induced Replication
Homology-dependent repair of DNA double-strand breaks (DSBs) by gene conversion involves short tracts of DNA synthesis and limited loss of heterozygosity (LOH). For DSBs that present only one end, repair occurs by invasion into a homologous sequence followed by replication to the end of the chromosome resulting in extensive LOH, a process called break-induced replication (BIR). We developed a BIR assay in Saccharomyces cerevisiae consisting of a plasmid with a telomere seeding sequence separated from sequence homologous to chromosome III by an I-SceI endonuclease recognition site. Following cleavage of the plasmid by I-SceI in vivo, de novo telomere synthesis occurs at one end of the vector, and the other end invades at the homologous sequence on chromosome III and initiates replication to the end of the chromosome to generate a stable chromosome fragment (CF). BIR was infrequent in wild-type cells due to degradation of the linearized vector. However, in the exo1Δ sgs1Δ mutant, which is defective in the 5′-3′ resection of DSBs, the frequency of BIR was increased by 39-fold. Extension of the invading end of the plasmid was detected by physical analysis two hours after induction of the I-SceI endonuclease in the wild-type exo1Δ, sgs1Δ, and exo1Δ sgs1Δ mutants, but fully repaired products were only visible in the exo1Δ sgs1Δ mutant. The inhibitory effect of resection was less in a plasmid-chromosome gene conversion assay, compared to BIR, and products were detected by physical assay in the wild-type strain. The rare chromosome rearrangements due to BIR template switching at repeated sequences were increased in the exo1Δ sgs1Δ mutant, suggesting that reduced resection can decrease the fidelity of homologous recombination.
Published in the journal:
. PLoS Genet 6(7): e32767. doi:10.1371/journal.pgen.1001007
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001007
Summary
Homology-dependent repair of DNA double-strand breaks (DSBs) by gene conversion involves short tracts of DNA synthesis and limited loss of heterozygosity (LOH). For DSBs that present only one end, repair occurs by invasion into a homologous sequence followed by replication to the end of the chromosome resulting in extensive LOH, a process called break-induced replication (BIR). We developed a BIR assay in Saccharomyces cerevisiae consisting of a plasmid with a telomere seeding sequence separated from sequence homologous to chromosome III by an I-SceI endonuclease recognition site. Following cleavage of the plasmid by I-SceI in vivo, de novo telomere synthesis occurs at one end of the vector, and the other end invades at the homologous sequence on chromosome III and initiates replication to the end of the chromosome to generate a stable chromosome fragment (CF). BIR was infrequent in wild-type cells due to degradation of the linearized vector. However, in the exo1Δ sgs1Δ mutant, which is defective in the 5′-3′ resection of DSBs, the frequency of BIR was increased by 39-fold. Extension of the invading end of the plasmid was detected by physical analysis two hours after induction of the I-SceI endonuclease in the wild-type exo1Δ, sgs1Δ, and exo1Δ sgs1Δ mutants, but fully repaired products were only visible in the exo1Δ sgs1Δ mutant. The inhibitory effect of resection was less in a plasmid-chromosome gene conversion assay, compared to BIR, and products were detected by physical assay in the wild-type strain. The rare chromosome rearrangements due to BIR template switching at repeated sequences were increased in the exo1Δ sgs1Δ mutant, suggesting that reduced resection can decrease the fidelity of homologous recombination.
Introduction
DNA double-strand breaks (DSBs) are highly cytotoxic lesions that arise spontaneously during cell growth or following exposure to DNA damaging agents, such as ionizing radiation (IR). The repair of DSBs is critical for cell survival and maintenance of genome integrity. There are two major pathways to repair DSBs: non-homologous end joining (NHEJ) and homologous recombination (HR) [1]. NHEJ involves the religation of the two ends of the broken chromosome, which can occur with high fidelity or be accompanied by gain or loss of nucleotides at the junction [2]. HR relies on the presence of a homologous duplex to template repair of the broken chromosome and is generally considered to be error-free. However, HR can lead to a local loss of heterozygosity (LOH) if the homologous sequences are not identical, and to extensive LOH if repair is associated with a crossover. Furthermore, if repeats are utilized as the sequence donor and recombination is associated with crossing over, translocations can occur [3], [4]. If both ends of the DSB share homology with the donor duplex sequence, HR proceeds by a two-ended mechanism, such as double strand break repair (DSBR) or synthesis dependent strand annealing (SDSA) [1]. However, if coordination of the two ends is not maintained or only one end of the break is available, such as at a critically short telomere, repair can occur by break-induced replication (BIR) [5], [6]. In this case, following strand invasion replication occurs to the end of the chromosome to generate a stable repaired product. This can cause very long gene conversion tracts and significant LOH, and non-reciprocal translocations if invasion occurs at a dispersed repeated sequence [5], [7]–[10].
The repair of DSBs by HR requires the 5′-3′ nucleolytic degradation of the DNA ends to form invasive 3′ single-stranded DNA (ssDNA) tails. This processing reaction occurs in two steps: the first is catalyzed by the Mre11-Rad50-Xrs2 (MRX) complex and Sae2, which function together to clip sequences from the 5′ ends in increments of around 20–100 nt, the second step involves processive resection of the 5′ ends by Exo1 or Sgs1-Dna2 to form long tracts of ssDNA [11]–[14]. In the absence of Exo1 and Sgs1, or Exo1 and Dna2, partially processed intermediates with 3′ ssDNA tails of about 100–700 nts accumulate [12], [14]. The 3′ ssDNA tails created by end resection are bound by Rad51 to form a nucleoprotein filament that searches for homology and promotes pairing between the ssDNA bound by Rad51 and complementary sequences in the donor duplex [1]. The invading 3′ end is then used to prime DNA synthesis templated by the donor sequence. To complete two-ended repair two short tracts of leading strand DNA synthesis are required, whereas BIR requires extensive leading and lagging strand DNA synthesis [15]–[18].
Telomeres represent a natural source of one-ended DSBs. These are normally protected from recombination by telomere capping proteins, but when telomeres become critically short, for example, in the absence of telomerase, telomere maintenance depends on homologous recombination and this is thought to occur by BIR [6]. The analysis of telomere maintenance in the absence of telomerase revealed the importance of the RAD52 epistasis group genes and POL32 for BIR at telomeres [17], [19]–[22].
Because two-ended DSBs are repaired primarily by gene conversion, specialized assays have been developed to study BIR at non-telomeric sites in which only one end of a DSB can undergo homology dependent strand invasion. In one assay system, repair of an HO endonuclease induced DSB at the MAT locus was forced to occur by BIR from the chromosome homologue because homologous sequences on one side of the DSB had been deleted in diploid cells, or haploid cells disomic for chromosome III [16], [23]. Strand invasion and DNA synthesis resulted in copying sequences from the donor chromosome to the telomere. Using this system it was shown that strand invasion and completed repair, monitored by PCR and Southern blot analysis, respectively, is slower than repair by gene conversion. Haploid assay systems have been developed using the HO cut site inserted at an ectopic site so that non-essential sequences centromere distal to the cut site can be lost without compromising viability, and strand invasion using homologous sequences on only one side of the DSB can restore a telomere by BIR [17], [24], [25]. In these assays, the kinetics of repair as determined by extension of the invading strand were slow, even though Rad51 loading and pairing with the donor sequence were rapid, suggesting cells sense whether DSBs have a second end to complete repair by gene conversion and delay initiation of DNA synthesis when only one end is present [25].
Another approach to study BIR involves transforming yeast cells with a linearized plasmid that contains a telomere seeding sequence on one end and a region of homology to unique sequences present on a yeast chromosome at the other end [26]. One end of the plasmid undergoes de novo telomere formation and the other is used for strand invasion and replication to the end of the chromosome. In this assay system, the strand invasion intermediate was shown to be unstable, as predicted for the SDSA model of recombination, and to undergo repeated cycles of strand invasion and dissociation before establishing a processing replication fork to complete replication of sequences downstream of the site of strand invasion [9]. These results suggest strand invasion during BIR is similar to gene conversion and extensive replication (BIR) occurs only after failure to capture the second end and repair by gene conversion. Template switching during BIR has also been shown to occur during repair of a chromosomal DSB resulting in triparental translocations [27].
These two hypotheses for how BIR initiates differ considerably in that one predicts there is a delay in the initiation of DNA synthesis from the strand invasion intermediate when the other end of the break to engage in repair is absent, the other predicts extension of the invading strand is independent of the other end and is terminated by dissociation of the extended invading strand pairing with the other side of the DSB. If there were no end to pair with then strand invasion would occur again and eventually lead to formation of a processive replication intermediate.
The transformation assay has several limitations: it cannot be used to study the kinetics of repair, or to identify repair intermediates, because less than one percent of cells are competent for transformation and the synchrony of DNA uptake is not known. In order to overcome these problems, we generated a new assay where the plasmid is cut in vivo by the I-SceI endonuclease. BIR occurs with low frequency in this system because the Exo1 and Sgs1-dependent resection pathways degrade the linearized plasmid. Inhibition of extensive end resection using the exo1Δ sgs1Δ double mutant enabled BIR products to be detected by physical assays. The inhibitory effect of resection was less in a plasmid-chromosome gene conversion assay, compared to BIR, and products were detected by physical assay in the wild type strain. By comparing the kinetics of strand invasion and extension of the invading strand in the plasmid BIR and gene conversion assays we observed no significant difference between BIR and gene conversion repair suggesting the initiation of both processes is similar.
Results
Plasmid-based BIR assay
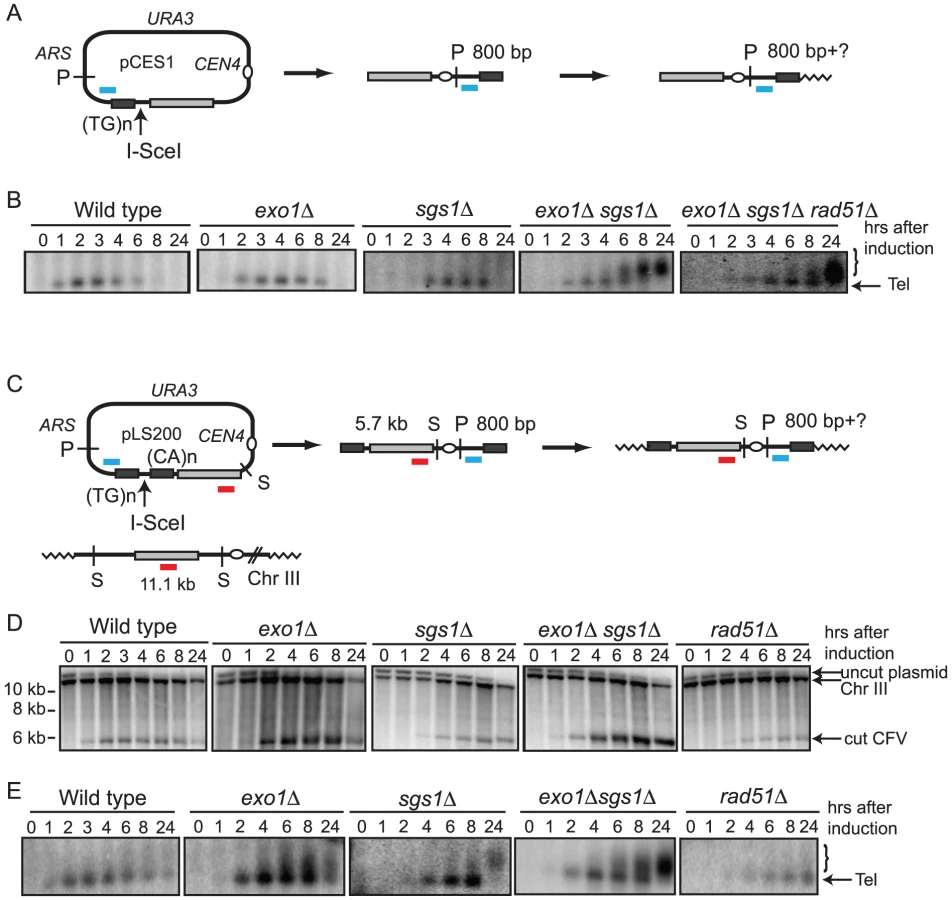
We previously used a plasmid transformation assay to study the mechanism and genetic control of BIR [9], [26], [28]. The chromosome fragmentation vector (CFV1) contains the URA3 selectable marker, SUP11, CEN4, an ARS, a tract of (G1–3T)n to provide a site for telomere addition and a unique DNA segment from the left arm of chromosome III (BUD3). The CFV is linearized between BUD3 and the telomere seeding sequences with a restriction endonuclease and used to transform yeast, selecting for Ura+ colonies. Most transformants (95%) arise by de novo telomere addition to heal one end of the CFV and strand invasion at the other end into the endogenous yeast locus to copy the entire chromosome arm yielding a stable 110-kb chromosome fragment (CF). This BIR assay involves transformation of chemically competent yeast cells with a linearized CFV and selection for completed repair events. This system is not useful for physical analysis of intermediates and products of BIR because only 0.1% of cells are competent for transformation, the synchrony of DNA uptake is not known and multiple copies of the linearized vector could enter the cell. Furthermore, because naked DNA is used as a substrate it is possible that the efficiency and mechanism of repair could be different to a chromatinized template.
To circumvent these limitations the CFV was modified by introduction of the I-SceI endonuclease recognition site between the telomere seeding sequences and the sequences with homology to the BUD3 region of chromosome III (Figure 1A). I-SceI was used instead of HO to overcome the requirement of using yeast strains with the native HO cut sites deleted. This plasmid (pCES1) was used to transform a yeast strain containing an integrated PGAL:I-SCEI cassette. The plasmid was maintained as an episome until induction of the I-SceI endonuclease. Cleavage of the plasmid and subsequent repair were detected by Southern blot analysis of SpeI-digested genomic DNA isolated from samples taken prior to I-SceI induction and at 1–2 hr intervals after addition of 2% galactose to the culture. pCES1 has a unique SpeI site resulting in a 12.6 kb fragment prior to I-SceI cleavage; of the two fragments produced by I-SceI digestion only the 5.7 kb fragment hybridizes to the BUD3 probe. Initiation of repair by BIR should result in a novel 6.4 kb SpeI fragment (Figure 1B). Linearization of the vector by I-SceI was detected at the 1 hr time point and the 5.7 kb cut fragment was visible until 6 hr (Figure 1B). However, we were unable to detect formation of the 6.4-kb SpeI fragment indicative of BIR. By 8 hr the cut fragment had disappeared and the band corresponding to uncut plasmid was very faint indicating most of the plasmids in the population had been cut and degraded. Plasmid loss following I-SceI induction was confirmed by PCR using primers specific for the vector (data not shown).

To determine whether BIR can occur at low frequency cells were plated after I-SceI induction on either synthetic complete (SC) medium or SC-URA medium to detect retention of the plasmid. Ura+ colonies from each time point were then tested for mitotic stability to distinguish between stable CFs formed by BIR and uncut or end-joined plasmids [26]. Although maintained under selection, only 30–50% of the cells were Ura+ before I-SceI induction. The plasmid was rapidly lost after induction of I-SceI with only 1% of the population remaining Ura+ 24 hr after I-SceI induction, and of these colonies about 10% were due to CF formation (verified by pulsed field gel electrophoresis (PFGE)).
5′-3′ resection by Exo1 and Sgs1 is inhibitory to BIR
Malkova et al [23] have shown that BIR is a slow repair process; thus, it is possible that the 12.6 kb linearized plasmid is completely degraded before BIR initiates. To determine whether degradation of the substrate was the cause for low BIR frequency we tested the exo1Δ, sgs1Δ and exo1Δ sgs1Δ mutants in the BIR assay. These mutants were chosen because resection is initiated by MRX-Sae2 forming a short 3′ ssDNA tailed intermediate that is a substrate for Rad51-catalyzed strand invasion, but long range resection is prevented in the exo1Δ sgs1Δ double mutant [12], [14]. While improvements in plasmid retention and the BIR frequency as determined by plating efficiency were observed in the single mutants, the double mutant showed the largest increase with 7.0% of the resulting colonies containing CFs, compared with 0.18% for wild type (Table 1). Furthermore, a faint band corresponding to the extension product was detected by Southern blot at 8 hr after I-SceI induction in the exo1Δ sgs1Δ double mutant (Figure 1B). Only one CF was detected among 171 rare Ura+ colonies formed 24 hr after I-SceI induction in the sgs1Δ exo1Δ rad51Δ mutant indicating that the increase in BIR was dependent on RAD51. To ensure that the Exo1 nuclease activity was responsible for the reduction in BIR, the exo1Δ mutant was transformed with plasmids expressing either wild type EXO1 or the exo1-D173A allele [29]. Only the strain expressing the nuclease-defective exo1-D173A allele showed elevated BIR confirming that the Exo1 nuclease is responsible for low frequency BIR (Table 1).

In a PCR assay to measure primer extension during BIR the product was barely detectable in the wild type strain and did not increase over time, but was readily detected in the resection defective exo1Δ, sgs1Δ and exo1Δ sgs1Δ strains (Figure 1C). A rad51Δ mutant was used as a control and no PCR product was detected in this strain. The PCR product diagnostic of extension of the invading 3′ end was detected 2 hr after I-SceI induction in the exo1Δ sgs1Δ double mutant, earlier than BIR was detected by Southern blot analysis. The PCR assay can detect extension of the 3′ end of the strand invasion intermediate as well as completed products, whereas the Southern blot assay requires the invading end become dsDNA to be cleaved by SpeI; therefore the delayed product formation in the Southern blot assay could be due to a delay in initiating lagging strand synthesis. However, it is more likely that the apparent difference in timing is due to the higher sensitivity of PCR compared with the Southern blot assay.
Analysis of 34 CFs recovered from the wild type strain by PFGE and Southern blot analysis revealed that all were of the expected size of 110 kb (Figure 2); in two of the samples, the mobility of chromosome III was altered suggesting strand invasion can result in chromosome III rearrangements. These have not been analyzed further. Because chromosome III rearrangements are rarely seen in the wild-type strain, and the two events recovered were of the same size and from the same induction, it is possible that these are clonal events. Two of 30 independent repair events analyzed for the exo1Δ mutant exhibited aberrant-sized CFs. From the sgs1Δ mutant, three clones had aberrant-sized CFs and two had chromosome III rearrangements (Figure 2). Twelve CFs analyzed from the exo1Δ sgs1Δ double mutant were of aberrant sizes and one clone had a chromosome III rearrangement (13 out of 30 total aberrant events), a significant increase compared with wild type (P = 0.0007) and the single mutants (P<0.05). The CFs of different sizes are likely to result from elevated template switching at the cluster of Ty/δ elements near the LEU2 locus [9]. Some of the Ura+ colonies had two different sized CFs; these could be due to two independent repair events in cells with two copies of the CFV or be formed by secondary recombination events. For samples with one prominent CF band and a fainter CF hybridizing signal we assume the latter is due to a secondary event and these are not marked as aberrant events.

The negative effect of resection on BIR was also seen in the transformation-based BIR assay. In this system the frequency of BIR is determined by the number of Ura+ colonies derived from transformation of cells with the linear CFV compared to an uncut replicating plasmid to account for transformation efficiency [26]. A derivative of the CFV lacking the ARS element (pLS192) was used to eliminate the background of Ura+ colonies due to NHEJ. The exo1Δ and exo1Δ sgs1Δ mutants both exhibited a significant increase in the BIR frequency compared with wild type (Table 2).

The sgs1-D664Δ mutation improves the efficiency BIR
One problem with using the exo1Δ sgs1Δ double mutant for further physical analysis of BIR is the increased number of aberrant CFs generated; in addition, the strain exhibits slow growth. Bernstein et al [30] described an sgs1 separation of function allele, sgs1-D664Δ, which suppresses the top3Δ slow growth defect, but does not exhibit spontaneous hyper-recombination or DNA damage sensitivity. We have analyzed DSB processing in the exo1Δ sgs1-D664Δ double mutant and found there is a defect in the processivity of resection resulting in ssDNA tails of around 5-kb (E. Mimitou and K. Bernstein, unpublished data). To determine whether this might provide the benefits of enhanced BIR by preventing complete degradation of the linear CFV, but without the hyper-recombination phenotype, we analyzed the kinetics and frequency of BIR in the exo1Δ sgs1-D664Δ double mutant. By Southern blot analysis and in the plating assay the frequency of BIR was similar to the exo1Δ sgs1Δ mutant (Figure 3A and Table 1). The 6.4 kb SpeI fragment, corresponding to the extended CFV, was clearly detected 6 hr after I-SceI induction and 4 hr after the appearance of the I-SceI cut fragment. The faster appearance of the BIR product compared with the exo1Δ sgs1Δ mutant could be because there is more resection creating a better substrate for Rad51-catalyzed strand invasion. High frequency BIR, comparable to the exo1Δ sgs1Δ mutant, was also observed in the transformation assay (Table 2). Analysis of CFs recovered from the exo1Δ sgs1-D664Δ double mutant revealed 6 aberrant-sized CFs of 29 events analyzed (Figure 3B). The number of total aberrant events recovered from the exo1Δ sgs1-D664Δ strain is not significantly different to the wild-type strain (P = 0.13) or the exo1Δ sgs1Δ mutant (P = 0.09), but if only the aberrant-sized CFs are considered then the difference between wild type and exo1Δ sgs1-D664Δ is significant (P = 0.007).

Because the exo1Δ sgs1-D664Δ mutant exhibits faster growth than the exo1Δ sgs1Δ mutant, but still shows increased frequency BIR, we tested whether completed BIR repair events were formed during the time course of I-SceI induction by PFGE. A faint band corresponding to the 110 kb CF was detected as early as 6 hr after I-SceI induction in the exo1Δ sgs1-D664Δ strain, but, as expected, was not seen in wild type. This result shows that the appearance of the 6.4 kb SpeI fragment by Southern blot hybridization correlates with the formation of completed repair products.
Telomere addition is more efficient in the exo1Δ sgs1Δ double mutant
CF formation requires strand invasion at one end of the vector and de novo telomere addition at the other end. Thus, the low frequency of CF formation could be due to a defect in one or both of these processes. To analyze telomere addition, DNA isolated from cells after I-SceI induction was digested with PstI to liberate the terminal fragment containing the telomere seeding sequence (Figure 4A). De novo telomere addition to this end of the linearized CFV should result in a shift to a higher molecular weight form [31]. Because telomere length is variable we expected to observe a heterogeneous population of fragments up to 300 bp longer than the original 800 bp fragment. We were not able to detect extended products resulting from telomere addition at the seeding site in wild type, exo1Δ or sgs1Δ strains. However, in the exo1Δ sgs1Δ double mutant telomere addition was detected 6–8 hr after linearization of the plasmid (Figure 4B). This raised the concern that inefficient telomere recognition and/or elongation might contribute to the low frequency CF formation in the wild-type strain. To test this, a plasmid with telomere seeding sequences on either side of the I-SceI site was constructed (Figure 4C). When this plasmid was linearized in vivo in the wild type, exo1Δ, sgs1Δ, and exo1Δ sgs1Δ strains the linear form was still detected at the 24 hr time point indicating the ends are recognized as telomeres and protected from degradation. However, it was still difficult to see addition at the telomere-seeding site in the wild type and the exo1Δ strains (Figure 4D). The improved telomere addition in the exo1Δ sgs1Δ mutant is most likely due to the reduced resection providing a short ssDNA tail that can be recognized by Cdc13 [31]. The end protection was not due to homologous recombination because it was also seen in the rad51Δ strain, which is defective for inter-chromosomal homologous recombination [26], [32].
The exo1Δ mutation improves the efficiency of plasmid gene conversion
To determine whether degradation of short linear substrates is a general problem in ectopic recombination assays, or specific to BIR, we created a plasmid substrate with an I-SceI cut site within the ADE2 gene to monitor repair by gene conversion from a chromosomal donor sequence (ade2-1) (Figure 5A). Following cleavage of the plasmid-borne ade2-IS allele, repair from the chromosomal allele restores the AatII site that was disrupted by the I-SceI cut site insertion. Cells from each plasmid-containing strain were plated onto SC and SC-URA media at different time points after induction of the I-SceI endonuclease. The number of Ura+ colonies formed 8 hr after induction of I-SceI was reduced by ten-fold in the wild-type strain, by five-fold in the sgs1Δ mutant, but only two to three-fold in the exo1Δ and exo1Δ sgs1Δ mutants. Only 4% of the rad51Δ cells retained the plasmid 8 hr after I-SceI induction indicating that plasmid loss is due to inefficient repair. Southern blot analysis of BamHI digested genomic DNA confirmed that I-SceI cleavage products are generated in all of the strains (Figure 5B). There was delay in I-SceI cutting in the exo1Δ sgs1Δ mutant and the characteristic smearing of the bands due to partial resection was observed [12], [14]. Repair of the plasmid DSB results in restoration of the BamHI fragment, and this band was present 8 hr after I-SceI induction in all the strains except the rad51Δ mutant.

To determine the kinetics of strand invasion, primers were designed to anneal to the plasmid upstream of the homology region and to the genomic locus downstream of the homology region. These primers should amplify the product formed by strand invasion and DNA synthesis beyond the region of homology shared by the plasmid and chromosome XV, and also completed events that result in plasmid integration. Studies of plasmid gap repair indicate that 20–50% of the repair events are associated with integration of the plasmid [33]–[35]. Although plasmid integration would lead to formation of an unstable dicentric chromosome, these events are likely to persist through the 8 hr time course. For the wild type and single mutants, the PCR product was detected 2 hr after induction of I-SceI, and at 3 hr in the exo1Δ sgs1Δ double mutant (Figure 5C). No PCR product was seen in a rad51Δ strain (data not shown).
Completed repair products can also be detected by PCR using primers that anneal to plasmid sequences upstream of the homology region and within ade2, downstream of the I-SceI cut site. These primers amplify the uncut plasmid allele, invasion intermediates and completed gene conversion products, but only intermediates and completed repaired product will be digested with AatII (Figure 5D). In contrast to BIR, gene conversion products were detected by the physical assay in the wild type strain, appearing 3 hr after inducing I-SceI, similar to the repair kinetics reported for other ectopic recombination assays [36]. Similar results were obtained using primers that anneal to plasmid sequences flanking the ade2 insert that are specific for uncut plasmid or completed repair events (data not shown). Repaired products were detected with similar kinetics in the exo1Δ and sgs1Δ mutants, but were delayed in the exo1Δ sgs1Δ double mutant. The delay in detection of strand invasion intermediates and completed products in the exo1Δ sgs1Δ mutant may be due to the delay in I-SceI cutting and reduced resection. I-SceI cutting is complete at 8 hr in the exo1Δ mutant and plasmid retention is around 30%, compared with 10% for the wild type strain; thus, the frequency of gene conversion is increased by three-fold in the absence of Exo1. The increased cleavage of the PCR product by AatII in the exo1Δ mutant is also diagnostic of increased repair compared with wild type and the sgs1Δ mutant (Figure 5D).
Discussion
We have developed a plasmid-based assay system to study BIR as an alternative to chromosomal systems. This assay utilizes features of the previously described plasmid transformation system except the initiating DSB is made in vivo instead of by transforming linearized plasmid DNA into cells. The goal of developing this system was to physically monitor BIR using an assay that could be easily moved into different strain backgrounds; however, we discovered that a major limitation to the assay was degradation of the linearized substrate. Although this could be overcome by eliminating the Exo1 nuclease and Sgs1 helicase, which control degradation of linear DNA [11], [12], [14], the absence of Exo1 and Sgs1 resulted in elevated chromosome rearrangements during BIR. The inhibitory effect of DNA end resection by Exo1 was also observed using a plasmid-chromosome gene conversion assay.
In this study we found a much lower frequency of BIR in the wild type strain (<1%) than observed in chromosomal ectopic BIR assays (10–30%) [17], [25]. The low frequency may be due to inefficient cleavage of the plasmid by I-SceI, the low copy number of the plasmid, and the requirement for telomere addition at the telomere seed sequence at one end of the linearized vector. Although a plasmid with two telomere seeding sequences flanking the DSB was maintained as a linear plasmid following linearization it was not 100% efficient (Figure 4). During the course of these studies we found that the SUP11 gene present on the vector is responsible for the low plasmid retention. Thus, it is possible that the system could be improved by using the HO cut site instead of I-SceI and by replacing the short telomere seed sequence (43 bp) with a longer one in a vector lacking SUP11.
Deleting the EXO1 and SGS1 genes resulted in a significant increase in the frequency of BIR, compared with the wild type strain (Table 1 and Table 2). We attribute this effect to the role of both proteins in regulating the resection of DSBs because the linearized plasmid was still present 8 hr after I-SceI cleavage in the exo1Δ sgs1Δ double mutant, but was undetectable in the wild-type strain (Figure 1). Furthermore, the Exo1 nuclease activity was responsible for reduced BIR and the resection-defective sgs1-D664Δ exo1Δ mutant behaved the same as the sgs1Δ exo1Δ mutant. The sgs1Δ mutation conferred less of a suppressive effect than the exo1Δ mutation, but together the two mutations synergized to increase the frequency of BIR. In the absence of Exo1 and Sgs1 partial resection of DSB ends occurs to form 3′ single-stranded DNA (ssDNA) tails of around 100–700 nucleotides [12], [14]. These intermediates can be used for gene conversion repair albeit with slight reduced efficiency compared with resection proficient strains [12], [14]. Thus the increased efficiency of BIR in the exo1Δ sgs1Δ double mutant appears to be due to sufficient resection to form an invasive ssDNA end, but without the extensive resection that completely degrades the plasmid. Although the resection defect of the exo1Δ sgs1-D664Δ mutant is not as severe as the exo1Δ sgs1Δ mutant (E. Mimitou and K. Bernstein, unpublished results), the more extensive resection appears to create a better substrate for Rad51-catalyzed strand invasion and the linearized plasmid is stable for long enough to allow BIR to occur. Preventing extensive resection was also found to increase the retention of a linearized plasmid that can repair by gene conversion. The increased stability of short linear DNA fragments in the exo1Δ sgs1Δ double mutant could also explain the increase in plasmid integration during transformation reported previously, and reduced resection might be more favorable for spontaneous recombination between short repeats [37], [38].
Extensive resection would be expected to be less problematic in the chromosomal assay systems compared with the plasmid assay because of the much larger substrates used. However, even when generating a DSB on chromosome III with extensive homology available on one side of the DSB to initiate BIR, more than 50% of the repair events occur by resection of about 30-kb from the HO cut site to a pair of inverted Ty elements proximal to the MAT locus (FS2) [10], [23]. The ssDNA formed at these elements undergoes intramolecular annealing, or intermolecular annealing between sister-chromatids, to form inverted dimers that undergo complex rearrangements before yielding viable products. In the ectopic assays involving short homologies, extensive resection could expose repeated sequences, such as δ and Ty elements, that could be used to initiate recombination with repeats elsewhere in the genome. Furthermore, the 3′ ends of resected intermediates are unstable several hours after induction of HO endonuclease [39]; thus, the small region of homology required for strand invasion could be lost resulting in lower frequency BIR. Indeed, elimination of Exo1 or Sgs1 also increases the efficiency of BIR in an ectopic chromosomal assay [40].
The sgs1 mutation has been shown to increase the percent of DSBR events that resolve as crossovers [41]. Thus, one possible explanation for the increase in events that result in CFs in the exo1Δ sgs1Δ double mutant would be increased resolution of the strand invasion intermediate to form a crossover linking the CFV to the left arm of chromosome III. We have previously shown that mutations in the Polymerase δ complex result in CFs from half-crossovers instead of BIR [18]. However, when a diploid exo1Δ sgs1Δ strain was used in the BIR assay we did not find increased loss of one copy of chromosome III as predicted from half crossovers (data not shown). Furthermore, a simple crossover between the CFV and left arm of chromosome III would generate CFs of uniform size, contrary to our findings (Figure 2). The other possible explanation for increased stability of the linear CFV would be de novo telomere addition at the I-SceI cut site, as shown to occur at an HO-induced DSB in exo1Δ sgs1Δ mutants [40], [42]. Short linear plasmids do not exhibit high mitotic stability in S. cerevisiae so these events would not have been scored as stable Ura+ colonies [43]. In addition, the physical analysis of CFs in the exo1Δ sgs1Δ and exo1Δ sgs1-D664Δ mutants showed most of the aberrant-sized CFs were larger than the expected size of 110-kb, instead of the 13.2-kb product expected from telomere addition at both ends of the linear vector. Furthermore, because there is no replication origin present on the vector used in the BIR transformation assay all the transformants must arise by strand invasion to copy or link a chromosomal origin to the vector and cannot arise by telomere addition at both ends of the vector. Thus, de novo telomere addition at the I-SceI site appears to be infrequent, and the large increase in events in the in vivo cutting and transformation assays in the exo1Δ sgs1Δ double mutant is consistent with stabilization of the short linear DNA resulting in more time for strand invasion to occur.
In the wild-type strain all of the CFs recovered were of the expected size from strand invasion at the BUD3 locus and replication to the end of chromosome III (Figure 2). Twenty to 40% of the CFs analyzed from the exo1Δ sgs1-D664Δ and exo1Δ sgs1Δ mutants, respectively, exhibited aberrant mobility by PFGE (Figure 2). Template switching at the Ty element or cluster of δ elements located 5-kb downstream of the region of homology is likely to be responsible for the novel CFs. If the end of the substrate is extruded as it passes through these repetitive elements, re-invasion might occur into any of the multiple copies of Ty or δ elements found scattered throughout the genome as reported previously using the plasmid transformation assay [9]. Consistent with this hypothesis, we did not recover unusual sized CFs from the exo1Δ sgs1-D664Δ mutant using a vector that invades a chromosome arm devoid of Ty and δ elements (data not shown).
Because we did not observe a significant increase in the number of aberrant CFs in the sgs1Δ single mutant, but the number was increased in the exo1Δ sgs1Δ and exo1Δ sgs1-D664Δ mutants, it appears to be due to the decrease in end resection instead of the increased crossovers [41]. However, another possibility is increased recombination between diverged sequences (homeologous) resulting in template switching to more diverged Ty/δ elements, a process suppressed by SGS1 [44], [45]. In a study of gross chromosome rearrangements, combining the sgs1Δ mutation with defects in checkpoint functions or chromatin assembly factors resulted in complex rearrangements between the diverged CAN1, LYP1 and ALP1 loci, interpreted as BIR and multiple cycles of template switching between homeologous sequences [46]. The exo1Δ mutant also exhibits a slight increase in homeologous recombination [47], but the exo1Δ sgs1Δ double mutant has not been tested to determine whether the mutations synergize to allow more promiscuous recombination.
Previous studies showed a delay in extension of the 3′ end of the strand invasion intermediate in BIR compared with a two-ended repair event [23], [25]. For both processes the kinetics of Rad51 loading to the resected end and pairing with donor sequences were the same, suggesting the delay is at the step of DNA synthesis from the invading end [25]. In our assay we are unable to assess the kinetics of repair in the wild-type strain because the process is too inefficient to detect by Southern blot analysis. However, a weak product was detected two hours (2 hr) after I-SceI induction in the PCR primer extension assay (Figure 1). In the exo1Δ sgs1Δ mutant the PCR primer extension product was detected at 2 hr and the novel SpeI fragment indicative of repair detected at 8 hr. It is possible that the SpeI fragment is present earlier, but not detected due to the low sensitivity of the Southern blot assay. The kinetics of primer extension seen here are faster than reported in studies from the Haber lab [17], [23], [25]. In our assay only one end generated by the DSB is available for strand invasion and the telomere seeding sequence at the other end should be recognized by telomere binding proteins. Thus, one possible explanation for the more rapid detection of strand invasion intermediates in the plasmid assay is that the telomere end is not recognized as a broken chromosome and only one end is “seen” to be in need of repair.
In the gene conversion assay primer extension was detected at 2 hr in the wild type, exo1Δ and sgs1Δ strains, and at 3 hr in the exo1Δ sgs1Δ double mutant (Figure 5). The delay in the double mutant was likely due to less efficient cleavage of the plasmid by I-SceI and reduced resection to form the substrate for Rad51. Completion of repair was detected in the wild type and mutant strains at 3–4 hr, similar to other ectopic recombination assays [36]. The lower efficiency of BIR compared with gene conversion is consistent with failure at a step downstream of strand invasion [17], [25]. We suggest that following extension of the invading 3′ end by a short tract of DNA synthesis, the invading end dissociates and in the absence of a second end to anneal to, the linear plasmid is susceptible to more extensive degradation of the 5′ end before initiating a second strand invasion event [5]. This cycle could occur several times before assembly of the replisome and completion of BIR. Long 3′ ssDNA tailed intermediates are unstable [43]; thus, reducing the length of the 3′ tail by preventing resection of the 5′ strand may help to preserve the intermediate formed by dissociation resulting in more frequent BIR in the exo1Δ sgs1Δ double mutant. Similarly, stabilization of the linear intermediate following dissociation of the invading strand at repeated sequences could result in the higher frequency of aberrant-sized CFs in the resection-defective exo1Δ sgs1Δ double mutant (Figure 2). In the gene conversion assay only one round of strand invasion and extension of the 3′ end by DNA synthesis, followed by dissociation and annealing, would be necessary to form a stable repaired product.
Materials and Methods
Media, growth conditions, and genetic methods
Rich medium (yeast extract-peptone-dextrose (YPD)), synthetic complete medium (SC) lacking the appropriate amino acids or nucleic acid bases, sporulation medium, and genetic methods were as described previously [48]. Synthetic deficient medium (SD) containing 2% raffinose and supplemented with adenine, histidine, tryptophan, lysine, and leucine was used for the galactose induction of I-SCEI. Transformation of yeast was by the lithium acetate method [49]. Standard procedures were used for genetic crosses [48].
Yeast strains and plasmids
S. cerevisiae strains used in this study were derived from W303 (his3–11,15 leu2–3,112 trp1–1 ura3–1 ade2–1 can1–100) and are listed in Table S1, only deviations from this genotype and the MAT allele are given [50]. The W303 derivative with the lys2::PGAL-I-SCEI cassette (Lev488) was a gift from S. Marcand. This strain was crossed to strains in the lab collection with exo1::HIS3, sgs1::HphMX4, sgs1-D664D or rad51::LEU2 alleles to create haploid derivatives with the lys2::PGAL-I-SCEI cassette and mutations in the relevant genes.
Plasmid pCES1 was constructed by inserting the annealed oligonucleotides, CES1-2F and CES1-2R, carrying the I-SceI recognition sequence adjacent to a telomere seeding sequence, into BglII/HinDIII digested pYCF/D8B plasmid [28] (Table S2). The annealed oligonucleotides BglII/Tel-ISceI/BglII and BglII/ISceI-tel/BglII (Table S2) were ligated to BglII-digested pCES1 generating a plasmid (pLS200) with the I-SceI site flanked by telomere seeding sequences in opposite orientations. The structures of the plasmids were confirmed by DNA sequencing. To construct the ARS− CFV, pADW17 (CFV2/D8B-tg) was cut with XmaI and sequences were degraded from the cut site using E. coli ExoIII to remove the ARS and SUP11 sequences. The digested plasmids were recircularized by ligation and sequenced to determine the extent of resection. One of the resulting plasmids, pSL192, was tested for ability of the SnaBI cut, but not uncut, to transform yeast to Ura+ prototrophy and the products shown to be exclusively CFs. pRS316:ADE2 was made by cloning the KpnI/Sac1 fragment from pKH5 [51] into the pRS316 vector. The 1.8 kb SalI/BglII fragment from pLS189, containing the I-SceI cut site inserted at the AatII site [52], was used to replace the SalI/BglII fragment of pRS316:ADE2, generating pLS199. The plasmids pSM502 (EXO1) and pSM638 (exo1-D173A) were described previously [29].
BIR assay and telomere seeding assay
Plasmids pCES1 or pLS200 were used to transform wild type or mutant strains, selecting for Ura+. Five mL SC-URA glucose cultures of Ura+ transformants were grown overnight at 30°C. Cells were diluted to a concentration of 1×105 cells/ml in 350 mL supplemented SD/raffinose medium. Cultures were grown overnight to a concentration of 3×106 cells/mL and galactose was added to the cultures for a final concentration of 2%. Fifty mL of cells were harvested at each indicated time point after galactose induction, DNA was isolated from each sample and digested with SpeI or PstI. DNA fragments were separated by electrophoresis through 0.8% agarose gels, transferred to nylon membranes and hybridized with a radiolabeled probe. Cutting efficiency and BIR was assayed with a probe generated by PCR from Chromosome III sequence (coordinates 100791–102072) with primers D8BF and D8BR (Table S2). A probe specific for the vector sequences adjacent to the telomere seeding sequence was generated by PCR from primers pADW17F and pADW17R (Table S2) and used for the telomere seeding assays. Samples of cells from each time point were serially diluted and plated on solid SC and SC-URA medium to determine plasmid retention. Because some Ura+ colonies are due to uncut plasmids or NHEJ resulting in an unstable plasmid, Ura+ colonies were struck onto non-selective YPD medium, then replica plated to SC-URA to determine the stability of the Ura+ phenotype. Stable clones were analyzed by PFGE to confirm the presence of the CF. The percent BIR was determined by the number of stable Ura+ colonies divided by the number of Ura+ colonies normalized to the percent Ura+ at 0 hr. The numbers presented in Table 1 are from at least three trials for each strain; significance was determined using an unpaired t test
Strand invasion from the CFV was detected by PCR of genomic DNA from cells collected at different times during the induction. PCR was performed with primers CFF2 and CFR3 (Table S2) with Turbo Pfu (Stratagene) and the reaction profile of 92°C for 45 seconds, 55°C for 15 seconds, 68°C for 8 minutes for 25 cycles. The control PCR utilized primers pADW17F and pADW17R.
To detect completed CF events in the time course, 350 ml cultures were grown as above and 50 mL aliquots were collected at each designated time point. Cells were harvested and then prepared for PFGE. These gels were transferred to nylon membrane and hybridized with radiolabled probe generated from D8BF and D8BR as above (Table S2).
To analyze independent repair events cells were grown in SC-URA, transferred to SD/raffinose as above, and then subject to a short (2 h) liquid induction of I-SceI before plating onto medium containing 2% raffinose and 1% galactose with appropriate selection (-URA), or plated directly onto this medium. Individual colonies that arose were tested for stable repair products as above and then prepared for PFGE. These gels were transferred to nylon membranes and hybridized with a radiolabeled probe generated from D8BF and D8BR as above. Statistical significance for aberrant sized CFs was performed using Fisher's exact test.
Transformation assay for BIR
The chromosome fragmentation vector, pLS192, containing a 5.2-kb insert from the left arm of chromosome III (SGD coordinates 96821–102096), was digested with SnaBI and 100 ng used to transform competent yeast cells, selecting for Ura+ transformants. The frequency of BIR presented in Table 2 is the number of Ura+ transformants per microgram of linearized DNA transformed divided by the number of Ura+ transformants per microgram of circular pRS416 transformed. The mean BIR frequencies (with standard deviations) presented are from at least 3 independent transformations of each strain.
DSB–induced gene conversion assay
Five mL SC-URA glucose cultures were grown overnight at 30°C. Cells were diluted to a concentration of 1×105 cells/ml in 350 ml supplemented SD raffinose medium. Cultures were grown overnight to a concentration of 3×106 cells/mL and galactose was added to the cultures for a final concentration of 2%. Fifty mL of cells were harvested at each indicated time point after galactose induction. Samples of cells from each time point were serially diluted and plated on solid SC and SC-URA medium to determine plasmid retention. The percent URA3 (plasmid) retention was determined by the fraction of Ura+ colonies 8 hours after induction normalized to the fraction of Ura+ colonies at initiation of the induction. DNA was isolated from each time point and used as template for PCR with primers 3GCA and 5GCA (Table S2) and GoTaq Flexi polymerase (Promega) and the reaction profile of 95°C for 1 minute, 55°C for 1 minute, 72°C for 2 minutes and 30 seconds for 35 cycles. A portion of the PCR reaction was then digested with AatII and analyzed on a 1% agarose gel.
Strand invasion from pLS199 was detected by PCR of genomic DNA from cells collected at different times during the induction. PCR was performed with primers pRS416-2147 and Ade2-2485 (Table S2) with Phusion polymerase (New England Biolabs) and a reaction profile of 98°C for 10 seconds, 55°C for 20 seconds, 72°C for 1 minute 15 seconds for 25 cycles. Reaction products were run on a 1% agarose gel.
Supporting Information
Zdroje
1. KroghBO
SymingtonLS
2004 Recombination proteins in yeast. Annu Rev Genet 38 233 271
2. DaleyJM
PalmbosPL
WuD
WilsonTE
2005 Nonhomologous end joining in yeast. Annu Rev Genet 39 431 451
3. Jinks-RobertsonS
PetesTD
1986 Chromosomal translocations generated by high-frequency meiotic recombination between repeated yeast genes. Genetics 114 731 752
4. FasulloMT
DavisRW
1988 Direction of chromosome rearrangements in Saccharomyces cerevisiae by use of his3 recombinational substrates. Mol Cell Biol 8 4370 4380
5. LlorenteB
SmithCE
SymingtonLS
2008 Break-induced replication: what is it and what is it for? Cell Cycle 7 859 864
6. McEachernMJ
HaberJE
2006 Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75 111 135
7. Voelkel-MeimanK
RoederGS
1990 Gene conversion tracts stimulated by HOT1-promoted transcription are long and continuous. Genetics 126 851 867
8. LemoineFJ
DegtyarevaNP
LobachevK
PetesTD
2005 Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120 587 598
9. SmithCE
LlorenteB
SymingtonLS
2007 Template switching during break-induced replication. Nature 447 102 105
10. VanHulleK
LemoineFJ
NarayananV
DowningB
HullK
2007 Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol 27 2601 2614
11. GravelS
ChapmanJR
MagillC
JacksonSP
2008 DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 22 2767 2772
12. MimitouEP
SymingtonLS
2008 Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455 770 774
13. NealeMJ
PanJ
KeeneyS
2005 Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436 1053 1057
14. ZhuZ
ChungWH
ShimEY
LeeSE
IraG
2008 Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134 981 994
15. WangX
IraG
TerceroJA
HolmesAM
DiffleyJF
2004 Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol 24 6891 6899
16. DeemA
BarkerK
VanhulleK
DowningB
VaylA
2008 Defective Break-Induced Replication Leads to Half-Crossovers in Saccharomyces cerevisiae. Genetics 179 1845 1860
17. LydeardJR
JainS
YamaguchiM
HaberJE
2007 Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448 820 823
18. SmithCE
LamAF
SymingtonLS
2009 Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol Cell Biol 29 1432 1441
19. ChenQ
IjpmaA
GreiderCW
2001 Two survivor pathways that allow growth in the absence of telomerase are generated by distinct telomere recombination events. Mol Cell Biol 21 1819 1827
20. LeS
MooreJK
HaberJE
GreiderCW
1999 RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152 143 152
21. LundbladV
BlackburnEH
1993 An alternative pathway for yeast telomere maintenance rescues est1 - senescence. Cell 73 347 360
22. TengSC
ChangJ
McCowanB
ZakianVA
2000 Telomerase-independent lengthening of yeast telomeres occurs by an abrupt Rad50p-dependent, Rif-inhibited recombinational process. Mol Cell 6 947 952
23. MalkovaA
NaylorML
YamaguchiM
IraG
HaberJE
2005 RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol 25 933 944
24. BoscoG
HaberJE
1998 Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics 150 1037 1047
25. JainS
SugawaraN
LydeardJ
VazeM
Tanguy Le GacN
2009 A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23 291 303
26. DavisAP
SymingtonLS
2004 RAD51-dependent break-induced replication in yeast. Mol Cell Biol 24 2344 2351
27. RuizJF
Gomez-GonzalezB
AguileraA
2009 Chromosomal Translocations Caused by Either Pol32-Dependent or -Independent Triparental Break-Induced Replication. Mol Cell Biol
28. MorrowDM
ConnellyC
HieterP
1997 “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147 371 382
29. MoreauS
MorganEA
SymingtonLS
2001 Overlapping functions of the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNA metabolism. Genetics 159 1423 1433
30. BernsteinKA
ShorE
SunjevaricI
FumasoniM
BurgessRC
2009 Sgs1 function in the repair of DNA replication intermediates is separable from its role in homologous recombinational repair. EMBO J 28 915 925
31. DiedeSJ
GottschlingDE
1999 Telomerase-mediated telomere addition in vivo requires DNA primase and DNA polymerases alpha and delta. Cell 99 723 733
32. SymingtonLS
2002 Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev 66 630 670
33. BartschS
KangLE
SymingtonLS
2000 RAD51 is required for the repair of plasmid double-stranded DNA gaps from either plasmid or chromosomal templates. Mol Cell Biol 20 1194 1205
34. Orr-WeaverTL
SzostakJW
1983 Yeast recombination: the association between double-strand gap repair and crossing-over. Proc Natl Acad Sci U S A 80 4417 4421
35. Welz-VoegeleC
Jinks-RobertsonS
2008 Sequence divergence impedes crossover more than noncrossover events during mitotic gap repair in yeast. Genetics 179 1251 1262
36. AylonY
LiefshitzB
Bitan-BaninG
KupiecM
2003 Molecular dissection of mitotic recombination in the yeast Saccharomyces cerevisiae. Mol Cell Biol 23 1403 1417
37. NagDK
CavalloSJ
2007 Effects of mutations in SGS1 and in genes functionally related to SGS1 on inverted repeat-stimulated spontaneous unequal sister-chromatid exchange in yeast. BMC Mol Biol 8 120
38. StafaA
SvetecI-K
ZgagaZ
2005 Inactivation of the SGS1 and EXO1 genes synergistically stimulates plasmid integration in yeast. Genes, Food Technol Biotechnol 43 103 108
39. ZierhutC
DiffleyJF
2008 Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J 27 1875 1885
40. LydeardJR
Lipkin-MooreZ
JainS
EapenVV
HaberJE
2010 Sgs1 and Exo1 Redundantly Inhibit Break-Induced Replication and de novo Telomere Addition at Broken Chromosome Ends. PLoS Genet 6 e1000973 doi:10.1371/journal.pgen.1000973
41. IraG
MalkovaA
LiberiG
FoianiM
HaberJE
2003 Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115 401 411
42. Tinline-PurvisH
SavoryAP
CullenJK
DaveA
MossJ
2009 Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J 28 3400 3412
43. DaniGM
ZakianVA
1983 Mitotic and meiotic stability of linear plasmids in yeast. Proc Natl Acad Sci U S A 80 3406 3410
44. MyungK
DattaA
ChenC
KolodnerRD
2001 SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet 27 113 116
45. SpellRM
Jinks-RobertsonS
2004 Examination of the roles of Sgs1 and Srs2 helicases in the enforcement of recombination fidelity in Saccharomyces cerevisiae. Genetics 168 1855 1865
46. SchmidtKH
WuJ
KolodnerRD
2006 Control of translocations between highly diverged genes by Sgs1, the Saccharomyces cerevisiae homolog of the Bloom's syndrome protein. Mol Cell Biol 26 5406 5420
47. NicholsonA
HendrixM
Jinks-RobertsonS
CrouseGF
2000 Regulation of mitotic homeologous recombination in yeast. Functions of mismatch repair and nucleotide excision repair genes. Genetics 154 133 146
48. ShermanF
FinkG
HicksJ
1986 Methods in Yeast Genetics. Cold Spring
Harbor, N.Y. Cold Spring Harbor Laboratory
49. ItoH
FukudaY
MurataK
KimuraA
1983 Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153 163 168
50. ZouH
RothsteinR
1997 Holliday junctions accumulate in replication mutants via a RecA homolog-independent mechanism. Cell 90 87 96
51. HuangKN
SymingtonLS
1994 Mutation of the gene encoding protein kinase C 1 stimulates mitotic recombination in Saccharomyces cerevisiae. Mol Cell Biol 14 6039 6045
52. MozlinAM
FungCW
SymingtonLS
2008 Role of the Saccharomyces cerevisiae Rad51 paralogs in sister chromatid recombination. Genetics 178 113 126
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 7
- Souvislost haplotypu M2 genu pro annexin A5 s opakovanými reprodukčními ztrátami
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
Nejčtenější v tomto čísle
- CHD7 Targets Active Gene Enhancer Elements to Modulate ES Cell-Specific Gene Expression
- Extensive DNA End Processing by Exo1 and Sgs1 Inhibits Break-Induced Replication
- Question and Answer: An Anniversary Interview with Jane Gitschier
- Multi-Variant Pathway Association Analysis Reveals the Importance of Genetic Determinants of Estrogen Metabolism in Breast and Endometrial Cancer Susceptibility
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy