
Competitive Repair by Naturally Dispersed Repetitive DNA during Non-Allelic Homologous Recombination
Genome rearrangements often result from non-allelic homologous recombination (NAHR) between repetitive DNA elements dispersed throughout the genome. Here we systematically analyze NAHR between Ty retrotransposons using a genome-wide approach that exploits unique features of Saccharomyces cerevisiae purebred and Saccharomyces cerevisiae/Saccharomyces bayanus hybrid diploids. We find that DNA double-strand breaks (DSBs) induce NAHR–dependent rearrangements using Ty elements located 12 to 48 kilobases distal to the break site. This break-distal recombination (BDR) occurs frequently, even when allelic recombination can repair the break using the homolog. Robust BDR–dependent NAHR demonstrates that sequences very distal to DSBs can effectively compete with proximal sequences for repair of the break. In addition, our analysis of NAHR partner choice between Ty repeats shows that intrachromosomal Ty partners are preferred despite the abundance of potential interchromosomal Ty partners that share higher sequence identity. This competitive advantage of intrachromosomal Tys results from the relative efficiencies of different NAHR repair pathways. Finally, NAHR generates deleterious rearrangements more frequently when DSBs occur outside rather than within a Ty repeat. These findings yield insights into mechanisms of repeat-mediated genome rearrangements associated with evolution and cancer.
Published in the journal:
. PLoS Genet 6(12): e32767. doi:10.1371/journal.pgen.1001228
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1001228
Summary
Genome rearrangements often result from non-allelic homologous recombination (NAHR) between repetitive DNA elements dispersed throughout the genome. Here we systematically analyze NAHR between Ty retrotransposons using a genome-wide approach that exploits unique features of Saccharomyces cerevisiae purebred and Saccharomyces cerevisiae/Saccharomyces bayanus hybrid diploids. We find that DNA double-strand breaks (DSBs) induce NAHR–dependent rearrangements using Ty elements located 12 to 48 kilobases distal to the break site. This break-distal recombination (BDR) occurs frequently, even when allelic recombination can repair the break using the homolog. Robust BDR–dependent NAHR demonstrates that sequences very distal to DSBs can effectively compete with proximal sequences for repair of the break. In addition, our analysis of NAHR partner choice between Ty repeats shows that intrachromosomal Ty partners are preferred despite the abundance of potential interchromosomal Ty partners that share higher sequence identity. This competitive advantage of intrachromosomal Tys results from the relative efficiencies of different NAHR repair pathways. Finally, NAHR generates deleterious rearrangements more frequently when DSBs occur outside rather than within a Ty repeat. These findings yield insights into mechanisms of repeat-mediated genome rearrangements associated with evolution and cancer.
Introduction
Human structural variation contributes to phenotypic differences and susceptibility to disease [1]. Recent studies suggest that many structural variants are mediated by non-allelic homologous recombination (NAHR) between dispersed repetitive DNA elements [2]–[5]. While the importance of NAHR in shaping genome structure is becoming more apparent, the mechanism of NAHR remains poorly understood.
NAHR (also known as ectopic recombination) utilizes the molecular pathways that mediate allelic homologous recombination (AHR) between sister chromatids or homologs. AHR and NAHR are both initiated by a double-strand break (DSB) that is processed by 5′-3′ DNA resection to generate 3′-OH tailed single-stranded DNA (ssDNA) intermediates [6]. The resected ssDNA, called the recipient, is activated to search for homologous sequences, called the donor, to be used as a template for repair. If the recipient is unique DNA, then the donor will be the homolog or sister chromatid, and AHR ensues. However, if the recipient is repetitive DNA, it may choose a non-allelic repeat as a donor, leading to NAHR and potentially a chromosome rearrangement. The establishment of this basic recipient-donor partnership during homologous recombination (HR) defines four fundamental parameters for NAHR that we address here.
The first parameter is the position of a DSB relative to repetitive and unique sequences. DNA resection starts from the DSB ends and is thought to activate break-proximal sequences before break-distal sequences [7]. Based on this model, break-proximal recipients (sequences at or near the break site) direct homology searches before break-distal recipients (sequences distal from the break site). Therefore, a DSB near or in a repetitive element should activate that repeat as a recipient, which may search for a non-allelic donor repeat to promote NAHR. Alternatively, a DSB in a large track of unique sequences should preferentially activate break-proximal unique sequences as recipients. In a diploid, these break-proximal recipients can repair efficiently using allelic donors on the sister chromatid or homolog. Therefore it has been assumed, but never tested directly, that a DSB in unique sequences in a diploid will rarely induce NAHR. However, a few studies in haploid yeast have observed a preference for recombination using more distal sequences over break-proximal recipients, suggesting that break-distal recipients can participate in homology searches [8]–[10].
The second important parameter of NAHR is the percent and length of identity shared between a recipient and potential donors. Introduction of ∼1% sequence divergence between model repeats decreases recombination rates 9 - to 25-fold [11], [12], suggesting that even very limited divergence may significantly affect NAHR rates. The minimum length of uninterrupted identity between two sequences needed for efficient recombination is called the minimal effective processing segment (MEPS) [13]. Using model repeats, the MEPS necessary for efficient NAHR is about 250 bp [14], [15]. This suggests that small retroelements, such as Alus (∼300 bp) and long terminal repeats (LTRs; ∼330 bp), are potentially sufficient to promote efficient NAHR. However, how homology between natural repeats relates to usage for NAHR has never been assessed at a genome-wide scale.
The third important parameter of NAHR is genomic position of a recipient and potential donors. Recipients and donors are more likely to recombine when they are on the same chromosome than when they are on different chromosomes [16]–[18]. Interchromosomal recombination between model repeats can also be influenced by their proximity to centromeres and telomeres [19], [20]. However, these NAHR position preferences have not been tested with natural repeats in an unbiased system, where the unrestricted choice of repair partners and pathways is allowed.
Finally, which HR pathway acts upon a recipient and donor may impact whether NAHR occurs. Single-strand annealing (SSA) can occur when resection from a DSB proceeds through flanking direct repeats, exposing complementary sequences that anneal to generate a deletion product [6]. In contrast, Rad51-dependent HR pathways involve strand invasion events where Rad51 polymerizes onto resected recipient DNA to mediate invasion into a homologous duplex donor. When recipient sequences on both sides of the DSB invade the same donor, repair can occur by gene conversion (GC). However, if the recipient shares identity with the donor on just one side of a DSB, then one-ended strand invasion events can repair through break-induced replication (BIR). GC is faster and more efficient at repairing DSBs than BIR [21]. In addition, GC competes effectively with SSA [22], [23]. While the competition between SSA, GC, and BIR can influence NAHR outcomes, little is known about the relative usage of these pathways during NAHR with natural repeats.
Thus the efficiency and outcome of NAHR are potentially influenced by its ability to compete with AHR, the sequence identity between recipients and donors and their genomic position, and the usage of HR pathways. Yet these potential influences remain untested or unresolved, particularly in the context of a family of naturally repeated sequences. To address these fundamental issues, we developed a new genome-wide system to study NAHR between the dispersed and divergent families of Ty retrotransposons in purebred and hybrid diploids of budding yeasts. We exploit this system to provide insight into the most important parameters that control NAHR in a eukaryotic diploid genome.
Results
A genome-wide system to study NAHR events between Ty1/Ty2 families of repeats
Ty1 and Ty2 represent the most abundant families of dispersed repetitive elements in S. cerevisiae. Our system to study Ty-mediated NAHR relies on three components: (1) knowledge of the sequence and position of all Ty1/Ty2 elements in the genome, (2) strains with genetic features for the recovery of Ty-mediated NAHR events, and (3) a protocol to measure these events out of all possible outcomes. Below we provide a brief description of each component.
As a first step, we completed the sequence of the S. cerevisiae unannotated chromosome III Ty elements (Figure S1). With the completed sequence, we generated a map of the distribution of 37 full length Ty1s and 13 full length Ty2s [which includes 98 Ty-associated 5′ and 3′ long terminal repeats (LTRs)], and 208 solo LTRs (Figure 1A). The sequence and positional information is critical since it defines all potential Ty1/Ty2 recipients and donors in the S. cerevisiae genome, allowing us to determine whether some repeats are used and others are not in NAHR.

The potential for Ty elements to act as recipients and donors in NAHR depends in part on their sequence identity. The average percent sequence identity is 95.7±2.4% between Ty1s, 95.9±4.8% between Ty2s, and 73.9±3.4% between Ty1 and Ty2 (Table S3). Previous work has determined that recombination between model repeats decreases 9-fold with 99% identity and 50-fold with 91–94% identity relative to identical model repeats [11]. Thus the sequence divergence of the Ty1/Ty2 family could dramatically reduce the pool of potential Ty recipients and donors, limiting the number of elements that participate in NAHR.
However, if the mismatches are clustered, rather than distributed evenly within the full length of Ty1/Ty2 (5.9 kb), then long stretches of identity may allow efficient NAHR. With this in mind, we analyzed the longest block of uninterrupted identity between all pairwise alignments of Ty1/Ty2, a parameter that has not been previously assessed for Ty elements. To evaluate the significance of these blocks, we categorized them according to the previously determined MEPS value of about 250 bp for NAHR [14], [15]. Recombination rates are predicted to significantly drop when lengths are below MEPS and proportionally increase when lengths are above MEPS [13].
Using our binning analysis, 73% of all Ty1/Ty2 alignments (891 out of 1225) have blocks of identity ≥250 bp (Figure 1B and Table S4). All pairwise comparisons between repeats within either the Ty1 or Ty2 family are above the MEPS value while 31% of pairwise comparisons between Ty1 and Ty2 repeats have a block of identity ≥250 bp. Thus, for the full length Ty1s and Ty2s, the shared blocks of uninterrupted identity strongly predict that a given Ty1/Ty2 recipient can undergo NAHR with many potential Ty1/Ty2 donors, thereby establishing a competition among donors. In contrast, only 1% of all LTR pairwise comparisons (544 out of 46,665) have a block of uninterrupted identity ≥250 bp (Figure 1C and Table S5). This limited length of uninterrupted identity between the LTRs predicts that they may be inefficient substrates for NAHR. In addition, sequence identity amongst pairwise comparisons of the 306 LTR elements widely range between 3%–100%, with an average of 59.6%±22.7% (Table S6). Thus the poor sequence identity between LTRs suggests that solo LTRs will be unfavorable substrates for NAHR.
The second component of our system is the use of specific strains to optimize the recovery of Ty-mediated NAHR events. In order to recover all possible NAHR events, we use diploid yeast where loss of genetic material can be complemented by homologs. In contrast, Ty-mediated rearrangements that occur in haploids may delete genes necessary for viability. Along with S. cerevisiae diploids (referred hereafter as “purebred”), we generated synthetic hybrid diploids by mating S. cerevisiae with a sequenced relative, S. bayanus (referred hereafter as “hybrid”) (Figure 2), which is largely devoid of Ty1/Ty2 elements [24], [25]. The diploids are genetically marked to allow identification of all cells that suffer an I-SceI site-specific DSB as well as the subset of cells in which the broken chromosome is repaired or lost (Figure 2 and see below). Like the purebreds, viability remains high after induction of an I-SceI-induced DSB in the hybrid diploids (Figure 2). In addition, the hybrid diploids grow well and are competent in DNA maintenance and repair like the purebred diploids (Figure S2). Since S. bayanus complements almost all the genes in S. cerevisiae [26], S. bayanus can also balance S. cerevisiae by suppressing any loss of gene function due to NAHR of the S. cerevisiae genome. However, in contrast to the purebred diploids, the hybrid diploids have three important advantages. The significant sequence divergence between the two genomes (62% intergenic, 80% genic) [27] suppresses AHR, favoring NAHR between the more homologous Ty1/Ty2 elements and thus enhancing the recovery of Ty-mediated NAHR events. The sequence divergence also facilitates the analysis of S. cerevisiae rearrangements by array comparative genomic hybridization (aCGH) and PCR. Finally, the comparison of NAHR between the purebred and hybrid diploids allows the assessment of NAHR with and without AHR competition (Figure 2).

The third component of our system is an unbiased clone-based assay to determine the frequencies of NAHR events among all possible outcomes (Figure 3A). An I-SceI recognition sequence [referred to as the I-SceI cut site (cs)], along with a Hygromycin-resistance gene (HYG), is integrated at different positions on the S. cerevisiae chromosome III homolog. We choose to initiate a DSB on the S. cerevisiae chromosome III since this chromosome has the highest density of Ty1/Ty2 elements relative to all other chromosomes (see Figure 1A), making it a good model for the repetitive-rich chromosomes of higher eukaryotes. We initiate the DSB with the addition of galactose to the media for two hours in exponentially growing cultures to induce expression of the I-SceI endonuclease fused to the galactose promoter. Galactose induction of I-SceI expression leads to formation of a DSB at the 163cs position on one S. cerevisiae chromosome III homolog (Figure 3B), which activates recipient sequences adjacent to the break site to undergo a homology search. The cells are then plated onto nonselective YEPD media for individual colonies (referred to as clones). These clones are then phenotyped to determine whether the I-SceI-induced DSB occurred (HygS, see Figure S3) followed by chromosome repair (Leu+Ura+ or Leu+Ura−) or loss (Leu−Ura−). We find that the majority of I-SceI-induced DSBs are repaired in both the purebred (99±2%) and hybrid (79±5%) diploids, although the hybrid diploids exhibit a significant increase in chromosome loss (20±5%) compared to the purebred diploids (1±2%) (Figure 3C). HR mediates almost all of this DSB repair in both diploids since repair is nearly abolished when the essential HR protein Rad52 is absent (Figure 3C).

To assess the structure of the repaired chromosome in the two genetic repair classes, a random subset of clones in each class are further analyzed by pulse-field gel electrophoresis (PFGE)/Southern analysis (Figure 3D). An I-SceI-induced DSB at the 163cs position that is repaired by AHR results in an unchanged chromosome III size whereas repair by NAHR results in a rearrangement with a changed chromosome III size (Figure 3D). Further aCGH and PCR characterization of the genetic repair classes reveals four types of chromosome III rearrangement structures with Ty elements localized to the recombination junctions (Figure S4 and see Materials and Methods). The Leu+HygSUra+ repair class I contains internal deletions, and the Leu+HygSUra− repair class II includes isochromosomes, rings, and translocations (see schematics in Figure 3A). The recovery of these distinct Ty-mediated NAHR rearrangements from one site-specific DSB reveals a competition between recipient and donor Ty elements for NAHR, validating our system as a means to study NAHR between complex families of natural repeats.
Recipient competition revealed by break-distal recombination (BDR)
A site-specific DSB in unique DNA allows us to assess the likelihood that break-distal repeats are activated as recipients in a homology search to facilitate NAHR. HR events that use a break-distal recipient for recombination are termed here as break-distal recombination (BDR). With 163cs positioned inside 18.1 kb of unique DNA on chromosome III (see map in Figure 3A), we tested the possibility for BDR by monitoring three potential Ty recipient loci (YCRCdelta6, YCRCdelta7, RAHS) at various distances distal from the break site. Because our assay employs no selection, we are able to calculate the frequencies of I-SceI-induced Ty-mediated rearrangements among all possible outcomes after the DSB (see Materials and Methods). Below we highlight the major points from the data compiled in Table 1 and Table 2.


In purebred diploids, 17% of cells after DSB at 163cs undergo NAHR through BDR to mediate rearrangements. Despite a sufficient length of unique sequences that can facilitate AHR with the identical homolog after the DSB, 15±6% of cells use the RAHS recipient, 0.3±0.3% of cells use YCRCdelta7, and 2±0.7% of cells use the YCRCdelta6 recipient located 11.7 kb, 28.9 kb, and 47.5 kb distal from the DSB, respectively (Figure 4A). To test the robustness of BDR, we changed a number of parameters. We eliminated the nonhomology immediately at the DSB ends (1.6 kb I-SceIcs/HYG construct) to test whether BDR is due to the presence of nonhomologous ends, which may inhibit the coordination of two-ended strand invasion events during GC [28]. However, with identity at the DSB ends, BDR is still observed, generating rearrangements (Figure S5). We further tested if BDR was specific to the 163cs position by moving the position of the DSB more centromere-proximal. With the I-SceI-induced DSB at 147cs, BDR-mediated Ty rearrangements occur in 3±3% of cells after DSB (Figure 4A). Interestingly, the frequency of YCRCdelta6/YCRCdelta7 usage is similar to when a DSB initiates at 163cs, suggesting that the usage of these LTR recipients is not determined by their distance from the break site. Lastly, we tested if BDR occurs when the I-SceI-induced DSB initiates on a different chromosome. BDR still occurs in 8±4% of cells after formation of a DSB on S. cerevisiae chromosome V to generate Ty-mediated rearrangements (Figure S6). Thus distal repeats mediate BDR despite the presence of break-proximal unique DNA that can effectively facilitate AHR. This result suggests that unique and repetitive recipient sequences at least 47.5 kb distal to a DSB can participate in recombination.

To test whether AHR competes with BDR, we analyzed BDR in the hybrid diploids. In the hybrid diploids, AHR is mostly suppressed compared to purebred diploids (3±4% of cells after DSB in hybrid compared to 82±6% of cells after DSB in purebred, Figure 4B), as expected from the extent of divergence between S. cerevisiae and S. bayanus genomes. Under these conditions of suppressed AHR, the frequency of BDR increases 4.5-fold compared to purebred diploids (increasing from 17% to 76%, Figure 4B), indicating that BDR competes with AHR. Furthermore, the distribution of different BDR-mediated rearrangements remains the same between hybrid and purebred diploids (compare Figure 4C to Figure 4A, and Table 1). Thus the presence of a divergent homolog at the break site enhances BDR-mediated rearrangements but does not alter preferences of Ty recipient and donors on chromosome III. This aspect of hybrid diploids makes them an excellent model to investigate the features of the recipients and donors that give rise to their preferred use.
To begin to define the parameters that influence the preferred use of recipient sequences to repair a DSB, we determined the largest block of uninterrupted identity between the recipient and its donor. The DSB at 163cs is positioned in the right arm of chromosome III distanced 57.4 kb from the centromere and 165.6 kb from the right telomere. Thus for AHR in purebred diploids, there is >50 kb of identity with the homolog on both sides of the DSB. In contrast, among the BDR events, the largest block of uninterrupted identity with the donors is 1,877 bp for the RAHS recipient, 29 bp for YCRCdelta7 recipient, and 98 bp for the YCRCdelta6 recipient. This reveals that the homology search in purebred diploids can be efficiently directed by 0.1%, 0.2%, or 3% (29, 98 or 1,877 bp out of 57,453 bp) of the potential recipient sequences activated by the DSB, and that this small fraction very distal to the break site generates rearrangements in a total of 17% of cells after DSB. In addition, the smaller and more break-distal solo LTRs, YCRCdelta6 and YCRCdelta7, compete effectively with the larger and more break-proximal RAHS cluster in both purebred and hybrid diploids (see Figure 4A and Figure 4C). These data are consistent with our analysis of AHR in hybrid diploids, where the recombinant junctions occur both proximal and distal to the break site (data not shown). Moreover, these hybrid allelic junctions do not coincide with the longest length of uninterrupted identity (138 bp) found between potential recipients through S. cerevisiae and S. bayanus chromosome III alignments. Thus the relative effectiveness of repetitive and unique recipient sequences competing next to the DSB is not solely predicted by length of uninterrupted identity or distance from the DSB.
Donor competition dictated by an intrachromosomal position bias
Our characterization of Ty-mediated NAHR events also allowed us to investigate the preferred usage of Ty donors with a DSB at 163cs. Intrachromosomal Ty sequences are used as donors in 75±4% of hybrid and 17±6% of purebred cells after DSB at 163cs, generating internal deletions, isochromosomes, or chromosome rings (intra-NAHR in Figure 5A and Table 1). In contrast, only 1±0.7% and 0.3±0.3% of cells after DSB at 163cs produce Ty-mediated interchromosomal translocations in hybrid and purebred diploids, respectively (inter-NAHR in Figure 5A and Table 1). Thus despite the greater number of potential inter - than intrachromosomal Ty donors (see Figure 1A), Ty donors on the same chromosome are preferred approximately 50 times more than Ty donors on a different chromosome.

Again as a first assessment, we wondered whether the NAHR biases for intra - over interchromosomal donors and amongst the two intrachromosomal donors (LAHS and FRAHS) are dictated by sequence identity between the donors with its Ty recipient. We generated a ranked list of sequence homology, comparing the three Ty recipient elements distal to 163cs (YCRCdelta6, YCRCdelta7, RAHS) with all potential Ty donor elements in the genome. We find that the intrachromosomal Ty donors (LAHS and FRAHS) are not among the most identical by either percent sequence identity or the longest block of uninterrupted identity (Figure 5B and Table S7, Table S8). Of the intra-NAHR Ty partners, we also find no correlation with the extent of sequence homology between the chosen Ty donors and their frequency of usage. For example, in the hybrid diploids, 61±3% of cells after DSB generate internal deletions between RAHS and YCRWTy1-5 at FRAHS (97% identity, 1,635 bp largest block of uninterrupted identity) whereas only 3±1% of cells after DSB generate a chromosome ring between the same RAHS recipient and the LAHS donor (97% identity, 1,877 bp largest block of uninterrupted identity). Furthermore, relaxing the stringency for sequence identity in NAHR using msh2Δ/msh2Δ, msh6Δ/msh6Δ, and sgs1Δ/sgs1Δ mutants in hybrid diploids does not abolish the intrachromosomal donor preference (Figure 5A), further suggesting that the preferred usage of donors is not due to sequence identity [29], but donor position. Similar to the findings for the usage of recipient sequences for NAHR, the preferred usage of Ty donors is neither dictated nor can be predicted by sequence homology. Thus the primary determinant of Ty donor choice during NAHR is genomic position, with ∼50-fold preference for intrachromosomal over interchromosomal donors.
Intrachromosomal position effect is due to the inefficiencies of NAHR pathways
Sequence homology between the Ty1/Ty2 families failed to dictate the recipient and donor competition during NAHR. One explanation is that each Ty-mediated rearrangement requires different genetic factors (Table 1), suggesting that they are generated through distinct NAHR pathways. Since HR pathways are known to compete after a DSB, we examined how this competition affected recipient and donor choice. In the hybrid diploids with the I-SceI-induced DSB in unique sequences at 163cs, 61±3% of cells form internal deletions between the RAHS recipient and the FRAHS donor (Table 1). These deletions form independent of RAD51 suggesting they occur through SSA (Table 1). RAHS also mediates isochromosomes (3±1%) and rings (3±1%) with the LAHS donor, and translocations with interchromosomal Ty donors (1±0.7%), all of which have Rad51-dependencies (Table 1). Thus the same RAHS recipient mediates internal deletions 20–40 fold higher than isochromosomes, rings, or translocations, suggesting that SSA dominates the NAHR pathway choice to generate Ty-mediated rearrangements when a DSB occurs in unique sequences.
With at least four NAHR pathways operating after the DSB at 163cs (suggested by the different genetic dependencies of the Ty-mediated BDR rearrangements, see Table 1), we then asked if these NAHR pathways were in competition with one another. To address pathway competition, we attempted to abolish or enhance particular NAHR pathways by removing their intrachromosomal donors and/or repositioning the I-SceIcs in the hybrid diploids. We then compared changes in the frequencies of the Ty-mediated rearrangement product as a readout of their NAHR pathway, where compensatory effects indicate competing pathways. In addition, since Rad51-independent SSA and Rad51-dependent pathways have been shown to compensate for each other after a DSB and hence compete [22], [30], our analysis groups the NAHR pathways into these two distinct HR mechanisms.
We first eliminated the dominant SSA pathway by deleting the FRAHS donor (FRAHSΔ, B in Figure 6) and looked for compensation through the remaining rearrangements. These rearrangements are grouped as Rad51-dependent NAHR since rings show full Rad51-dependency while isochromosomes and translocations have partial Rad51-dependency (Table 1). While some Rad51-dependent rearrangements show a modest increase (rings increase 3±1% to 11±3%, Table 2), the majority of cells cannot repair the DSB at 163cs without SSA, resulting in chromosome loss (71±3% loss, Figure 6). One possibility for this repair inefficiency is that the DSB is too far from the Ty recipients (at least 11.7 kb from the break site) to effectively activate the recipients in Rad51-dependent NAHR pathways. This would be consistent with evidence that Rad51 binding is limited to about 5 kb on either side of a DSB [31]. We then repositioned the I-SceIcs at 151cs, within 0.1 kb of the RAHS recipient in the FRAHSΔ strain (C in Figure 6), in order to enhance Rad51 presynaptic filament assembly onto RAHS. Although a modest increase in Rad51-dependent rearrangements was observed, the majority of cells after the DSB at 151cs with FRAHSΔ cannot efficiently repair the chromosome in the absence of SSA (58±2% loss, Figure 6). These data reveal that Rad51-dependent NAHR pathways induced by a DSB in unique sequences (163cs or 151cs) are inherently inept at repairing the DSB using Ty1/Ty2 elements. Taken together, for a DSB in unique DNA, the efficiency of the SSA pathway coupled with the inefficiency of Rad51-dependent NAHR pathways generates the intrachromosomal position bias and preferential usage of Ty recipients and donors.

Mutagenic potential of DSBs in the genome
Our findings show that the I-SceI-induced DSB in unique DNA (147cs, 151cs, or 163cs) generates substantial NAHR between Ty repeats, giving rise to a broad spectrum of rearrangements through BDR in the purebred diploids. This is in contrast to current models that propose that break-proximal sequences determine the outcome, where DSBs in unique DNA lead to AHR (between sisters or homologs) and DSBs in repetitive DNA can lead to NAHR [32]. To assess the relative consequence of DSBs in unique versus repetitive DNA, we repositioned the I-SceIcs into the RAHS locus (called RAHScs, Figure 7) and used our nonselective assay to measure all possible outcomes after the DSB at RAHScs in hybrid and purebred diploids. From the repair clones generated in our assay, we further characterized two Ty-mediated products that exclusively arise with the DSB at RAHScs, intra-Ty deletions and Ty GC. These Leu+HygSUra+ repair clones are distinguished from each other by assaying RAHS locus size using PFGE/Southern analysis (Figure S7). In comparison to the wild-type RAHS size, we observe a smaller RAHS size for intra-Ty deletion events and a similar RAHS size (with only the removal of the small nonhomologous 1.6 kb I-Scecs/HYG ends) for Ty GC events.

Similar to results with the DSB at 163cs, SSA dominates the NAHR pathway competition, with 66% and 61% of cells after DSB at RAHScs generating Ty-mediated deletions in hybrid and purebred diploids, respectively (Table 2). SSA again imposes a strong intrachromosomal position bias, dictating recipient and donor preferences. The internal deletions from RAHScs, however, can be generated between the RAHS recipient and two different Ty donors, sequences within RAHS itself (referred to as intra-Ty) and FRAHS (now referred to as inter-Ty). All of the internal deletions in purebred diploids are intra-Ty events (61±9%) whereas in hybrid diploids, 59±9% are intra-Ty and 7±5% are inter-Ty (Figure 7 and Table 2). This is consistent with previous work describing a proximity effect during SSA using model repeat donors, with break-proximal donors preferred over break-distal donors [7].
In addition to the events observed with a DSB at 163cs, we find that the second most frequent event after DSB at RAHScs is Ty GC. 22±8% and 33±10% of cells after DSB at RAHScs lead to Ty GC events in hybrid and purebred diploids, respectively (Figure 7). The lower frequency of Ty GC relative to intra-Ty deletions measured in our diploids are in agreement with those events measured using an HO-induced DSB inside Ty1 in S. cerevisiae haploids [33]. Ty GC occurs through the coordination of a two-ended strand invasion event into a Ty donor, which is not a possibility when the DSB initiates in unique DNA (as for 163cs). These GC events in the hybrid diploids must be mediated by a non-allelic Ty donor from the S. cerevisiae genome (since S. bayanus lacks Ty1/Ty2), which likely occurs in purebred diploids as well [16]. Thus, paradoxically, NAHR efficiently mediates conservative repair when a DSB occurs in repetitive DNA.
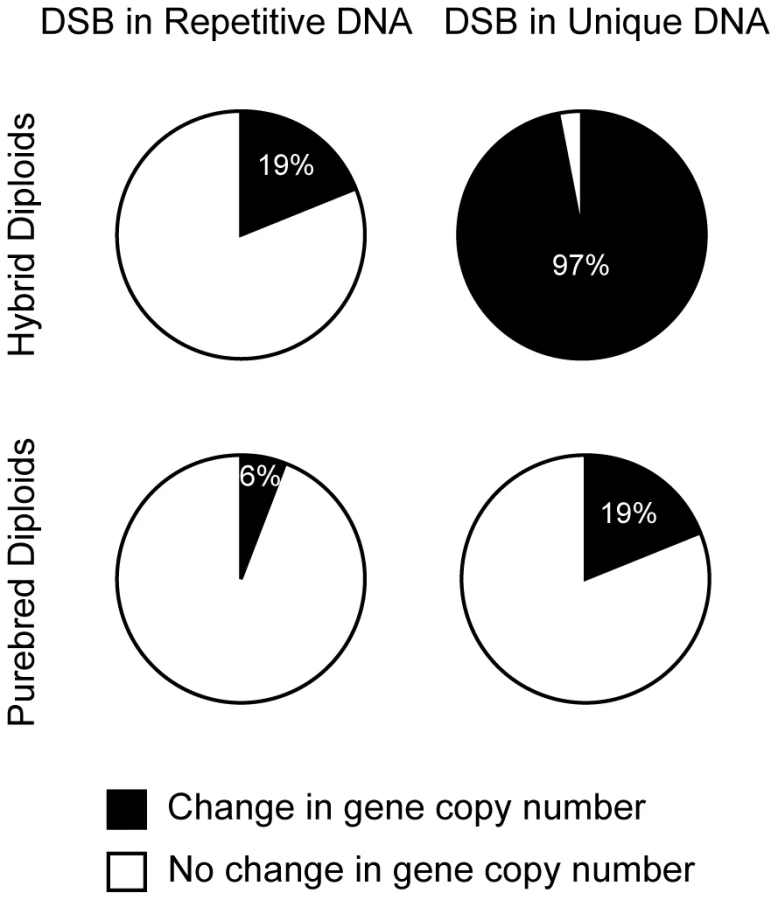
Having completed our analyses of a DSB within a Ty1 repeat, we can now compare its impact to a DSB in unique DNA on genome integrity. We categorized the outcomes of the I-SceI-induced DSB at RAHScs and at 163cs into two groups: (1) change in gene copy number (inter-Ty deletion, isochromosome, ring, translocation, and chromosome loss) and (2) no change in gene copy number (intra-Ty deletion, Ty GC, and allelic). This comparison reveals that the DSB in unique DNA is 3 to 5-fold more likely to cause a change in gene copy number than the DSB in repetitive DNA (increases from 19% to 97% in hybrid diploids and 6% to 19% in purebred diploids, Figure 8). Thus, distinct from models that highlight the role of DSBs inside repeats in mediating genome rearrangements, our results suggest that the relative mutagenic potential of a DSB in the genome actually decreases when the break occurs within repetitive DNA. Furthermore, this finding suggests that DSBs in unique DNA are more likely to lead to mutagenic rearrangements than DSBs in repetitive DNA.
Discussion
We report a novel genome-wide system in budding yeast to study non-allelic homologous recombination (NAHR) between natural repeats. While previous assays isolate aspects of competitive repair addressed here, our system gauges the competition between all parameters concurrently, as what naturally transpires in a cell. The value of this new approach is evidenced by the surprising features of NAHR our system reveals. Remarkably, in purebred diploids, DSBs within a long stretch of unique sequences are not always repaired by allelic homologous recombination (AHR) as previously assumed. Rather, 17% of these DSBs repair by NAHR. This NAHR arises because the DSB activates Ty recipients 12 to 48 kb distal from the break site to recombine with non-allelic Ty donor sequences. Robust NAHR through break-distal recombination (BDR) is supported by a previous study of bridge-breakage-fusion in diploid budding yeast by Malkova and colleagues [34].
In this and the previous study, competition between BDR-dependent NAHR and AHR occurs after an endonuclease-induced DSB. In diploids, endonucleases can cleave one homolog prior to DNA replication and both its sister chromatids after DNA replication, thereby eliminating the sister chromatid as a donor for AHR. Therefore, the only AHR donor is the uncut homolog. However, a homolog is also the only AHR donor for repair of spontaneous DSBs that occur on unreplicated DNA in G1 or S. Indeed, recent evidence suggests that spontaneous DSBs occur on unreplicated DNA [35]. We suggest that spontaneous DSBs in unique unreplicated DNA are also likely to induce robust BDR-dependent NAHR.
The fact that break-distal Ty sequences undergoes frequent NAHR reveals two surprising features of recombination that have important mechanistic implications for current models of recipient activation and choice. The first surprise is that distal Ty repeats are activated as recipients at all (presumably by becoming single-stranded) when break-proximal ssDNA can undergo AHR. Indeed, a recent study in diploid yeast suggests that ssDNA is generated at least 10 kb from a DSB before its repair is complete [36]. To explain this extensive break-distal resection, we suggest that a step after resection must be slow, such as the homology search for donor sequences. A slow homology search would provide time for break-distal sequences to be resected and compete with previously resected break-proximal sequences. Such a slow homology search is consistent with studies suggesting the slow diffusion of chromosomal sequences [37].
The second surprise is the disproportionate use of very small break-distal Ty sequences as recipients for NAHR. They would represent only a very small proportion of the entire block of resected DNA, which can all act as a recipient for AHR. We suggest that the smaller Ty recipients encounter their potential Ty donors first because chromosome territories [38] generate a high local concentration of potential intrachromosomal Ty donors. In contrast, the larger allelic recipients must travel further to partner with allelic donors on the homolog. Consistent with this model, almost all NAHR rearrangements through break-distal Ty recipients result from pairing with intrachromosomal Ty donors.
Along with recipient usage, our genome-wide system reveals the role sequence homology and genomic position play in NAHR donor choice. We find that the Ty donors chosen by a recipient are not among the most homologous in the genome by the criteria of either percent identity or longest block of uninterrupted identity. Rather the primary determinant of NAHR donor choice is local proximity. We observe a ∼50-fold preference for Ty repeat donors on the same chromosome over different chromosomes. This intrachromosomal NAHR preference is consistent with previous studies [16]–[19], although the magnitude of this preference differs, possibly due to specific configurations of repeats relative to a break site, as observed in our studies. However, in contrast to previous work, our study shows this intrachromosomal bias occurs under conditions that allow unrestricted choice of repair pathways and partners amongst a natural repetitive family. Interestingly, Ty1/Ty2 elements are preferentially inserted within 750 bp upstream of tRNA genes [39], and dispersed tRNA genes cluster together [40]. Our results suggest that possible Ty interchromosomal contacts mediated by tRNA clustering is not sufficient to overcome an intrachromosomal bias. It will be interesting to see whether higher-order chromosome organization may influence donor repair choice of natural repeats when only interchromosomal donors are available for NAHR.
Our system also provides insights into the preferred repair pathways that act on a family of natural repeats. We show that NAHR occurs mostly by the SSA pathway whether DSBs occur in unique sequences or a Ty repeat. The robustness of SSA is consistent with previous studies using model repeats [18], [23], [30], [41], [42]. Since repair of a single DSB by SSA will occur through an intrachromosomal donor, the predominance of SSA helps explain the preferential usage of intrachromosomal donors and the resulting preference for intrachromosomal NAHR.
Importantly, our pathway analysis of NAHR also helps explain one of the most surprising and striking observations of this study: DSBs that occur outside repeat clusters are more mutagenic than DSBs that occur inside repeat clusters. This seemingly counterintuitive observation arises because DSBs that occur inside a Ty have better options for repair, both in efficiency of pathways and favorably positioned donors. DSBs within the Ty predominately repair through two highly efficient pathways, SSA within the Ty locus or GC with preferred intrachromosomal Ty donors [16]. These types of repair preserve gene copy number since neighboring unique genes are unaffected. Since SSA and GC are compensatory pathways [22], it is possible that DSBs inside repetitive elements that cannot undergo SSA (i.e. solo insertion of LINE-1) efficiently repair through GC events [43]. A recombination execution checkpoint has been suggested to maintain genome integrity by ensuring the coordination of two-ended strand invasion events during GC for conservative repair [28]. Consistent with this, our results suggest that NAHR through GC between natural repeats is a major mechanism that limits changes in genome structure.
In contrast, DSBs in unique sequences that repair predominately through GC with the homolog is not as effective in limiting detrimental rearrangements. As the search for the interchromosomal homolog allows for more time to activate a break-distal Ty as a recipient, BDR occurs more frequently through SSA between distinct Ty loci or one-ended events through the BIR pathway. In this situation, SSA always, and BIR often times, change the copy number of neighboring unique genes. Hence, this opens up the possibility that DSBs in unique sequences, rather than repeats, may generate spontaneous or irradiation-induced NAHR-dependent rearrangements observed in yeast [32], [44]. Similarly, NAHR-dependent rearrangements in the human genome may also occur by a DSB in the surrounding unique DNA followed by BDR-dependent NAHR. If so, then the recombinant junction would not coincide with the site of the initiating lesion. Therefore, analysis of NAHR junctions alone may miss underlying mechanisms for genome rearrangements. Examining broad regions around NAHR junctions could potentially identify fragile sites that predispose a locus to recurrent instability, contributing to genetic diversity and disease.
Materials and Methods
Yeast strains
Standard yeast genetic and molecular biology methods were used [45]. All S. cerevisiae strains were derived from BY4700 (MATa ura3Δ0), BY4716 (MATα lys2Δ0), or BY4704 (MATa ade2Δ::hisG his3Δ200 leu2Δ0 lys2Δ0 met15Δ0 trp1Δ63) [46]. All S. bayanus strains were derived from a S. bayanus prototroph received as a gift from Ed Louis. Deletion of the HO gene and auxotrophic markers were introduced by transformation to generate a number of haploid S. bayanus strains for laboratory use, including MH3399 (MATa hoΔ::hisG ura3Δ::NAT leu2Δ::NAT ade2Δ::hisG), YZB9-4B1 (MATa hoΔ::KAN ura3Δ::NAT leu2Δ::NAT), YZB5-102 (MATα hoΔ::KAN lys2-1) (this study, [47]). Since S. bayanus is sensitive to high temperatures, the following modifications were made to the high efficiency yeast transformation protocol [48] for S. bayanus and hybrid diploids strains: room temperature incubation of transformation mix for 30 minutes, 5 minute heat shock at 42°C, and 5 minute rest at room temperature following heat shock.
Except for some noted below, insertion/knockout constructs were generated through one-step transformation of a PCR amplified linear construct. Each primer for these constructs included ∼50 bp of homology to target for genomic integration and ∼20 bp that anneal to a plasmid template for the amplification of a selectable marker [pAG32-hphMX4 (Hygromycin B), pAG25-ClonatMX4 (Clonat), pFA6a-kanMX4 (Kanamycin), or pMPY-ZAP (hisG-URA3-hisG pop-in/pop-out construct)]. One primer of each of the I-SceI cut site primer pairs also included the 30 bp I-SceI recognition sequence from [49]. For RAHScs, the primers included linkers to amplify an AgeI-I-SceIcs/HYG-ClaI fragment, which was digested and ligated into AgeI-ClaI site of pFT1 (derived from p150Ty, this study). The resulting plasmid, called pFT1-SceIcs, was double-digested with NotI and KpnI and a 10.2 kb purified NotI-KpnI fragment was used for transformation to create RAHScs. For FRAHSΔ::hisG, three primer pairs (FRAHSΔ-left, FRAHSΔ-middle, FRAHSΔ-right) were used to generate three overlapping fragments that were co-transformed. Sequences for gene knockout primers are available upon request. All other strain construction primers included in Table S2. All genome manipulations were performed in haploid strains, and all constructs were verified by Southern blot analysis. Pairs of S. cerevisiae and S. bayanus haploids were mated to generate the desired purebred and hybrid diploids, and then transformed with the I-SceI expression plasmid (see below). All experiments in this study were performed at 23°C unless noted otherwise.
Media and reagents
Yeast strains were grown in YEP, SC-ADE, SC-ADE-URA media supplemented with 2% dextrose (D), 2% lactic acid 3% glycerol (LAG), 0.3 mg/ml Hygromycin B (HYG), as indicated. YEPD media was supplemented with 10 µg/ml adenine. Glucose and glycerol was purchased from EMD Biosciences, lactic acid (40% v/v stock, [pH 5.7]) from Fisher Scientific, and Hygromycin B (HYG) from Roche. SC dropout powders were homemade from amino acids purchased from Sigma-Aldrich.
I-SceI expression plasmids
The GALp-I-SceI construct was from pWJ1320 [49], a gift from Rodney Rothstein. pMH5 was derived from pWJ1320 (2 micron-based) by deleting a 2.0 kb EcoO109I fragment containing URA3 marker. pMH6 (2 micron-based) and pMH7 (CEN-based) were created by ligating the 2.0 kb SalI fragment from pWJ1320 (containing the GALp-I-SceI expression construct) into the unique SalI site of pRS422 and pRS412, respectively. pMH6 and pMH7 were generated to include a larger promoter sequence for the ADE2 marker, however, all plasmids yielded similar results.
Induction of I-SceI site-specific DSB
A single colony from SC-ADE-URA+D+HYG plates [to select for GALp-I-SceI expression plasmid (Ade+) and no DSB (HygRUra+)] was used to inoculate SC-ADE-URA+D for a 5 ml starter culture that was grown to saturation. A small volume of the starter was used to inoculate SC-ADE+LAG cultures and these cultures were grown for more than two doubling to exponential phase [OD(600) ∼1.0]. For the uninduced control, immediately before DSB induction, an aliquot was appropriately diluted in water and plated onto YEPD for individual colonies (uninduced frequencies are subtracted out of induced frequencies, see below). To induce the DSB, galactose (20% v/v stock) was added to a final of 2% and after two hours, the cultures were diluted in water and plated onto YEPD for individual colonies (referred to as clones). Plates were incubated at 23°C for 3–5 days.
Determine frequencies of chromosome III repair or loss after an I-SceI–induced DSB
YEPD platings from uninduced and induced were first replica plated onto YEPD or 2% agar plates. This replica plate was then immediately used on a fresh velvet to replica onto YEPD+HYG, SC-URA+D, and SC-LEU+D plates. These marker plates were incubated at 23°C for 2–4 days. Each colony from the original YEPD plate was scored for the presence or absence of chromosome III markers (LEU2, HYG, URA3) by growth or no growth on marker plates. Assessment of the heterozygous markers (present on the S. cerevisiae homolog with the I-SceIcs) determines whether the founding cell had experienced an I-SceI-induced DSB (leading to the HygS phenotype) followed by chromosome repair [HygS and Leu+Ura+ (class I) or HygS and Leu+Ura− (class II)] or chromosome loss [HygS and Leu−Ura− (class III)]. The HygS phenotype most likely occurs through the removal of the nonhomologous ends (1.6 kb I-SceIcs/HYG construct), which is a natural and efficient step during HR repair [50], [51].
The following three steps were used to calculate frequencies of repair and loss events. First, the numbers of clones that fell into each genetic class (I, II, III) out of the total number of clones scored were calculated as percentages for both uninduced and induced cultures. Second, uninduced percentages were subtracted from induced percentages to eliminate events that occurred before galactose addition. Occasionally, cultures with high background frequencies (>50% of clones were HygS in uninduced cultures) were observed and not used. HygS phenotypes before galactose induction are due to leakiness of the galactose promoter during nonrepressive growth (see Figure S3). Third, the total percentage (class I + class II + class III) was normalized to 100%. A third potential repair class, HygS and Leu−Ura+, arose so infrequently (<1% in wild-type purebred and hybrid diploids) that it was omitted from these calculations.
Determining type of repair after an I-SceI–induced DSB
Single repair clones (class I and II) from SC-LEU+D marker plates were restruck for individual isolates onto fresh SC-LEU+D plates to ensure clonality (i.e. possible mixing during replica plating process). One isolate from this restreak was used to inoculated YEPD media and grown to saturation for the subsequent isolation of genomic DNA for PFGE/Southern analysis using a LEU2 probe (see below). Hybridization that resulted in wild-type chromosome III size (purebred diploids at 341 kb, hybrid diploids at 320 kb) was identified as AHR and those with an altered chromosome III size, indicative of a rearrangement, were classified as potential NAHR. The structures of the chromosome III rearrangement structures were first determined in wild-type hybrid diploids (MH3360) due to the advantage of no signal from an uncut homolog.
Internal deletions
Rearrangements in genetic class I from MH3360 were determined to be internal deletions mediated by RAHS and FRAHS and based on three pieces of evidence: 1) 18 repair clones analyzed by PFGE/Southern, which indicated a ∼20–30 kb decrease in S. cerevisiae chromosome III size compared wild-type (341 kb) as would be predicted for an internal deletion between RAHS and FRAHS, 2) Same 18 repair clones were subjected to PCR analysis using S. cerevisiae specific primers that flank RAHS (RAHS-L and RAHS-R) and FRAHS (FRAHS-L and FRAHS-R), which resulted in PCR products at the two outer sides of RAHS and FRAHS and no PCR product at the inner two sides (whereas all bands appear in the wild-type control) (primer sequences in Table S2). At least one hybrid and purebred internal deletion clone was further analyzed by long-range GeneAmp XL-PCR (Applied Biosystems) with primers that amplified the predicted RAHS-FRAHS deletion junction (primer sequences in Table S2) and 3) FRAHSΔ in MH3524/MH3572/MH3573 (eliminates donor) nearly abolished genetic repair class I (<4% of cells after DSB).
Isochromosomes, chromosome rings, and translocation
Rearrangements in genetic class II were determined to be mainly composed of three structures. 52 repair clones in class II from MH3360 were classified into three groups based on PFGE/Southern hybridization pattern: Group W for hybridization in well, Group L for larger (>340 kb), and Group S for smaller (210–280 kb). The recipient Ty loci used to mediate the rearrangements were localized to the recombinant junction by PCR analysis on 21 repair class II clones from MH3360 using primer pairs that flank YCRCdelta6 (YCRCdelta6-L and YCRCdelta6-R), YCRCdelta7 (YCRCdelta7-L and YCRCdelta7-R), and RAHS (RAHS-L and RAHS-R) (primers sequences in Table S2). Group W was further determined to be chromosome rings mediated by RAHS and LAHS based on the following observations: (1) Leu+ phenotype, yet PFGE/Southern analysis indicated no LEU2 probe hybridization in the lane, but strong hybridization in well, (2) Unlike control samples, Southern analysis on four clones from MH3346 (same as MH3360, but I-SceIcs/HYG construct is inverted) showed an absence of signal from probes that hybridize to restriction fragments near telomere ends, (3) Digestion of four PFG agarose plugs with PacI from MH3346 followed by PFGE/Southern analysis resulted in the release of an ∼80 kb fragment that hybridizes to LEU2 probe concomitant with loss of hybridization signal to the well, (4) aCGH on one clone generated from MH3346 showed sequence loss of all left and right telomere-proximal sequences adjacent to LAHS and RAHS, (5) MH3398 (LAHSΔ, eliminates ring donor) and MH3471 (147cs, eliminates ring recipient) abolished Group W by PFGE/Southern analysis, and (6) at least one hybrid and purebred ring clone was further analyzed by long-range GeneAmp XL-PCR using primers that amplified the predicted RAHS-LAHS ring junction (primer sequences in Table S2). For Group L, PFGE/Southern analysis was repeated on 12 clones from strain MH3360 and MH3398 (LAHSΔ enriches for translocations in class II) under conditions that separated all S. cerevisiae chromosomes. Majority of clones (9 out of 12) were ∼485 kb and aCGH on two of these clones suggested a translocation mediated between RAHS and YJRWTy1-1/YJRWTy1-2 locus from chromosome X. For Group S, PCR analysis localized to the recombinant junction three different Ty recipient loci, YCRCdelta6, YCRCdelta7, and RAHS corresponding to Group S size subclasses of 210–230 kb, 240–255 kb, and 260–280 kb, respectively. At least one hybrid and purebred isochromosome clone was further analyzed by long-range GeneAmp XL-PCR using primers that amplified the predicted YCRCdelta6-LAHS and YCRCdelta7-LAHS junction (primer sequences in Table S2). Group S were further determined to be isochromosomes based on (1) aCGH on one clone from MH3346 indicated a 2-fold increase to of left arm adjacent to LAHS and loss of all sequences to right of YCRCdelta6. (2) MH3398 (LAHSΔ, eliminates isochromosome donor) abolished Group S by PFGE/Southern analysis. (3) MH3471 (147cs, eliminates RAHS recipient) abolishes 260–280 kb-sized clones (RAHS-mediated isochromosomes) by PFGE/Southern analysis.
These aCGH and PCR analyses of chromosome III rearrangements revealed that many specific rearrangements reoccur and have signature mobility on PFGs. Representative clones were subjected to aCGH and PCR analyses to validate the use of signature mobilities as a diagnostic tool for rearrangements. These signature mobilities matched the mobilities of the rearranged chromosome III from repair clones found in the mutant hybrids as well as wild-type and mutant purebreds. Therefore in these other diploids, we could use the mobility of the rearrangement to identify the type of rearrangement as well as the specific recipient and donor loci.
Calculation of frequencies of outcomes after an I-SceI–induced DSB
Frequencies were calculated in three steps. 1) Frequencies of genetic classes (I, II, III) of uninduced cultures were subtracted from frequencies of induced cultures to eliminate events that occurred prior to galactose addition (described in more detail above, frequency of chromosome loss determined here). 2) For the repair events, the fraction of each type of repair (i.e. allelic, internal deletion, etc) among the total PFG plugs analyzed from its corresponding genetic class (I or II) was calculated. 3) For the repair events, the genetic class frequency (step one) was multiplied by the fraction of each repair type in that genetic class (step two). For example, in wild-type purebred diploids (MH3359), 85.7% of HygS clones (n = 1062) were class I (Leu+HygSUra+). 5 out of 32 random repair clones of class I were classified as internal deletions by PFGE/Southern analysis, so the frequency of internal deletions in MH3359 is 5/32(85.7%) = 13.4%.
PFGE/southern analysis
Yeast genomic DNA was prepared in 1% low-melting agarose plugs (SeaPlaque 50100) as previously described [52] and resolved on 1% agarose gel (Bio-Rad 162-0138) in 0.5XTBE using Bio-Rad CHEF-DR III System. To optimize resolution between S. cerevisiae and S. bayanus chromosome III the following parameters were used: 6 V/cm, 120° angle, 1–25 s switch times, 24 hours at 14°C. To assess yeast whole genome karyotypes (i.e. for translocations), the parameters were the same except for 60–120 s switch times. Gels were blotted using GeneScreen Plus membrane (Perkin Elmer NEF988) and probed with a 1.3 kb fragment from the S. cerevisiae LEU2 locus amplified using the U2-FOR/U2-REV primer pair (Table S2).
Calculation of standard error of the mean (SEM)
To calculate SEMs for the repair outcomes, the following numbers were used: (a) average frequency of Leu+HygSUra+ genetic class I, (b) average frequency of Leu+HygSUra− genetic class II, (c) total number of Leu+HygSUra+ (class I) plugs analyzed by PFGE/Southern analysis, (d) total number of Leu+HygSUra− (class II) plugs analyzed using PFGE/Southern analysis, (e) number of Leu+HygSUra+ (class I) plugs of a particular repair outcome (i.e. allelic, internal deletion), (f) number of Leu+HygSUra− (class II) plugs of a particular outcome (i.e. ring, translocation, isochromosome). SEM was calculated in two steps. First, the initial SEM was calculated using the formula SQRT(pq/n), where p = fraction of a particular repair outcome observed by PFGE/Southern analysis over total analyzed from that class (e or f divided by c or d, respectively), q = 1-p, and n = total number of repair clones analyzed by PFGE/Southern analysis from that corresponding class (c or d). Second, the final SEM was calculated by weighting the SEM with the corresponding genetic class frequency (initial SEM multiplied by a or b).
The rationale for this method was to be most stringent by using the smallest n (d or e). In the following cases e or f was assigned the number 1: (1) when all Leu+HygSUra+ plugs were deletions (i.e. in hybrid diploids), (2) no products appear in any plugs analyzed (i.e. rings in rad51Δ/rad51Δ mutant), (3) genetic class is 0 (i.e. Leu+HygSUra− class II in rad52Δ/rad52Δ hybrid diploids), (4) when no plugs analyzed (i.e. Leu+HygSUra− class II in rad52Δ/rad52Δ purebred diploids). For case 1, the error was estimated by assuming the next plug would not be that particular outcome. For case 2, 3, and 4, the upper bound was estimated by assuming the next plug would be that particular outcome. In the case where repair outcomes came from both the Leu+HygSUra+ and Leu+HygSUra− genetic classes (i.e. other, allelic in purebred diploids), “final SEMs” were calculated as described above and then “final SEMs” from each class was added together for the reported SEM. To calculate SEMs for chromosome loss, the formula SD/SQRT(n) was used where SD (standard deviation) = SD of the frequency of Leu−HygSUra− clones from different isolates and/or DSB-inductions (same experiment used to generate numbers for a and b above) and n = total number of different DSB-inductions performed for that particular strain (ranging between 2 to 8).
Viability
Exponential cultures in –ade +2% lactic acid +3% glycerol were appropriately diluted in water and the same volume was plated on –ade +2% galactose and –ade +2% glucose. Plates were incubated at 23°C. Percent viability was calculated as the number of colony forming units on galactose divided by the number of colony forming units on glucose.
Array CGH
aCGH methods were performed as previously described [53]. S. cerevisiae/S. bayanus hybrid microarrays were custom designed and printed by Lewis-Sigler Institute Microarray Facility at Princeton University.
Sequencing of LAHS, RAHS, FRAHS from S288C background
Numerous studies have brought to light unannotated Ty elements on chromosome III [34], [44], [54]–[56], with a few studies publishing a limited restriction digest map of the Ty structure in these regions [44], [54], [55]. These unannotated Ty clusters were sequenced here. Each cluster was cloned from strain MH3303 (MATa lys2Δ0 ura3Δ0, derived from BY4716 [46]) by gap repair to create p85Ty, p150Ty, and p169Ty (see Figure S1). Each plasmid was subjected to transposon bombing using the Finnzymes Template Generation System (TGS). For each plasmid, 192 clones with different random transposon insertions were picked and sequenced with a pair of primers located at the edges of the TGS transposon to produce pairs of oppositely directed reads. 384 attempted reads were performed per yeast clone. Sequence data were processed, assembled and edited using the Phred/Phrap/Consed suite of programs [57]. Each assembly was reviewed and edited to ensure there were no discrepancies due to misplaced reads or low quality regions. The automated assembler resulted in collapses of repeats, and these were manually resolved. 16.8 kb of sequence at LAHS, 14.5 kb at RAHS, and 14.7 kb at FRAHS were deposited into GenBank with accession number GU224294, GU220389, and GU220390, respectively. The sequence included five additional full length Ty1s and a solo LTR, complementing the LAHS reference sequence in SGD and almost entirely replacing the RAHS and FRAHS reference sequence. The new sequence changes chromosome III size from 316,617 bp (in SGD) to 341,823 bp.
Sequence comparisons of Ty1 and Ty2 elements
Sequences for all previously described Ty1, Ty2 and LTRs (delta) elements were obtained from the SGD “Non-ORF dataset” (http://downloads.yeastgenome.org/, timestamp January 5, 2010). Several corrections were made based on our resequencing and analysis: (1) addition of five Ty1 elements on chrIII (Ty1–1 through Ty1–5) (2) addition of nine delta elements on chrIII (delta16 through delta24) (3) removal of three delta elements on chrIII (YCRWdelta8, YCRWdelta9, and YCRWdelta10) (4) addition of one unannotated Ty1 element on chrXII (encompassing YLR035C-A) (5) addition of two unannotated delta elements on chrIV (LTRs for YDRCTy1-2).
The “Overall Identity (%)” between two sequences was determined by creating a global sequence alignment using the Needleman-Wunsch algorithm (gapopen = 10, gapextend = 0.5) as implemented in needleall v6.2.0 [58].
The “Longest Block of 100% Identity (nt)” was determined by first creating a local sequence alignment using the NCBI BLAST algorithm (match = 1, mismatch = −3, gapopen = −1, gapextend = −1) as implemented in bl2seq v2.2.18 [59]. Custom Perl scripts using BioPerl v1.6.1 iterated through each set of hits to identify the longest contiguous block of matching nucleotides [60].
Finally, the contribution of sequence similarity to donor usage is likely more complex than either overall identity or longest block of perfect identity. We therefore calculated bit scores using the BLAST heuristic, which attempts to balance length and perfect identity when searching for a shared region between two sequences that has the “most” similarity. This “Local Identity (bitscore)” was determined using blastall.
Source code and data files can be found at: http://dl.getdropbox.com/u/547386/code.zip
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. StankiewiczP
LupskiJR
2010 Structural variation in the human genome and its role in disease. Annu Rev Med 61 437 455
2. KorbelJO
UrbanAE
AffourtitJP
GodwinB
GrubertF
2007 Paired-end mapping reveals extensive structural variation in the human genome. Science 318 420 426
3. KiddJM
CooperGM
DonahueWF
HaydenHS
SampasN
2008 Mapping and sequencing of structural variation from eight human genomes. Nature 453 56 64
4. HanK
LeeJ
MeyerTJ
RemediosP
GoodwinL
2008 L1 recombination-associated deletions generate human genomic variation. Proc Natl Acad Sci U S A 105 19366 19371
5. XingJ
ZhangY
HanK
SalemAH
SenSK
2009 Mobile elements create structural variation: analysis of a complete human genome. Genome Res 19 1516 1526
6. PaquesF
HaberJE
1999 Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 63 349 404
7. SugawaraN
HaberJE
1992 Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol 12 563 575
8. RudinN
HaberJE
1988 Efficient repair of HO-induced chromosomal breaks in Saccharomyces cerevisiae by recombination between flanking homologous sequences. Mol Cell Biol 8 3918 3928
9. RayA
MachinN
StahlFW
1989 A DNA double chain break stimulates triparental recombination in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 86 6225 6229
10. InbarO
KupiecM
1999 Homology search and choice of homologous partner during mitotic recombination. Mol Cell Biol 19 4134 4142
11. DattaA
HendrixM
LipsitchM
Jinks-RobertsonS
1997 Dual roles for DNA sequence identity and the mismatch repair system in the regulation of mitotic crossing-over in yeast. Proc Natl Acad Sci U S A 94 9757 9762
12. LarocqueJR
JasinM
2010 Mechanisms of recombination between diverged sequences in wild-type and BLM-deficient mouse and human cells. Mol Cell Biol
13. ShenP
HuangHV
1986 Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics 112 441 457
14. WaldmanAS
LiskayRM
1988 Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol Cell Biol 8 5350 5357
15. Jinks-RobertsonS
MichelitchM
RamcharanS
1993 Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol Cell Biol 13 3937 3950
16. RoederGS
SmithM
LambieEJ
1984 Intrachromosomal movement of genetically marked Saccharomyces cerevisiae transposons by gene conversion. Mol Cell Biol 4 703 711
17. LichtenM
HaberJE
1989 Position effects in ectopic and allelic mitotic recombination in Saccharomyces cerevisiae. Genetics 123 261 268
18. AgmonN
PurS
LiefshitzB
KupiecM
2009 Analysis of repair mechanism choice during homologous recombination. Nucleic Acids Res 37 5081 5092
19. BurgessSM
KlecknerN
1999 Collisions between yeast chromosomal loci in vivo are governed by three layers of organization. Genes Dev 13 1871 1883
20. SchlechtHB
LichtenM
GoldmanAS
2004 Compartmentalization of the yeast meiotic nucleus revealed by analysis of ectopic recombination. Genetics 168 1189 1203
21. MalkovaA
NaylorML
YamaguchiM
IraG
HaberJE
2005 RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51-mediated gene conversion. Mol Cell Biol 25 933 944
22. WuX
WuC
HaberJE
1997 Rules of donor preference in saccharomyces mating-type gene switching revealed by a competition assay involving two types of recombination. Genetics 147 399 407
23. LiangF
HanM
RomanienkoPJ
JasinM
1998 Homology-directed repair is a major double-strand break repair pathway in mammalian cells. Proc Natl Acad Sci U S A 95 5172 5177
24. NeuvegliseC
FeldmannH
BonE
GaillardinC
CasaregolaS
2002 Genomic evolution of the long terminal repeat retrotransposons in hemiascomycetous yeasts. Genome Res 12 930 943
25. LitiG
PeruffoA
JamesSA
RobertsIN
LouisEJ
2005 Inferences of evolutionary relationships from a population survey of LTR-retrotransposons and telomeric-associated sequences in the Saccharomyces sensu stricto complex. Yeast 22 177 192
26. LeeHY
ChouJY
CheongL
ChangNH
YangSY
2008 Incompatibility of nuclear and mitochondrial genomes causes hybrid sterility between two yeast species. Cell 135 1065 1073
27. KellisM
PattersonN
EndrizziM
BirrenB
LanderES
2003 Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423 241 254
28. JainS
SugawaraN
LydeardJ
VazeM
Tanguy Le GacN
2009 A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev 23 291 303
29. SpellRM
Jinks-RobertsonS
2004 Examination of the roles of Sgs1 and Srs2 helicases in the enforcement of recombination fidelity in Saccharomyces cerevisiae. Genetics 168 1855 1865
30. Fishman-LobellJ
RudinN
HaberJE
1992 Two alternative pathways of double-strand break repair that are kinetically separable and independently modulated. Mol Cell Biol 12 1292 1303
31. SugawaraN
WangX
HaberJE
2003 In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell 12 209 219
32. ArguesoJL
WestmorelandJ
MieczkowskiPA
GawelM
PetesTD
2008 Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci U S A 105 11845 11850
33. ParketA
InbarO
KupiecM
1995 Recombination of Ty elements in yeast can be induced by a double-strand break. Genetics 140 67 77
34. VanHulleK
LemoineFJ
NarayananV
DowningB
HullK
2007 Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol 27 2601 2614
35. LeePS
PetesTD
2010 From the Cover: mitotic gene conversion events induced in G1-synchronized yeast cells by gamma rays are similar to spontaneous conversion events. Proc Natl Acad Sci U S A 107 7383 7388
36. ChungWH
ZhuZ
PapushaA
MalkovaA
IraG
2010 Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet 6 e1000948 doi:10.1371/journal.pgen.1000948
37. MarshallWF
StraightA
MarkoJF
SwedlowJ
DernburgA
1997 Interphase chromosomes undergo constrained diffusional motion in living cells. Curr Biol 7 930 939
38. DuanZ
AndronescuM
SchutzK
McIlwainS
KimYJ
2010 A three-dimensional model of the yeast genome. Nature 465 363 367
39. KimJM
VanguriS
BoekeJD
GabrielA
VoytasDF
1998 Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res 8 464 478
40. ThompsonM
HaeuslerRA
GoodPD
EngelkeDR
2003 Nucleolar clustering of dispersed tRNA genes. Science 302 1399 1401
41. Johnson-SchlitzDM
FloresC
EngelsWR
2007 Multiple-pathway analysis of double-strand break repair mutations in Drosophila. PLoS Genet 3 e50 doi:10.1371/journal.pgen.0030050
42. ElliottB
RichardsonC
JasinM
2005 Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell 17 885 894
43. TremblayA
JasinM
ChartrandP
2000 A double-strand break in a chromosomal LINE element can be repaired by gene conversion with various endogenous LINE elements in mouse cells. Mol Cell Biol 20 54 60
44. UmezuK
HiraokaM
MoriM
MakiH
2002 Structural analysis of aberrant chromosomes that occur spontaneously in diploid Saccharomyces cerevisiae: retrotransposon Ty1 plays a crucial role in chromosomal rearrangements. Genetics 160 97 110
45. GuthrieC
FinkGR
1991 Guide to Yeast Genetics and Molecular Biology. San Diego, California Academic Press, Inc.
46. BrachmannCB
DaviesA
CostGJ
CaputoE
LiJ
1998 Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14 115 132
47. MartinOC
DeSevoCG
GuoBZ
KoshlandDE
DunhamMJ
2009 Telomere behavior in a hybrid yeast. Cell Res 19 910 912
48. GietzRD
SchiestlRH
2007 High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2 31 34
49. LisbyM
MortensenUH
RothsteinR
2003 Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat Cell Biol 5 572 577
50. Fishman-LobellJ
HaberJE
1992 Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 258 480 484
51. HaberJE
1992 Mating-type gene switching in Saccharomyces cerevisiae. Trends Genet 8 446 452
52. SchwartzDC
CantorCR
1984 Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell 37 67 75
53. DunhamMJ
BadraneH
FereaT
AdamsJ
BrownPO
2002 Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 99 16144 16149
54. WicksteedBL
CollinsI
DershowitzA
StatevaLI
GreenRP
1994 A physical comparison of chromosome III in six strains of Saccharomyces cerevisiae. Yeast 10 39 57
55. LemoineFJ
DegtyarevaNP
LobachevK
PetesTD
2005 Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120 587 598
56. ShibataY
MalhotraA
BekiranovS
DuttaA
2009 Yeast genome analysis identifies chromosomal translocation, gene conversion events and several sites of Ty element insertion. Nucleic Acids Res 37 6454 6465
57. GordonD
AbajianC
GreenP
1998 Consed: a graphical tool for sequence finishing. Genome Res 8 195 202
58. RiceP
LongdenI
BleasbyA
2000 EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16 276 277
59. AltschulSF
GishW
MillerW
MyersEW
LipmanDJ
1990 Basic local alignment search tool. J Mol Biol 215 403 410
60. StajichJE
BlockD
BoulezK
BrennerSE
ChervitzSA
2002 The Bioperl toolkit: Perl modules for the life sciences. Genome Res 12 1611 1618
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 12
- Souvislost haplotypu M2 genu pro annexin A5 s opakovanými reprodukčními ztrátami
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
Nejčtenější v tomto čísle
- Functional Comparison of Innate Immune Signaling Pathways in Primates
- Expression of Linear and Novel Circular Forms of an -Associated Non-Coding RNA Correlates with Atherosclerosis Risk
- Genome-Wide Interrogation of Mammalian Stem Cell Fate Determinants by Nested Chromosome Deletions
- Histone H2A C-Terminus Regulates Chromatin Dynamics, Remodeling, and Histone H1 Binding
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy