
FACT Prevents the Accumulation of Free Histones Evicted from Transcribed Chromatin and a Subsequent Cell Cycle Delay in G1
The FACT complex participates in chromatin assembly and disassembly during transcription elongation. The yeast mutants affected in the SPT16 gene, which encodes one of the FACT subunits, alter the expression of G1 cyclins and exhibit defects in the G1/S transition. Here we show that the dysfunction of chromatin reassembly factors, like FACT or Spt6, down-regulates the expression of the gene encoding the cyclin that modulates the G1 length (CLN3) in START by specifically triggering the repression of its promoter. The G1 delay undergone by spt16 mutants is not mediated by the DNA–damage checkpoint, although the mutation of RAD53, which is otherwise involved in histone degradation, enhances the cell-cycle defects of spt16-197. We reveal how FACT dysfunction triggers an accumulation of free histones evicted from transcribed chromatin. This accumulation is enhanced in a rad53 background and leads to a delay in G1. Consistently, we show that the overexpression of histones in wild-type cells down-regulates CLN3 in START and causes a delay in G1. Our work shows that chromatin reassembly factors are essential players in controlling the free histones potentially released from transcribed chromatin and describes a new cell cycle phenomenon that allows cells to respond to excess histones before starting DNA replication.
Published in the journal:
. PLoS Genet 6(5): e32767. doi:10.1371/journal.pgen.1000964
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000964
Summary
The FACT complex participates in chromatin assembly and disassembly during transcription elongation. The yeast mutants affected in the SPT16 gene, which encodes one of the FACT subunits, alter the expression of G1 cyclins and exhibit defects in the G1/S transition. Here we show that the dysfunction of chromatin reassembly factors, like FACT or Spt6, down-regulates the expression of the gene encoding the cyclin that modulates the G1 length (CLN3) in START by specifically triggering the repression of its promoter. The G1 delay undergone by spt16 mutants is not mediated by the DNA–damage checkpoint, although the mutation of RAD53, which is otherwise involved in histone degradation, enhances the cell-cycle defects of spt16-197. We reveal how FACT dysfunction triggers an accumulation of free histones evicted from transcribed chromatin. This accumulation is enhanced in a rad53 background and leads to a delay in G1. Consistently, we show that the overexpression of histones in wild-type cells down-regulates CLN3 in START and causes a delay in G1. Our work shows that chromatin reassembly factors are essential players in controlling the free histones potentially released from transcribed chromatin and describes a new cell cycle phenomenon that allows cells to respond to excess histones before starting DNA replication.
Introduction
The FACT complex plays an important role by allowing RNA polymerase II (Pol II) to transcribe through chromatin (reviewed by [1], [2]), the only factor known to date that is able to stimulate Pol II-dependent transcription elongation through chromatin in a highly purified system [3], [4]. Yeast FACT is required in vivo to transcribe genes with highly positioned nucleosomes at the 5′ end of the transcribed region [5], and several lines of evidence of other organisms also support that FACT plays an important role in transcription elongation in vivo [6]–[10]. In spite of its role in elongation, several in vivo and in vitro approaches indicate an additional role of yFACT in establishing transcription initiation complexes by promoting TBP binding to core promoters in a TFIIA-dependent manner [11], [12]). Finally and in addition to its role in transcription, FACT also plays an important function during DNA replication [13]–[15].
In humans, the FACT complex is composed of two proteins, p140 and SSRP1, which are highly homologous to the essential yeast proteins Spt16/Cdc68/Ssf1 (hereafter referred to as Spt16) and Pob3, respectively [16]. SPT16 had been previously identified as both a CDC gene [17], and also as a recessive suppressor of the deletion of SWI4, a transcription factor required for the high-level expression of the G1 cyclin genes, CLN1 and CLN2 [18]. Besides, Spt16 had also been described as a protein involved in transcription since several spt16 alleles suppress the transcriptional effects of Ty insertions in yeast (Spt- phenotype) [19].
yFACT has been reported to interact physically or genetically with other factors related to histone modifications and chromatin remodeling, like the Paf complex, the ATP-dependent chromatin factor Chd1 and the NuA3 histone acetyltransferase complex [11], [20]–[22]. A reciprocal regulation of the FACT function by H2B ubiquitination has also been described [23]. In agreement with these findings, yFACT and the HMG-box protein Nhp6 have been shown to form a heterodimer capable of binding nucleosomes [24] and of reorganizing them in vitro [25], [26]. Both the yFACT subunits are able to bind H3/H4 tetramers and H2A/H2B dimers, sometimes in a functionally redundant manner [27], [28]. These interactions are thought to allow FACT to destabilize nucleosomes during transcription [26], [29].
Some spt16 alleles are synthetically lethal with mutations affecting chromatin assembly [30]. Moreover, they lead to the activation of cryptic transcription initiation sites within coding regions, indicating that FACT, together with other factors like Spt6, also plays a role in maintaining the integrity of the chromatin structure during transcription [9], [31]–[34].
Several spt16 mutants show defects while progressing through START, the main regulatory event in the G1 phase of the cell cycle [17], [35]. At a non-permissive temperature, the G1 phenotype of these spt16 mutants has been accounted for by the drastic reduction in the expression of CLN1, CLN2 and CLN3, the genes encoding the three G1 cyclins [35], [36]. CLN1 and CLN2 are able to self-regulate their expression by a positive feed-back mechanism [37], but the regulation of G1 length requires the activation of the cyclin-dependent kinase Cdc28 (Cdk1) by Cln3 [38]–[41]. Cln3-associated Cdk1 binds SBF (Swi4-Swi6) to the CLN1 and CLN2 promoters where it phosphorylates the negative regulator of START, Whi5 [42]. This phosphorylation promotes its release from SBF and leads to the activation of the CLN1 and CLN2 promoters [43], [44]. SBF-dependent recruitment of FACT plays an important role in this activation, which promotes the G1/S transition [45]. Notably, the kinase activity of Cln1,2-Cdk1 triggers the degradation of the cyclin-dependent kinase inhibitor Sic1 which no longer inhibits the S phase-promoting complex Clb5,6-Cdc28 [46], [47].
Another key regulatory process during the G1/S transition is the induction of histone genes, which allows the coupling of bulk histone synthesis to ongoing DNA replication. In proliferating cells, the synthesis of the vast majority of histones occurs during the S-phase of the cell cycle. The tight cell cycle regulation of the histone genes results from their transcriptional repression in phases G1 and G2, their transcriptional activation just before the S-phase and the post-transcriptional regulation of their mRNAs. During the S-phase, histone genes can also respond to changes; for instance, the accumulation of histones in response to the genotoxic agents interfering with DNA replication induces their repression (reviewed by [48]).
In recent years, a novel mechanism in budding yeast preventing the accumulation of free histones and which is superimposed upon the regulation of histone gene transcription and mRNA stability has been described [49]. This mechanism involves the use of the DNA damage checkpoint protein kinase Rad53 as part of a surveillance process that not only monitors the accumulation of excess histones, but also induces their degradation. This degradation is controlled by phosphorylation and is carried out by the proteosome in an ubiquitylation-mediated manner [50]. Excess histones are thought to be generated at the end of the normal S-phase or in response to an abrupt decrease of DNA synthesis following DNA damage. This mechanism is dependent on neither the checkpoint kinases Mec1 and Tel1 nor other DNA-damage checkpoint proteins (reviewed in [51]).
Some aspects of the G1 phenotype of the spt16 mutants remain unknown. It is not clear whether FACT plays a direct role in the regulation of CLN3. Alternatively, the decreased expression of CLN3 might be a physiological response to FACT inactivation. We investigated this question and found that the inactivation of FACT down-regulates the CLN3 promoter in START. This phenomenon coincides with the accumulation of free histones and is enhanced by the mutation of the free histones controller, RAD53. We also found that the forced entry of FACT-deficient cells into the S-phase lowers their viability. Finally, we discovered that the overexpression of histones in the wild-type cells decreases CLN3 transcription in START and leads to a delay in G1. We propose that the accumulation of free histones triggers the down-regulation of CLN3, thereby contributing to control the excess histones before starting DNA replication. We further propose that the main potential source of free histones is transcribed chromatin and that chromatin reassembly factors play an essential protective role in this respect.
Results
FACT inactivation causes a down-regulation of the CLN3 promoter in START
The arrest of yeast cells in G1 after Spt16 inactivation has been suggested to be a possible direct consequence of a very strict requirement of Spt16 for CLN1, CLN2 and CLN3 transcription [35]. FACT has been shown to participate directly in the activation of the CLN1 and CLN2 promoters after START [45]. In order to explore the effect of FACT inactivation on the previous step, we quantified the mRNA levels of CLN3 in alpha factor-synchronized spt16-197 cells at a non-permissive temperature (see Materials and Methods for the ranges of permissive and non-permissive temperatures ranges). We treated cells for two hours with the pheromone at 30°C, then for one a further hour at either 30°C or 35°C in the continuous presence of alpha-factor. Next we released the cells from the arrest and analyzed CLN3 mRNA by Northern blotting (Figure 1). In agreement with previously published results based on asynchronous cells [35], the CLN3 mRNA levels in the pheromone-treated spt16-197 cells at 35°C were clearly lower than in the wild-type cells (see time 0 in Figure 1B). When cultures were released from alpha-factor, the wild-type cells progressed into the S-phase at any temperature, whereas spt16-197 cells entered the S-phase only at 30°C, with most spt16-197 cells remaining in G1 at the restrictive temperature (see times 10, 20 and 45 min in Figure 1A). The CLN3 mRNA levels in the spt16-197cells released from alpha-factor at 37°C remained very low. In contrast, the mRNA levels of ADH1, a constitutive non-cycling gene, were only partially affected by Spt16 inactivation (Figure 1B), which is in agreement with the limited effect of spt16-197 at restrictive temperatures in a broad set of non-cyclin genes [35], [36]. The occupancy of CLN3 and ADH1 by the RNA polymerase II (RNApol II) paralleled their mRNA levels. As Figure 1C depicts, the amount of RNApol II bound to the CLN3 transcribed region at 35°C was significantly lower in spt16-197 (52%) than in the wild type, whereas the variation of RNApol II binding to ADH1 was slighter.

The fact that the CLN3 expression markedly reduced in response to Spt16 inactivation might be due to either a direct effect of FACT dysfunction on CLN3 transcription or a yet unidentified signaling pathway targeting the CLN3 promoter in response to FACT inactivation. In order to distinguish between these possibilities, we first analyzed the response of a reporter fused to the CLN3 promoter upon Spt16 inactivation in alpha-factor-synchronized cells. To construct this fusion, we combined the entire intergenic region between the neighboring CLN3 and CYC3 coding regions and the coding region of E. coli lacZ (Figure 2A). We chose lacZ since we had previously showed that the transcription elongation of this reporter gene is not sensitive to FACT dysfunction [5]. As shown in Figure 2B, lacZ mRNA was hardly detectable when the synchronized spt16-197 cells were incubated at the restrictive temperature, indicating that the CLN3pr::lacZ reporter (fusion 1) behaved similarly to the endogenous CLN3 mRNA. In contrast, the mRNA levels of an ADH1pr::lacZ transcriptional fusion did not lower under the same conditions. This result indicates that the promoter region of CLN3 mediates the drop in its mRNA levels after FACT inactivation.

We further constructed additional CLN3pr::lacZ fusions containing different segments of the CLN3-CYC3 intergenic region (Figure 2A). No difference in copy number among the reporter plasmids was detected by quantitative PCR (Figure S1). We measured the lacZ mRNA levels expressed by these fusions in the spt16-197 cells synchronized in START. The expression patterns of the complete CLN3pr::lacZ (fusion 1) and ADH1pr::lacZ were in agreement with the experiments described above (Figure 2C). Fusion 2, which contains the entire intergenic region, except the CLN3 5′UTR and the CYC3 termination region (−999, −339) (Figure 2A), retained sensitivity to Spt16 inactivation as it dropped as the lacZ mRNA levels did after Spt16 inactivation (Figure 2C). A significant influence of mRNA instability in this decrease was ruled out because two completely different mRNAs (lacZ and CLN3) responded similarly to FACT inactivation when they were driven by the same promoter (CLN3pr). In contrast, when lacZ mRNA was driven by the ADH1 promoter, it did not show any significant variation in spt16-197 at the restrictive temperature (Figure 2C). In short, spt16-197 does not influence the expression of CLN3 at a post-initiation level. However, Spt16 may also play a role in transcription initiation by facilitating TBP binding to the core promoters. We constructed CLN3pr::lacZ fusion number 3 (−472, −389), which contains the minimal core promoter of CLN3 (Figure 2A). In spite of the weak expression of this fusion (five fold lower than fusion number 2 in asynchronous cultures), its mRNA level was not significantly influenced by Spt16 inactivation (Figure 2C). These results, in addition to the absence of FACT binding to the CLN3 promoter during START (David Stillman, personal communication), indicate that the effect of Spt16 inactivation on the expression of CLN3 in START is not due to the specific involvement of FACT in the transcription of this gene in this particular step of the cell cycle. Instead, our results suggest the existence of a control mechanism mediated by the promoter of CLN3 which regulates the G1/S transition in response to FACT dysfunction. Such a mechanism might protect the cell from the deleterious effect of entering the S-phase under these conditions. We tested this hypothesis by forcing the entry of spt16-197 cells into the S-phase. First, we went about this by overexpressing CLN3 with a Tetoff::CLN3 construct (negatively regulated by doxycycline) which suppressed the accumulation of spt16-197 cells in G1 at a restrictive temperature (Figure 3A). We observed a negative effect of the overexpression of CLN3 on the viability of spt16-197 cells at a semi-permissive temperature (Figure 3B). Similar results were obtained in those cells lacking Sic1, the inhibitor of the Cdc28-Clb complexes that negatively regulates entry into the S-phase (Figure 3C and 3D). These data reveal that the forced progression of spt16-197 cells into the S-phase at semi-permissive temperatures is deleterious; therefore, the G1 delay triggered by the inactivation of FACT is cell-protective.
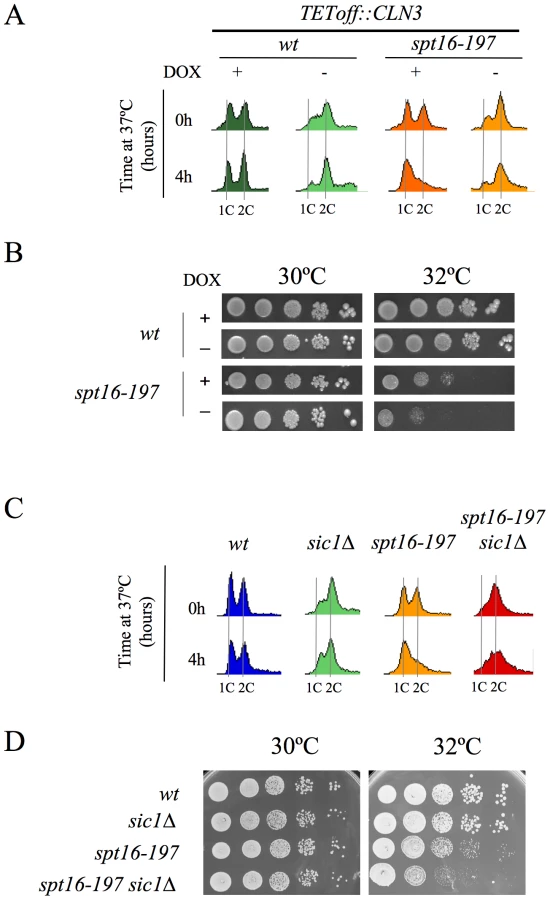
The lethality associated with FACT dysfunction relates to the accumulation of the free histones evicted during transcription
The association between impairment of transcription elongation and genome instability is well established [52]–[54]. Since FACT plays a role during elongation, we wondered whether the G1 delay induced by FACT inactivation could be due to the action of the canonical DNA-damage checkpoint. To test this hypothesis, we investigated the possible implication of Rad9, a component of the DNA-damage sensor machinery operating in G1 [55], [56]. We performed a FACS analysis with strains carrying the spt16-197 and rad9Δ mutations. The profile of the spt16-197 rad9Δ double mutant after four hours at a restrictive temperature was almost identical to that of the single spt16-197 mutant (Figure S2A), indicating that the G1 arrest produced by FACT inactivation is proficient in the absence of Rad9. Accordingly, no genetic interaction between spt16-197 and rad9Δ was detected when we analyzed the thermosensitivity of the double mutant (Figure S2B). We further tested whether spt16-197 exhibited any aggravation of its ts phenotype in the absence of Mec1, the key kinase of the DNA-damage checkpoint pathway [57]. As with rad9Δ, the deletion of MEC1 in spt16-197 did not affect its thermosensitive (ts) phenotype (Figure 4A).
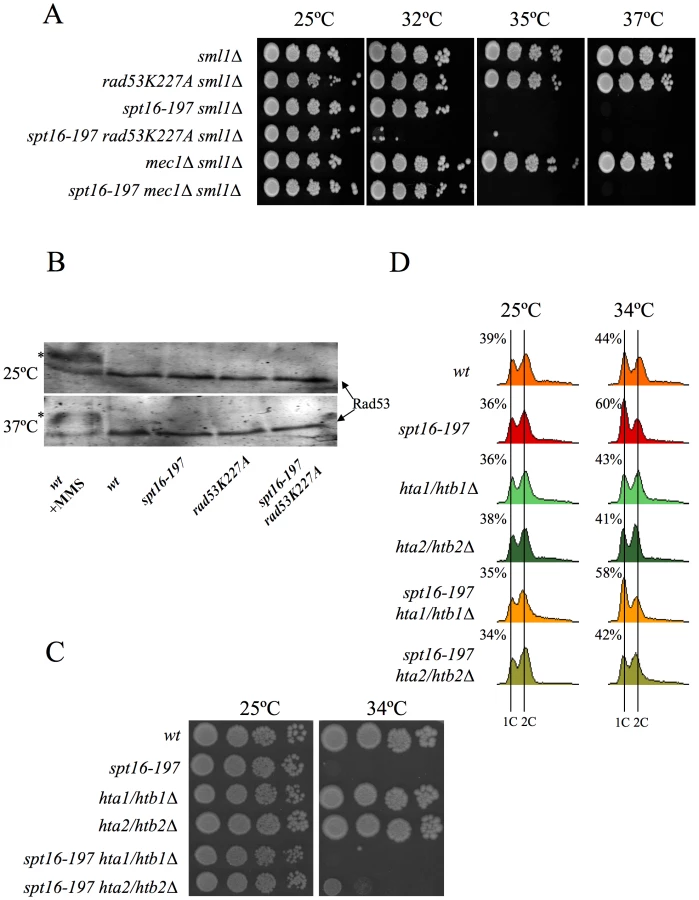
It was intriguing to note that of all the DNA-damage checkpoint genes we tested, only the mutation of RAD53, which encodes another essential protein kinase for the DNA-damage checkpoint [58], led to a clear genetic interaction with spt16-197. Indeed the rad53K227A allele, which abolishes most Rad53 kinase activity, dramatically enhanced the thermosensitive phenotype of spt16-197. While the growth of the spt16-197 and rad53K227A single mutants was virtually not affected at 32°C, the double spt16-197 rad53K227A mutant was unable to grow at this temperature (Figure 4A). Similar results were obtained with the complete deletion of RAD53 (Figure S3). Unlike what happens following DNA damage, Rad53 was not hyperphosphorylated in the spt16-197 mutant at a restrictive temperature (Figure 4B). Hence we conclude that Rad53 kinase activity is required to alleviate the deleterious effects of the spt16-197 mutation, irrespectively of the role it plays in the DNA damage response.
As we mentioned in the Introduction, Rad53 is involved in the detection and subsequent degradation of excess histones without becoming phosphorylated and independently of its role in the DNA damage checkpoint [49]. Since FACT is involved in chromatin transactions during transcription, we hypothesized that the dysfunction of Spt16 might cause an increase in free histones, which would need to be targeted for degradation by Rad53. The mutations lowering the H2A–H2B dosage have been described to enhance the viability of the spt16 mutants [30]. Accordingly, we observed how the deletion of HTA2–HTB2, one of the two loci encoding H2A and H2B, partially suppressed the ts phenotype of spt16-197 at restrictive (Figure 4C) and semi-restrictive temperatures (Figure S4A). The hta2Δhtb2Δ deletion also suppressed the accumulation of spt16-197 cells in G1 at a restrictive temperature (Figure 4D). In contrast, the deletion of the HTA1–HTB1 locus caused no suppression in either the ts phenotype (Figure 4C and Figure S4A) or the G1 delay (Figure 4D). The HTA1–HTB1 locus has been shown to be essential for viability and the hta1Δhtb1Δ strain we used (FY710) is only alive because of an extra-chromosomal copy of HTA2–HTB2 [59]. We confirmed the presence of this extra-chromosomal copy of HTA2–HTB2 not only in FY710, but in the isogenic spt16-197 hta1/htb1Δ double mutant (Figure S4B). The main difference between these two histone loci lies in their regulation. The expression of HTA1–HTB1 is sensitive to the levels of histones, whereas HTA2–HTB2 is not [60]. In order to properly compare the effect of the two loci on the ts phenotype of spt16-197, we engineered new strains containing all the viable combinations of the H2A/H2B-encoding loci. We found that the presence of two copies of the HTA2–HTB2 (non responsive to free H2A and H2B) led to a more severe thermosensitivity than the presence of two copies of the HTA1–HTB1 locus (repressible by free histones) (Figure S4A). Moreover the deletion of the regulatory sequence (NEG), which mediates the repression of HTA1–HTB1 in response to histone levels, enhanced the ts phenotype of spt16-197 (Figure S4A). Taken together, these results suggest that the accumulation of free H2A and H2B contributes to the lethality of spt16-197 at high temperatures and that the accumulation of spt16-197 cells in G1 responds to free histones.
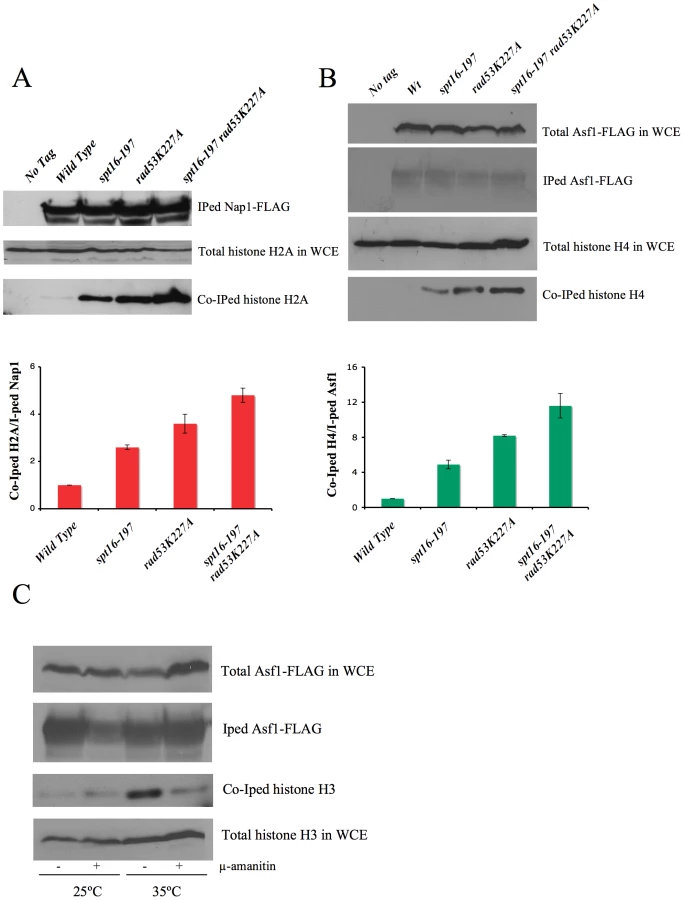
In order to confirm this hypothesis, we analyzed the amount of non chromatin-associated histones in spt16-197 in either the absence or presence of Rad53 kinase activity. The huge amount of histones present in the chromatin fraction makes it extremely difficult to quantify reproducibly free histones pools in a direct manner. Instead, we measured the amount of histones associated with soluble histone chaperones, a well-characterized procedure that allows a reproducible measurement of free histones [49], [50]. We performed co-immunoprecipitation assays using the H2A–H2B chaperone Nap1 (Figure 5A) and the H3–H4 histone chaperone Asf1 (Figure 5B) fused to the FLAG epitope. The spt16-197 mutation did not produce a significant effect on the levels of Nap1-FLAG and Asf1-FLAG detected in the extracts (Figure S5A and S5B). We quantified the amount of H2A co-immunoprecipitated with Nap1 in exponentially growing cells after switching them to the non-permissive temperature. We saw a clear increase in the accumulation of free H2A in Nap1 in the spt16 and rad53 mutants compared to the wild-type cells, and an even higher level of co-immunoprecipitated H2A in the double mutant (Figure 5A). Next we performed a similar experiment with Asf1-FLAG. Wild-type cells exhibit very little H4 associated with Asf1 under normal growth conditions at 25°C (Figure 5B). We found that, even at this permissive temperature, the spt16-197 mutation increased the amount of histones associated with Asf1 up to levels close to those shown by rad53K227A (Figure 5B). One again, the accumulation of free H4 in the double mutant exceeded the levels of the single mutants (Figure 5B). To test whether this increase of free histones in FACT-deficient cells was taking place in G1, independently of the histone synthesis that takes place during the S-phase, we incubated alpha factor-synchronized spt16-197 cells for two hours at either 25°C or 35°C. As expected, the amount of free histones associated with Asf1 in the spt16-197 mutant increased at the restrictive temperature. We also observed that inhibiting RNApol II transcription with alpha-amanitin prevented this increase (Figure 5C). Northern blot experiments showed no misregulated expression of the histone genes in spt16-197 during START (Figure S5D). Taken together, these results are compatible with a scenario in which Pol II-dependent transcription in the absence of active FACT causes an accumulation of the evicted histones, which become toxic to the cell if not targeted for degradation by Rad53.
Excess histones induce a G1 cell cycle delay
Our results pose an intriguing question about a possible link between the CLN3-dependent G1 delay and the presence of excess histones, both induced by FACT dysfunction. We addressed this question by testing whether the enhancement of the spt16-197 thermosensitivity caused by rad53K227A correlated with the observed delay in G1. As Figure 6A depicts, an asynchronous culture of the double mutant exhibited a stronger and faster accumulation of cells in G1 compared to the single spt16-197 mutant when they were shifted to a restrictive temperature. Moreover, the alpha-factor-synchronized spt16-197 rad53K227A cells also displayed a much slower entry into the S-phase than the single spt16-197 or rad53K227A mutants when the mating pheromone was removed from the medium at a semi-permissive temperature. Even at 25°C, the double mutant displayed a slower exit from G1, although the double mutant exhibited a proportionally stronger defect in the G1-S progression at 32°C, as compared to the single mutants (Figure 6B).
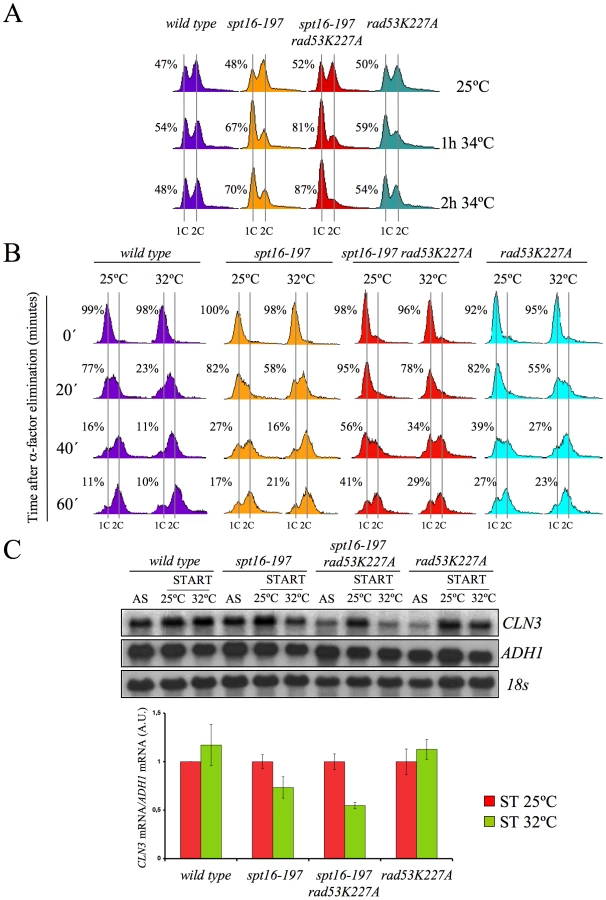
In order to establish a correlation between the excess histones and the CLN3 mRNA levels in START, we measured them under conditions in which the mutant's general transcriptional capacity is not significantly affected, but the level of free histones increases. To do this, we pre-synchronized the spt16-197 rad53K227A cells with alpha-factor and analyzed the CLN3 mRNA levels one hour after switching them to 32°C in the continued presence of pheromone. The results show a more marked reduction of CLN3 mRNA after one hour at 32°C in the double spt16-197 rad53K227A mutant than in the single spt16-197 mutant (Figure 6C). Therefore, the absence of Rad53 kinase activity enhances the down-regulation of CLN3 produced by the dysfunction of the spt16-197 allele.
If our hypothesis that transcriptionally evicted histones trigger the G1 delay of spt16-197 is true, we should then expect the other mutants affected in chromatin reassembly to also exhibit similar cell cycle defects. In addition to FACT, another important factor that participates in chromatin reassembly during transcription is Spt6 [61]. We found that the viability of cells bearing the mutant allele spt6-1004 clearly lowered in the absence of Rad53 kinase activity, but remained unchanged in the absence of Mec1 (Figure 7A). We also found that spt6-1004 cells clearly accumulated in G1 when shifted to a restrictive temperature in both asynchronous and alpha factor-synchronized cells (Figure 7B and 7C). As in the spt16-197 cells, the CLN3 mRNA levels, or the lacZ mRNA levels when driven by the CLN3 promoter, decreased when alpha factor-synchronized spt6-1004 cells were shifted to a restrictive temperature (Figure 7D).

Given these results, we thought it would be interesting to determine whether the overexpression of histones in G1 induces a cell cycle delay in wild-type cells. We found that an additional copy of the HTA1–HTB1 locus produced a very slight delay in cell cycle progression when alpha factor-synchronized cells were released from the pheromone. In contrast, a copy of the same locus lacking the regulatory sequence that mediates its repression in response to histone levels (ΔNEG) led to a clear accumulation of cells in G1 (asynchronous culture) and a more marked delay in the entry of synchronized cells into the S-phase (Figure 8A). Accordingly, we detected a more significant decrease of the CLN3 mRNAs in START, in those cells bearing the deregulated HTA1–HTB1 copy (ΔNEG) than in those transformed with the intact allele (Figure 8B). Since all the cells in this experiment were held in START by the presence of the mating pheromone, the drop in CLN3 mRNA could not be an indirect consequence of free histones inhibiting cell cycle progression. We conclude, therefore, that excess histones downregulate CLN3 in the wild-type cells during G1 and subsequently delay their progression through the G1/S transition.

Discussion
The primary objective of our study was to understand the defects in the G1/S progression exhibited by the spt16-197 mutant. The genetic and molecular evidence described in the Results section indicates that the dysfunction of Spt16 down-regulates the expression of CLN3 in START by triggering a regulatory mechanism that specifically represses the CLN3 promoter. Our results also reveal that the G1 delay undergone by the spt16 mutant is not mediated by the DNA-damage checkpoint, although the rad53K227A mutation enhances both the thermosensitivity of spt16-197 and its G1 phenotype. This result, in combination with the lack of phosphorylation of Rad53 after Spt16 inactivation, indicates that excess histones are involved in this phenomenon. We confirm that this hypothesis is true by showing that the Spt16 dysfunction produces an accumulation of free histones associated with histone chaperones in G1 (Figure 5C), and that excess histones induce a delay in the otherwise wild-type cells during the G1/S transition concomitantly with CLN3 downregulation.
Chromatin as a potential source of free histones
It is well known that the accumulation of non-nucleosomal histones in the cell is toxic [62] and that this toxicity is normally avoided by regulating the histone gene expression at both the transcriptional and posttranscriptional levels [60]. Obviously, these mechanisms are incapable of controlling excess histones when they originate by eviction from chromatin due to a dysfunction in chromatin reassembly during transcription. We demonstrate herein that Spt16 inactivation results in the accumulation of non-chromatin bound histones in G1. The only way for the cell to avoid the toxic effects of these excess histones is to degrade them in a process that is mediated by Rad53 [49]. Our results show that the absence of Rad53 kinase activity lowers the viability of the spt16-197 mutant and suggest that one of the roles of the Rad53-dependent histone degradation mechanism is the elimination of those histones evicted by the transcriptional activity that are not reassembled into chromatin. Beyond the S-phase, transcribed chromatin is probably the main source of free histones in yeast cells, presumably due to minor imbalances between histone supply and demand during chromatin reassembly. The general similarities between histone trafficking during all the chromatin transactions suggests that DNA repair or DNA replication might also result in the excess of free histones when chromatin assembly is dysfunctional [63].
Our results suggest that FACT, in addition to avoiding initiation from cryptic promoters [9], [31], is a protective factor against the toxic risk represented by evicted histones. A recent publication reports how Spt16 promotes the redeposition of the original H3 and H4 histones evicted by elongating Pol II [34]. Our results agree with this conclusion since we have detected a clear accumulation of free H3 and H4 in Spt16-deficient cells. However, we have also noted an increase in the free H2A pool. This result and the genetic interactions between spt16-197 and the H2A–H2B loci also indicate that Spt16 plays a role in preventing the accumulation of evicted H2A and H2B. In fact, a high H2A–H2B/H3–H4 gene ratio impairs the spt16-11 growth, whereas a low H2A–H2B/H3–H4 gene ratio improves it [30], suggesting that the accumulation of free H2A–H2B caused by Spt16 dysfunction may be more relevant for the phenotypes observed.
It has been recently demonstrated that FACT promotes the transition between the canonical nucleosome configuration and a looser, more dynamic structure that involves changes in the interaction of the four core histones with the DNA [26]. According to this view, FACT would be essential for the maintenance of this altered configuration during transcription elongation by limiting the amount of the four histones joining the free pools. One prediction of this model is that those histone mutations which destabilize this alternative nucleosomal configuration would promote free histone accumulation. Some H4 mutations affecting H3–H4 tetramer/H2A–H2B dimer interactions show delayed G1/S transition and reduced CLN3 expression levels [64]. Accordingly, we detected a negative synthetic interaction between one of these mutations (hhf1-36, bearing the H4-Y72G mutation) and rad53K227A (Figure S6).
The protective role against evicted histones is probably not an exclusive function of FACT, but is also a function of the other factors that cooperate during chromatin reassembly, like Spt6, for which we show some evidence. It is likely that Asf1, Nap1, the Hir complex or Nsr1 also protect against evicted histones. Asf1 has been seen to act as an intermediary in the parental histone disassembly/reassembly during replication as it associates with the MCM proteins at the replication fork [65]. An analogous situation might exist during transcription whereby Asf1 or the HIR complex, both of which have been shown to be capable of chromatin assembly outside the S-phase [66], [67], may act as an intermediary in the histone eviction and reassembly process in conjunction with FACT. Nap1 promotes nucleosome assembly by eliminating nonnucleosomal H2A- and H2B-DNA interactions and it prevents aberrant transcription by avoiding excessive H2A–H2B binding to DNA [68].
In this study we also show how the overexpression of H2A–H2B results in G1 delays in the wild-type cells. Similar effects, but to a lesser extent, were obtained by overproducing H3–H4 (data not shown). These results demonstrate that a histone-mediated G1 delay can be obtained in a background with no possible indirect effect mediated by either the role of FACT in the expression of the G1-S regulators, or the function of Rad53 in the control of early replication.
CLN3-dependent G1 delay in response to free histones
Our results show that the G1-delay caused by Spt16 dysfunction correlates with a down-regulation of the transcriptional activity of the CLN3 promoter. Cln3 shortage can only explain a transient G1-delay as cells can enter the S-phase after a prolonged G1 in the absence of Cln3. Therefore it is likely that the permanent G1 arrest shown by spt16-197 cells at high temperatures (over 37°C) is caused by a combination of the CLN3-mediated transient delay and additional defects on the expression of other G1/S regulators caused by Spt16 inactivation. For instance, the MluI cell-cycle boxes (MCB)-mediated activation of SWI4 and SWI6 is also negatively affected by FACT dysfunction [18]. The CLN1 and CLN2 promoters are two other clear targets of Spt16 inactivation, since FACT binds them during G1 and participates in their activation after START [45]. However, the marked reduction in the CLN3 expression following FACT inactivation cannot be caused by a direct involvement of FACT in the transcriptional activity of the CLN3 promoter during G1 because it does not bind this promoter during this phase of the cell cycle (David Stillman, personal communication). We show herein that a CLN3pr::lacZ fusion lacking any sequence in common with CLN3 mRNA is also responsive to the inactivation of Spt16 or Spt6 under conditions in which a constitutively expressed ADH1pr::lacZ is not. Furthermore, rad53K227A enhances the level of CLN3 down-regulation caused by spt16-197. Finally, the overexpression of H2A–H2B in an otherwise wild-type strain decreases the CLN3 expression. Taken together, these results support the existence of a mechanism which down-regulates the CLN3 promoter during G1 in response to the accumulation of free histones caused by either defects in cotranscriptional chromatin reassembly or histone genes deregulation.
CLN3 mediates several regulatory pathways by coupling the cell-cycle progression in G1 to physiological conditions, including nitrogen deprivation [69], daughter cell G1-delay [70], [71] and changes in glucose levels [72]. In the last case, the up-regulation of the CLN3 promoter is due to the binding of the Azf1 transcriptional activator. Another transcription factor shown to regulate the CLN3 promoter is the Mcm1-containing early cell-cycle box (ECB) binding complex [73]. Homeodomain repressors, Yox1 and Yhp1, restrict the ECB-dependent activation of the CLN3 transcription to the M/G1 phase [74]. The deletion of AZF1, YOX1 or YHP1 does not alter the accumulation of cells in G1, as indicated by spt16-197 at the restrictive temperature (data not shown). Moreover, the binding of Mcm1 to the CLN3 promoter, measured by chromatin immunoprecipitation, was not affected by Spt16 inactivation (data not shown). We conclude that the transcriptional regulation of CLN3 in response to the accumulation of free histones is not mediated by any of the known transcription factors operating on the CLN3 promoter. It is even conceivable that the promoter itself acts as a sensor of free histone concentration and is repressed in response to the excess histones.
In mammalian cells, histone overexpression slows down entry into and progression through the S-phase [65]. Interestingly, depletion of human Spt16 leads to the repression of the H1, H2A and H2B genes [75], which could be the result of the accumulation of the free histones in human cells after FACT dysfunction. Given the analogy between the G1-S regulators in yeast (Cln3-SBF-Whi5-Rpd3) and mammals (CyclinD1-E2F-Rb-HDAC1) [42], [45], the functional link between the accumulation of free histones and the regulation of the G1-S transition may be evolutionarily conserved.
Chromatin repair
The free histones evicted by the transcriptional activity of cells can potentially associate non-specifically with DNA via electrostatic interactions, and may give rise to aberrant chromatin structures which can be considered a form of chromatin damage. As with DNA damage, which enhances the risk of genome instability, excess free histones may have serious implications for the normal progression of DNA replication since the toxicity of free histones is maximal in the S-phase [49]. Consistently with this hypothesis, the experimental conditions under which the spt16-induced G1 delay is overcome (CLN3 overexpression, SIC1 deletion) involve an overall decrease in cell viability. The G1 delay should allow cells to reduce the free histone levels through the Rad53-mediated histone degradation pathway before entering the S-phase. The persistence of excess histones, as in the spt16-197 rad53K227A double mutant, would lead to severe replication dysfunctions. Accordingly, chromatin repair would be the combination of DNA repair, chromatin reassembly and excess histone degradation. In this sense, it is interesting to note that Rad53, which participates in both the DNA damage checkpoint and the excess histone degradation pathway, may act as a super-integrator of chromatin repair functions. This new concept could serve as a convenient framework to gain a better understanding of global genomic defects.
Materials and Methods
For further details, see Text S1.
Yeast strains and general procedures
All the yeast strains used in this study were derived from the S288C genetic background, unless otherwise indicated, and are listed in Table S1. In our background, temperatures over 33°C were restrictive for spt16-197 growth, whereas temperatures below 31°C were permissive. All the experiments including spt16-197 mutants were performed at several temperatures. For each experiment shown in the Results section, we chose the maximal restrictive temperature at which specific reproducible results were obtained. Standard procedures were followed for cell culturing, synchronization at START and flow cytometry [76], [77].
Northern blot analyses
The Northern blot analyses were performed as previously described [5]. Six micrograms of total RNA prepared from yeast cells underwent electrophoresis on formaldehyde-agarose gels transferred to Hybond–N filters and UV crosslinked prior to hybridization at 65°C in 0.5M sodium phosphate buffer pH7 7% SDS with a [32P]dCTP-labeled DNA probe. Quantification of the mRNA levels was performed in a phosphorimager (FLA-3000, FujiFilm); the data are provided in arbitrary units. All the values were normalized in relation to the amount of 25S rDNA detected by hybridization with a 32P-oligolabeled 589 bp 25S rRNA internal fragment obtained by PCR and by using the 19-mer oligonucleotides TTGGAGAGGGCAACTTTGG and CAGGATCGGTCGATTGTGC. For the mRNA histone analysis, the whole coding regions of HTA1 and HHT1 were used as probes.
Chromatin immunoprecipitation
Pol II ChIPs were performed as in [78], using the 8WG16 monoclonal antibody. Amplicons for Q-PCR quantification extended from +110 to +193 for CLN3, and from +6 to +95 for ADH1, in relation to the transcription start sites.
Rad53p phosphorylation assay
Yeast cultures were grown at 25°C to OD = 1. Cultures were kept at 25°C, or shifted at 37°C and incubated for two hours. Protein extracts for the Western blot analyses were prepared from trichloroacetic acid (TCA)-treated yeast cells. Protein extracts were resolved on a 7.7% SDS-PAGE (35:0.2 acrylamide/bis-acrylamide). Immunoblots were done with the goat anti-Rad53 polyclonal antibody from Santa Cruz Biotechnology.
Detection of non chromatin-bound “free” histones associated with the histone chaperones Nap1 and Asf1
For the determination of histones associated with Nap1 and Asf1 in Figure 5A and 5B, one-liter cultures of the indicated strains carrying Nap1-FLAG [79] or Asf1-FLAG were grown exponentially in YPD media at 25°C. Cells were then harvested as such at a density of 2.5×10E7 cells/ml for the Asf1-FLAG experiment shown in Figure 5B. For the Nap1-FLAG experiment shown in Figure 5A, cells were grown at 25°C until they reached a density of 1.5×10E7 cells/ml at which point they were switched to the restrictive temperature of 37°C for two hours prior to harvesting the cells at a density of 2.5×10E7 cells/ml. Whole cell extracts (WCEs) were prepared as previously described [49], and FLAG-tagged Nap1 (pRS316-Flag-yNap1) or Asf1-FLAG was immunoprecipitated using FLAG M2 agarose (Sigma). The immunoprecipitated material was resolved on precast 4–12% polyacrylamide gradient gels in MES buffer (BioRad), and were processed for Western blotting as previously described [49]. FLAG M2 antibodies (Sigma) were used to detect Nap1-FLAG and Asf1-FLAG, while histone H4 and H2B were detected using the previously described polyclonal antibodies [49]. Histone H2A was detected using an H2A antibody from Millipore (Cat. # 07-146).
The influence of transcription on free histone accumulation shown in Figure 5C was tested as follows. One liter of overnight culture of the spt16-197 (FY348) cells carrying FLAG-tagged Asf1 was grown in YPD at 25°C. Once the cells had reached a density of 1.5×10E7 cells/ml, they were treated with alpha-factor for two hours at 25°C. Cells were treated with additional amounts of alpha-factor, divided into four equal aliquots, and were treated with or without 200µg/ml alpha-amanitin for 2 hours at 25°C or 35°C in the continued presence of alpha-factor. Afterward, cells were harvested and processed as described before, except histone H3, which was detected using a polyclonal antibody directed against the C-terminus of histone H3, as previously described [49]. The inhibition of Pol II by alpha-amanitin was controlled by monitoring the mRNA levels of ACT1 in relation to ribosomal RNA levels (that are unaffected at the alpha-amanitin concentration used) using quantitative RT-PCR (Figure S5C).
Supporting Information
Zdroje
1. ReinbergD
SimsRJ3rd
2006 de FACTo nucleosome dynamics. J Biol Chem 281 23297 23301
2. FormosaT
2008 FACT and the reorganized nucleosome. Mol Biosyst 4 1085 1093
3. OrphanidesG
LeRoyG
ChangCH
LuseDS
ReinbergD
1998 FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92 105 116
4. PavriR
ZhuB
LiG
TrojerP
MandalS
2006 Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 125 703 717
5. Jimeno-GonzalezS
Gomez-HerrerosF
AlepuzPM
ChavezS
2006 A gene-specific requirement for FACT during transcription is related to the chromatin organization of the transcribed region. Mol Cell Biol 26 8710 8721
6. BiswasD
Dutta-BiswasR
MitraD
ShibataY
StrahlBD
2006 Opposing roles for Set2 and yFACT in regulating TBP binding at promoters. Embo J 25 4479 4489
7. FormosaT
2003 Changing the DNA landscape: putting a SPN on chromatin. Curr Top Microbiol Immunol 274 171 201
8. LindstromDL
SquazzoSL
MusterN
BurckinTA
WachterKC
2003 Dual roles for Spt5 in pre-mRNA processing and transcription elongation revealed by identification of Spt5-associated proteins. Mol Cell Biol 23 1368 1378
9. MasonPB
StruhlK
2003 The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Mol Cell Biol 23 8323 8333
10. SaundersA
WernerJ
AndrulisED
NakayamaT
HiroseS
2003 Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301 1094 1096
11. BiswasD
Dutta-BiswasR
StillmanDJ
2007 Chd1 and yFACT act in opposition in regulating transcription. Mol Cell Biol 27 6279 6287
12. BiswasD
YuY
PrallM
FormosaT
StillmanDJ
2005 The Yeast FACT Complex Has a Role in Transcriptional Initiation. Mol Cell Biol 25 5812 5822
13. O'DonnellAF
BrewsterNK
KurniawanJ
MinardLV
JohnstonGC
2004 Domain organization of the yeast histone chaperone FACT: the conserved N-terminal domain of FACT subunit Spt16 mediates recovery from replication stress. Nucleic Acids Res 32 5894 5906
14. SchlesingerMB
FormosaT
2000 POB3 is required for both transcription and replication in the yeast Saccharomyces cerevisiae. Genetics 155 1593 1606
15. VanDemarkAP
BlanksmaM
FerrisE
HerouxA
HillCP
2006 The structure of the yFACT Pob3-M domain, its interaction with the DNA replication factor RPA, and a potential role in nucleosome deposition. Mol Cell 22 363 374
16. OrphanidesG
WuWH
LaneWS
HampseyM
ReinbergD
1999 The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400 284 288
17. PrendergastJA
MurrayLE
RowleyA
CarruthersDR
SingerRA
1990 Size selection identifies new genes that regulate Saccharomyces cerevisiae cell proliferation. Genetics 124 81 90
18. LycanD
MikesellG
BungerM
BreedenL
1994 Differential effects of Cdc68 on cell cycle-regulated promoters in Saccharomyces cerevisiae. Mol Cell Biol 14 7455 7465
19. MaloneEA
ClarkCD
ChiangA
WinstonF
1991 Mutations in SPT16/CDC68 suppress cis- and trans-acting mutations that affect promoter function in Saccharomyces cerevisiae. Mol Cell Biol 11 5710 5717
20. JohnS
HoweL
TafrovST
GrantPA
SternglanzR
2000 The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30-containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)-FACT complex. Genes Dev 14 1196 1208
21. KroganNJ
KimM
AhnSH
ZhongG
KoborMS
2002 RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol 22 6979 6992
22. SquazzoSL
CostaPJ
LindstromDL
KumerKE
SimicR
2002 The Paf1 complex physically and functionally associates with transcription elongation factors in vivo. Embo J 21 1764 1774
23. FlemingAB
KaoCF
HillyerC
PikaartM
OsleyMA
2008 H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol Cell 31 57 66
24. FormosaT
ErikssonP
WittmeyerJ
GinnJ
YuY
2001 Spt16-Pob3 and the HMG protein Nhp6 combine to form the nucleosome-binding factor SPN. Embo J 20 3506 3517
25. RhoadesAR
RuoneS
FormosaT
2004 Structural features of nucleosomes reorganized by yeast FACT and its HMG box component, Nhp6. Mol Cell Biol 24 3907 3917
26. XinH
TakahataS
BlanksmaM
McCulloughL
StillmanDJ
2009 yFACT Induces Global Accesibility of Nucleosomal DNA without H2A–H2B Displacement. Molecular Cell 35 365 376
27. StuweT
HothornM
LejeuneE
RybinV
BortfeldM
2008 The FACT Spt16 “peptidase” domain is a histone H3–H4 binding module. Proc Natl Acad Sci U S A 105 8884 8889
28. VanDemarkAP
XinH
McCulloughL
RawlinsR
BentleyS
2008 Structural and functional analysis of the Spt16p N-terminal domain reveals overlapping roles of yFACT subunits. J Biol Chem 283 5058 5068
29. BelotserkovskayaR
SaundersA
LisJT
ReinbergD
2004 Transcription through chromatin: understanding a complex FACT. Biochim Biophys Acta 1677 87 99
30. FormosaT
RuoneS
AdamsMD
OlsenAE
ErikssonP
2002 Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics 162 1557 1571
31. KaplanCD
LapradeL
WinstonF
2003 Transcription elongation factors repress transcription initiation from cryptic sites. Science 301 1096 1099
32. CheungV
ChuaG
BatadaNN
LandryCR
MichnickSW
2008 Chromatin- and transcription-related factors repress transcription from within coding regions throughout the Saccharomyces cerevisiae genome. PLoS Biol 6 e277 doi:10.1371/journal.pbio.0060277
33. VantiM
GallasteguiE
RespaldizaI
Rodriguez-GilA
Gomez-HerrerosF
2009 Yeast genetic analysis reveals the involvement of chromatin reassembly factors in repressing HIV-1 basal transcription. PLoS Genet 5 e1000339 doi:10.1371/journal.pgen.1000339
34. JamaiA
PuglisiA
StrubinM
2009 Histone Chaperone Spt16 Promotes Redeposition of the Original H3–H4 Histones Evicted by Elongating RNA Polymerase. Molecular Cell 35 377 383
35. RowleyA
SingerRA
JohnstonGC
1991 CDC68, a yeast gene that affects regulation of cell proliferation and transcription, encodes a protein with a highly acidic carboxyl terminus. Mol Cell Biol 11 5718 5726
36. XuQ
JohnstonGC
SingerRA
1993 The Saccharomyces cerevisiae Cdc68 transcription activator is antagonized by San1, a protein implicated in transcriptional silencing. Mol Cell Biol 13 7553 7565
37. SkotheimJM
Di TaliaS
SiggiaED
CrossFR
2008 Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454 291 296
38. DirickL
BohmT
NasmythK
1995 Roles and regulation of Cln-Cdc28 kinases at the start of the cell cycle of Saccharomyces cerevisiae. Embo J 14 4803 4813
39. KochC
SchleifferA
AmmererG
NasmythK
1996 Switching transcription on and off during the yeast cell cycle: Cln/Cdc28 kinases activate bound transcription factor SBF (Swi4/Swi6) at start, whereas Clb/Cdc28 kinases displace it from the promoter in G2. Genes Dev 10 129 141
40. StuartD
WittenbergC
1995 CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev 9 2780 2794
41. TyersM
TokiwaG
FutcherB
1993 Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2 and other cyclins. Embo J 12 1955 1968
42. WangH
CareyLB
CaiY
WijnenH
FutcherB
2009 Recruitment of Cln3 cyclin to promoters controls cell cycle entry via histone deacetylase and other targets. PLoS Biol 7 e1000189 doi:10.1371/journal.pbio.1000189
43. CostanzoM
NishikawaJL
TangX
MillmanJS
SchubO
2004 CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 117 899 913
44. de BruinRA
McDonaldWH
KalashnikovaTI
YatesJ3rd
WittenbergC
2004 Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 117 887 898
45. TakahataS
YuY
StillmanDJ
2009 The E2F functional analogue SBF recruits the Rpd3(L) HDAC, via Whi5 and Stb1, and the FACT chromatin reorganizer, to yeast G1 cyclin promoters. Embo J 28 3378 3389
46. SchneiderBL
YangQH
FutcherAB
1996 Linkage of replication to start by the Cdk inhibitor Sic1. Science 272 560 562
47. SchwobE
BohmT
MendenhallMD
NasmythK
1994 The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in S. cerevisiae. Cell 79 233 244
48. GunjanA
PaikJ
VerreaultA
2005 Regulation of histone synthesis and nucleosome assembly. Biochimie 87 625 635
49. GunjanA
VerreaultA
2003 A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 115 537 549
50. SinghRK
KabbajMH
PaikJ
GunjanA
2009 Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat Cell Biol 11 925 933
51. SinghRK
PaikJ
GunjanA
2009 Generation and management of excess histones during the cell cycle. Front Biosci 14 3145 3158
52. ChavezS
AguileraA
1997 The yeast HPR1 gene has a functional role in transcriptional elongation that uncovers a novel source of genome instability. Genes Dev 11 3459 3470
53. LunaR
JimenoS
MarinM
HuertasP
Garcia-RubioM
2005 Interdependence between transcription and mRNP processing and export, and its impact on genetic stability. Mol Cell 18 711 722
54. NouraniA
RobertF
WinstonF
2006 Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol Cell Biol 26 1496 1509
55. GeraldJN
BenjaminJM
KronSJ
2002 Robust G1 checkpoint arrest in budding yeast: dependence on DNA damage signaling and repair. J Cell Sci 115 1749 1757
56. TohGW
LowndesNF
2003 Role of the Saccharomyces cerevisiae Rad9 protein in sensing and responding to DNA damage. Biochem Soc Trans 31 242 246
57. PellicioliA
FoianiM
2005 Signal transduction: how rad53 kinase is activated. Curr Biol 15 R769 771
58. AllenJB
ZhouZ
SiedeW
FriedbergEC
ElledgeSJ
1994 The SAD1/RAD53 protein kinase controls multiple checkpoints and DNA damage-induced transcription in yeast. Genes Dev 8 2401 2415
59. LibudaDE
WinstonF
2006 Amplification of histone genes by circular chromosome formation in Saccharomyces cerevisiae. Nature 443 1003 1007
60. OsleyMA
1991 The regulation of histone synthesis in the cell cycle. Annu Rev Biochem 60 827 861
61. BortvinA
WinstonF
1996 Evidence that Spt6p controls chromatin structure by a direct interaction with histones. Science 272 1473 1476
62. Meeks-WagnerD
HartwellLH
1986 Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell 44 43 52
63. De KoningL
CorpetA
HaberJE
AlmouzniG
2007 Histone chaperones: an escort network regulating histone traffic. Nat Struct Mol Biol 14 997 1007
64. SantistebanMS
ArentsG
MoudrianakisEN
SmithMM
1997 Histone octamer function in vivo: mutations in the dimer-tetramer interfaces disrupt both gene activation and repression. Embo J 16 2493 2506
65. GrothA
CorpetA
CookAJ
RocheD
BartekJ
2007 Regulation of replication fork progression through histone supply and demand. Science 318 1928 1931
66. GreenEM
AntczakAJ
BaileyAO
FrancoAA
WuKJ
2005 Replication-independent histone deposition by the HIR complex and Asf1. Curr Biol 15 2044 2049
67. SchwabishMA
StruhlK
2006 Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol Cell 22 415 422
68. AndrewsAJ
ChenX
ZevinA
StargellLA
LugerK
2010 The histone chaperone Nap1 promotes nucleosome assembly by eliminating nonnucleosomal histone DNA interactions. Mol Cell 26 834 842
69. GallegoC
GariE
ColominaN
HerreroE
AldeaM
1997 The Cln3 cyclin is down-regulated by translational repression and degradation during the G1 arrest caused by nitrogen deprivation in budding yeast. Embo J 16 7196 7206
70. LaabsTL
MarkwardtDD
SlatteryMG
NewcombLL
StillmanDJ
2003 ACE2 is required for daughter cell-specific G1 delay in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 100 10275 10280
71. Di TaliaS
WangH
SkotheimJM
RosebrockAP
FutcherB
2009 Daughter-specific transcription factors regulate cell size control in budding yeast. PLoS Biol 7 e1000221 doi:10.1371/journal.pbio.1000221
72. NewcombLL
HallDD
HeidemanW
2002 AZF1 is a glucose-dependent positive regulator of CLN3 transcription in Saccharomyces cerevisiae. Mol Cell Biol 22 1607 1614
73. MaiB
MilesS
BreedenLL
2002 Characterization of the ECB binding complex responsible for the M/G(1)-specific transcription of CLN3 and SWI4. Mol Cell Biol 22 430 441
74. PramilaT
MilesS
GuhaThakurtaD
JemioloD
BreedenLL
2002 Conserved homeodomain proteins interact with MADS box protein Mcm1 to restrict ECB-dependent transcription to the M/G1 phase of the cell cycle. Genes Dev 16 3034 3045
75. LiY
ZengSX
LandaisI
LuH
2007 Human SSRP1 has Spt16-dependent and -independent roles in gene transcription. J Biol Chem 282 6936 6945
76. RoseMD
WinstonF
HieterP
1990 Methods in Yeast Genetics: A Laboratory Course Manual Cold Spring Harbor, NY Cold Sprong Harbor Laboratory Press
77. WineyM
GoetschL
BaumP
ByersB
1991 MPS1 and MPS2: novel yeast genes defining distinct steps of spindle pole body duplication. J Cell Biol 114 745 754
78. PelechanoV
Jimeno-GonzalezS
Rodriguez-GilA
Garcia-MartinezJ
Perez-OrtinJE
2009 Regulon-specific control of transcription elongation across the yeast genome. PLoS Genet 5 e1000614 doi:10.1371/journal.pgen.1000614
79. Miyaji-YamaguchiM
KatoK
NakanoR
AkashiT
KikuchiA
2003 Involvement of nucleocytoplasmic shuttling of yeast Nap1 in mitotic progression. Mol Cell Biol 23 6672 6684
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 5
- Primární hyperoxalurie – aktuální možnosti diagnostiky a léčby
- Mateřský haplotyp KIR ovlivňuje porodnost živých dětí po transferu dvou embryí v rámci fertilizace in vitro u pacientek s opakujícími se samovolnými potraty nebo poruchami implantace
- Intrauterinní inseminace a její úspěšnost
- Akutní intermitentní porfyrie
- Srdeční frekvence embrya může být faktorem užitečným v předpovídání výsledku IVF
Nejčtenější v tomto čísle
- Common Genetic Variants near the Brittle Cornea Syndrome Locus Influence the Blinding Disease Risk Factor Central Corneal Thickness
- All About Mitochondrial Eve: An Interview with Rebecca Cann
- The Relationship among Gene Expression, the Evolution of Gene Dosage, and the Rate of Protein Evolution
- SMA-10/LRIG Is a Conserved Transmembrane Protein that Enhances Bone Morphogenetic Protein Signaling