
Cystická fibróza dospělých
Cystic fibrosis in adults
Cystic fibrosis (CF) is an inherited disease caused by mutations in the transmembrane conductance regulator (CFTR) gene. The disease leads to dysfunction of the exocrine glands with high concentration of chloride in the sweat and formation of abnormally viscous mucus in the respiratory, digestive and reproductive tract. Chronic sinopulmonary disease, exocrine pancreatic insufficiency, liver disease, intestinal obstruction, impaired nutritional status, salt loss syndrome and male infertility dominates in the clinical presentation. The examination of sweat chloride concentration and mutations in the CFTR gene is used in CF diagnostics for detection of CFTR protein dysfunction. The treatment comprises especially respiratory physiotherapy with mucolytics inhalations, aggressive antibiotic therapy and high-calorie diet together with adequate pancreatic enzymes substitution. The prevention of airway infection with resistant bacterial pathogens, particularly Pseudomonas aeruginosa, is a fundamental measure. Significant recent progress include the use of newborn screening of CF and drugs targeted to individual CFTR gene mutations in the clinical practise. The prognosis of patients has improved due to using of modern therapeutic methods in CF treatment centres. Children born at present time have survival probability 40–50 years.
Key words:
adults – cystic fibrosis – diagnostics – therapy
Autoři:
Libor Fila
Působiště autorů:
Pneumologická klinika 2. LF UK a FN Motol, Praha
Vyšlo v časopise:
Vnitř Lék 2017; 63(11): 834-842
Kategorie:
Přehledné referáty
Souhrn
Cystická fibróza (CF) je vrozené onemocnění vyvolané mutacemi genu pro transmembránový regulátor vodivosti (CFTR). Onemocnění vede k dysfunkci žláz s vnitřní sekrecí s vysokou koncentrací chloridů v potu a s tvorbou abnormálně vazkého hlenu v dýchacím, trávicím a rozmnožovacím ústrojí. Klinicky se projevuje především chronickým sinopulmonálním onemocněním, insuficiencí zevní sekrece pankreatu, hepatopatií, poruchami střevní pasáže a výživy, syndromem ztráty solí a mužskou neplodností. V diagnostice se k průkazu dysfunkce CFTR proteinu využívá vyšetření koncentrace chloridů v potu a mutací genu CFTR. V léčbě se uplatňuje především respirační fyzioterapie s inhalacemi mukolytik, agresivní antibiotická léčba a vysokokalorická strava spolu s adekvátní substitucí pankreatickými enzymy. Zcela zásadní je prevence infekce dýchacích cest rezistentními bakteriálními patogeny, zejména Pseudomonas aeruginosa. Mezi významné pokroky posledních let pak můžeme zahrnout využití novorozeneckého screeningu CF a léků cílených na jednotlivé mutace genu CFTR v klinické praxi. S využitím moderních léčebných metod a centrové péče se podařilo zlepšit prognózu nemocných tak, že v současnosti narozené děti s CF mají předpokládané přežití 40–50 let.
Klíčová slova:
cystická fibróza – diagnostika – dospělý věk – léčba
Úvod
Cystická fibróza (CF) je vrozené onemocnění vyvolané mutacemi genu pro transmembránový regulátor vodivosti (CFTR) projevující se především chronickým sinopulmonálním onemocněním, postižením trávicího ústrojí s poruchou stavu výživy, vysokou koncentrací chloridů v potu a obstruktivní azoospermií. Název tato nemoc získala od strukturálních změn pankreatu popsaných americkou patoložkou Dorothy Andersenovou v roce 1938. V Česku publikovala první malý soubor pediatrických nemocných prof. Dagmar Benešová již v roce 1946. Důležitý byl objev zvýšené koncentrace chloridů v potu Paulem di Sant‘ Agnesem v roce 1953 využitý posléze Gibsonem a Cookem jako diagnostický test. V té době byly diagnostikovány jen těžší formy onemocnění a takto postižené děti umíraly obvykle již v předškolním věku. Zlepšující se diagnostické a léčebné možnosti postupně vedly k rozpoznávání lehčích forem CF a k prodlužování přežití nemocných spolu se zvyšováním jejich počtu. Skutečnost, že se přežití nemocných s CF prodlužovalo, vedla k zakládání poraden pro dospělé s CF. První takové pracoviště vzniklo v roce 1965 ve Velké Británii. V Česku se jako první začal o dospělé s CF starat doc. Jaromír Musil v roce 1987. Narůstající počet poraden pro dospělé s CF dobře ilustrují data ze Spojených států amerických, v nichž v roce 1990 existovalo 5 takových pracovišť, ale v roce 2001 již 68. Rovněž v Česku se počet poraden pro dospělé s CF do roku 2001 zvýšil, a to na 5. Tyto poradny pracují při plicních klinikách fakultních nemocnic v Praze-Motole, Brně-Bohunicích, Hradci Králové, Olomouci a Plzni.
Epidemiologie
Výskyt CF je nejčastější v bělošských populacích s incidencí 1 : 2 500–4 500 živě narozených. V Česku je výskyt udáván mezi 1 : 2 700 (epidemiologická a genetická studie) a 1 : 4 000 (prenatální diagnostika a novorozenecký screening) živě narozených. Do budoucna však u nás lze očekávat pokles incidence, podobně jako v dalších evropských zemích. Důvodem je rostoucí podíl novorozenců neevropského původu. Dle registru americké CF Foundation vzrostl v letech 2000 a 2015 medián predikovaného přežití nemocných z 33,3 na 41,7 roku a podíl dospělých pacientů z 38,7 na 51,6 % [1]. V Česku pak zastoupení dospělých v letech 1998, 2007 a 2017 činilo 33,5, 38,2 a 44,9 % [2].
Etiologie
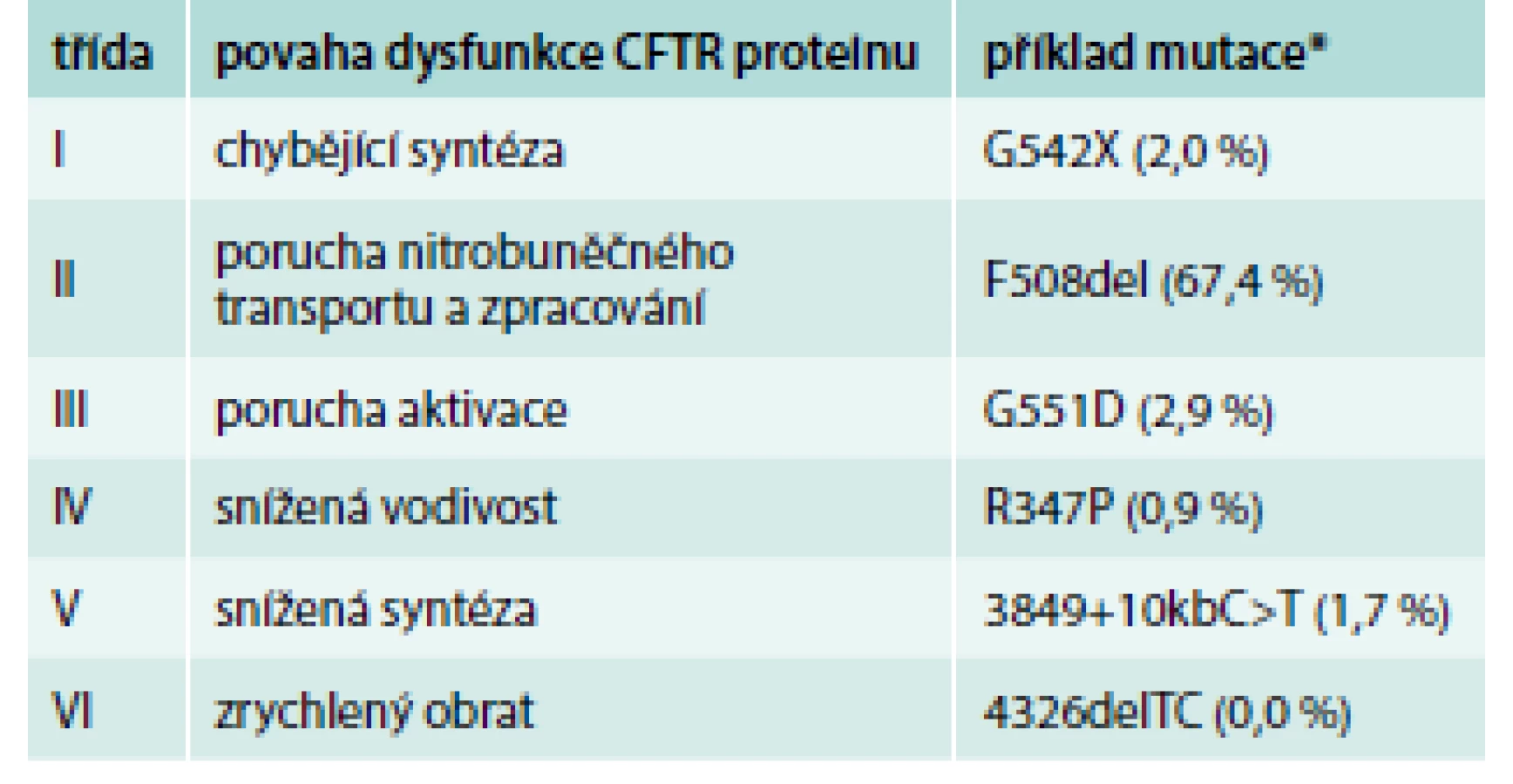
Mutace genu CFTR vedou k poruše tvorby a/nebo funkce CFTR proteinu. V aktuální databázi CFTR2 (www.cftr2.org) je zaznamenáno 281 mutací vyvolávajících CF („CF-causing“) a 21 mutací s variabilním klinickým dopadem („varying clinical consequence“). Celosvětově je nejčastější mutace F508del. Druhou nejčastější mutací v Česku je „slovanská“ mutace CFTRdele2,3 se zastoupením 5,8 %. Dle povahy dysfunkce CFTR proteinu lze mutace genu CFTR rozdělit do 6 tříd. Třídy I–III zahrnují mutace spojené s těžkým průběhem onemocnění a třídy IV–VI mutace s mírnějším fenotypem (tab. 1) [3,4].
Patogeneze
CFTR protein je cAMP regulovaný chloridový kanál, jehož dysfunkce vede k postižení žláz s vnější sekrecí. Potní žlázy nejsou schopny adekvátně absorbovat NaCl, v ostatních orgánech dochází k tvorbě abnormálně vazkého hlenu následkem nadměrné absorpce sodíkových iontů (a sekundárně i vody) epiteliálním natriovým kanálem (ENaC), jehož činnost je za normálních okolností prostřednictvím CFTR tlumena. V dýchacím ústrojí tak dochází mukostáze, na kterou nasedá chronická bakteriální infekce (typické patogeny reprezentují Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa a komplex Burkholderia cepacia) a neutrofilní zánět. Proteázy a kyslíkové radikály uvolňované z leukocytů poškozují stěnu dýchacích cest s rozvojem bronchiektázií, obstrukční ventilační poruchy a respirační insuficience [5]. V trávicím ústrojí se kromě vazkého hlenu uplatňuje i porucha sekrece hydrogenuhličitanů. Postižení pankreatu vede k fibróze a cystické přestavbě spolu s destrukcí Langerhansových ostrůvků a rozvojem diabetes mellitus. Pankreatická insuficience má za následek steatoreu, malnutrici a podílí se na kostní nemoci. Hepatobiliární postižení vede především k fibróze až cirhóze jater a cholelitiáze, střevní postižení ohrožuje pacienty rozvojem ileózního stavu při syndromu obstrukce distálního střeva (distal intestinal obstruction syndrome – DIOS) vazkým hlenem v oblasti ileocékálního přechodu. Obstrukční azoospermie je způsobena kongenitální bilaterální absencí vas deferens (congenital bilateral absence of the vas deferens – CBAVD) a vede k mužské neplodnosti.
Klinický obraz
Klinický obraz CF je značně variabilní. Klasická forma CF je obvykle diagnostikována v raném dětství s výraznými klinickými projevy, patologickou koncentrací chloridů v potu (≥ 60 mmol/l) a přítomností 2 těžkých mutací genu CFTR. Oproti tomu atypické formy CF bývají zjišťovány v adolescenci či v dospělosti, fenotyp je méně výrazný, chloridy v potu dosahují spíše hraničních hodnot (30–59 mmol/l) a jedna z mutací genu CFTR může patřit mezi mírné. Mezi tzv. CFTR-related diseases pak patří monosymptomatická onemocnění jako diseminované bronchiektázie, idiopatická recidivující/chronická pankreatitida a obstruktivní azoospermie, u kterých může být přítomna pouze jedna mutace genu CFTR a chloridy i v pásmu normy (< 30 mmol/l).
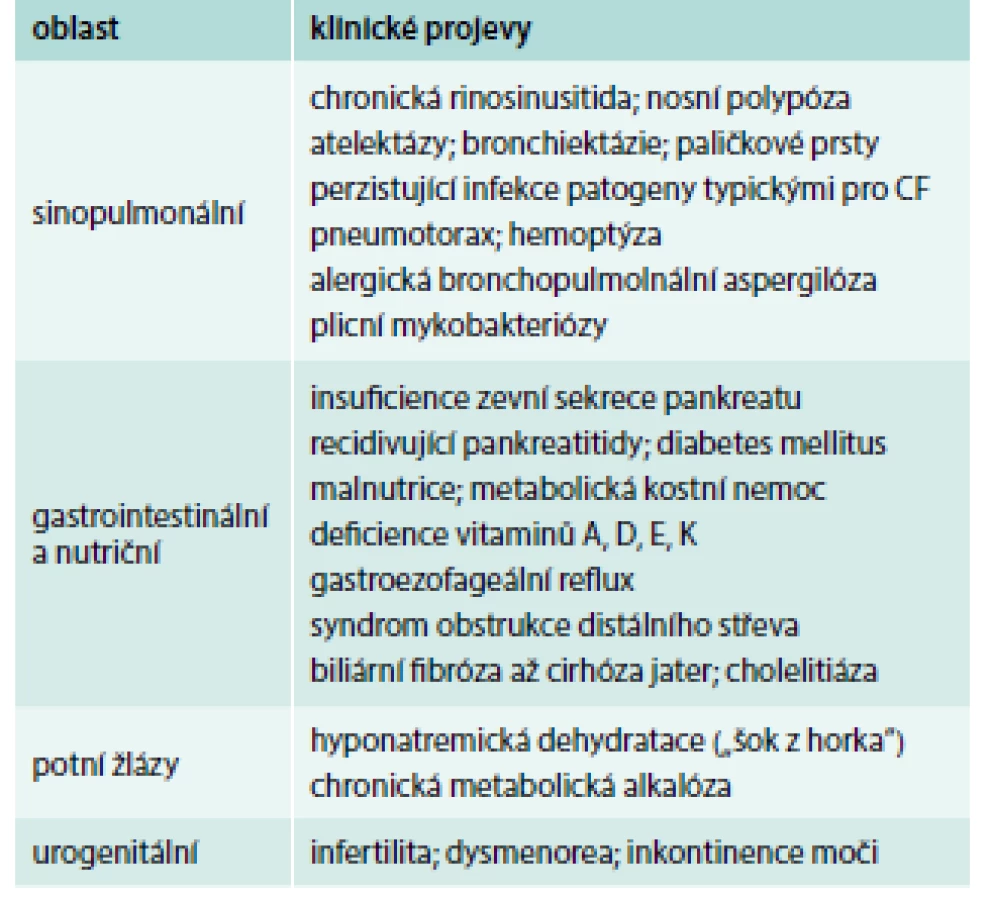
Klinický obraz může být u individuálních pacientů různě vyjádřen (od obstruktivní azoospermie jako jediného projevu až po např. těžké bronchopulmonální postižení u nemocného s pankreatickou insuficiencí, diabetem a cirhózou jater) a může se během života vyvíjet (např. muž s obstruktivní azoospermií zjištěnou ve 20 letech prodělá ve 28 letech poprvé pankreatitidu a ve 35 letech se u něj rozvine insuficience zevní sekrece pankreatu s nutností substituční léčby). Přehled hlavních projevů CF v dospělém věku je uveden v tab. 2 [6].
U bronchopulmonálního onemocnění u CF se typicky střídají období stability a období plicních exacerbací. I u stabilních nemocných je obvykle v různé míře přítomen produktivní kašel s hlenohnisavou expektorací a u pokročilejších stavů i námahová dušnost. Plicní exacerbace má obvykle infekční původ. Dochází ke zhoršení obtíží, z celkových příznaků mohou být přítomny zvýšené teploty, únavnost, nechutenství a váhový úbytek. Pomocná vyšetření detektují zvýšení systémových markerů zánětu, pokles ve spirometrii a eventuálně nový nález na skiagramu hrudníku. Nejzávažnější formou plicní exacerbace u nemocných s infekcí komplexem Burkholderia cepacia je tzv. cepacia syndrom. Jedná se o život ohrožující septický stav při nekrotizující pneumonii. Typické jsou pro něj vysoké horečky, nové multifokální plicní infiltráty na skiagramu hrudníku a pozitivita hemokultur. K této obvykle fatální komplikaci může dojít kdykoli po nákaze uvedeným patogenem. Funkčně se bronchopulmonální onemocnění projevuje progredující obstrukční ventilační poruchou a posléze rozvojem respirační insuficience a cor pulmonale.
Pokročilejší plicní onemocnění u CF se může komplikovat pneumotoraxem (PNO) nebo hemoptýzou. V obou případech je výskyt častější v dospělém než v dětském věku: incidence 1,4 % vs 0,2 % u pneumotoraxu, resp. 1,8 % vs 0,1 % u masivních hemoptýz. V dospělosti je rovněž častější pseudomonádová infekce dýchacích cest s prevalencí 79 % vs 47 % u dětí [7]. Pneumotorax se projevuje především náhle vzniklou dušností, malé pneumotoraxy mohou být asymptomatické. K potvrzení je zapotřebí provést skiagram hrudníku. Hemoptýza může kolísat od přítomnosti pouhých „žilek krve“ ve sputu až po masivní, život ohrožující krvácení.
Značně obtížná může být diagnostika alergické bronchopulmonální aspergilózy (ABPA) a plicních mykobakterióz, a to vzhledem k překrývání klinických obrazů CF a uvedených jednotek. Prevalence ABPA je uváděna okolo 10 % u osob ve věku 6 a starších, prevalence záchytu netuberkulózních mykobakterií byla v největších studiích mezi asi 7–14 % s převahou záchytu komplexu Mycobacterium avium a komplexu Mycobacterium abscessus. Výskyt ABPA i plicních mykobakterióz je častější u osob s horšími hodnotami plicních funkcí [8,9].
V oblasti horních cest dýchacích se běžně vyskytuje chronická rinosinusitida, častá je i nosní polypóza (u asi třetiny nemocných). Postižení může být asymptomatické, jindy je přítomna nosní sekrece, pocit ucpaného nosu či porucha čichu.
Insuficience zevní sekrece pankreatu se v průběhu života rozvine u asi 85 % nemocných CF. Pro maldigesci živin dochází k malnutrici, deficitu vitaminů rozpustných v tucích a průjmům (steatorea). Zhoršování stavu výživy je obvykle spojeno s poklesem plicních funkcí a stejně tak zhoršování bronchopulmonálního onemocnění vede k prohloubení malnutrice. Pankreaticky suficientní osoby jsou naproti tomu v riziku recidivujících a chronických pankreatitid, jejichž klinický obraz, diagnostika a léčba se neliší od pankreatitid jiného původu. Tato komplikace postihne v průběhu života asi 10 % pankreaticky suficientních nemocných [10].
Pokročilé postižení pankreatu má za následek destrukci Langerhansových ostrůvků a rozvoj diabetes mellitus. Jedná se o častou komplikaci dospělého věku, po které je třeba aktivně pátrat. Klinicky se projevuje zhoršováním stavu plicních funkcí anebo stavu výživy bez jiné detekovatelné příčiny; polyurie a polydipsie jsou příznaky pozdní. Diabetes mellitus je zjišťován u 40–50 % dospělých s CF [11].
Malnutrice a deficience vitaminu D jsou hlavními příčinami metabolické kostní nemoci. Patologickou kostní denzitu lze zjistit u 50–75 % dospělých – opět se jedná o projev CF, který je třeba aktivně vyhledávat. Osteoporóza v tomto případě vede k hyperkyfóze hrudní páteře a k riziku fraktur obratlů a žeber. Tyto komplikace pak negativně ovlivňují ventilaci plic a toaletu dýchacích cest [12].
Gastroezofageální reflux (GER) je popisován až u 80 % dospělých s CF. Na jeho vzniku se významně podílí kašel, dokumentován je i v průběhu dechové rehabilitace. Obtíže zahrnují bolesti v epigastriu, pyrózu, zvracení, dysfagie a nechutenství; část nemocných je však asymptomatických [13].
Obdobou mekoniového ileu novorozenců je u adolescentů a dospělých syndrom obstrukce distálního střeva (DIOS). Jedná se o komplikaci vyvolanou stázou zahuštěného střevního obsahu (včetně nestráveného hlenu) v oblasti ileocékálního přechodu u pankreaticky insuficientních osob s prevalencí asi 10 %. Obtíže mohou kolísat od nechutenství a pobolívání v pravé jámě kyčelní až po rozvinutý ileus [14].
Hepatopatie se u CF rozvíjí typicky v dětském věku (mezi 5.–10. rokem života) u asi třetiny nemocných. Jedná se o fokální biliární fibrózu jater, která u 5–10 % pacientů progreduje do multilobulární biliární cirhózy jater. U těchto nemocných se v dospělosti setkáváme s projevy portální hypertenze a selhání jater, především s krvácením z jícnových varixů. Hepatopatie je u CF příčinou úmrtí u 2,5 % případů. Mezi další hepatobiliární projevy CF patří steatóza jater (25–60 % případů) a cholelitiáza (15 % případů) [15].
Důležitými klinickými projevy CF jsou abnormality v mineralogramu a acidobazické rovnováze. Akutní hyponatremická dehydratace („šok z horka“) byla popsána u dětí s CF v období vlny veder v USA v roce 1948 Kesslerem a Andersenovou (Heat prostration in fibrocystic disease of the pancreas and other conditions. Pediatrics 1951) a vedla posléze k objevu potní anomálie u této choroby. Klinické projevy zahrnují anorexii, zvracení a váhový úbytek, u těžších případů i poruchy vědomí a křeče. Chronická metabolická alkalóza s hypokalemií pak byla popsána jako první příznak CF u dospělého pacienta [16].
CF postihuje rovněž reprodukční orgány. 97–98 % mužů s CF je neplodných z důvodu obstrukční azoospermie při CBAVD. Jedná se o nejkonstantnější klinický projev CF, který podstatně usnadňuje diagnostiku atypických forem CF u mužů. Existují však mutace se zachovalou mužskou plodností, např. 3849+10kbC>T. K neplodnosti či snížené plodnosti dochází i u žen s CF z důvodu vazkého cervikálního hlenu a poruch menstruačního cyklu při malnutrici [17,18].
Rozmanitost klinických projevů CF doplňují další příznaky plynoucí z projevů atelektáz, CF-astmatu, dekompenzovaného cor pulmonale, dilatační kardiomyopatie, apendicitidy, klostridiové kolitidy, fibrotizující kolonopatie, vývojových anomálií žlučových cest, artropatií, vaskulitid a dalších vzácnějších projevů. Podezření na CF budí rovněž nález paličkovitých prstů.
Diagnostika
Zlatým standardem v diagnostice CF je vyšetření chloridů v potu po stimulaci pocení polikarpinovou iontoforézou. Klasický sběr potu do filtračního papírku je dnes nahrazován sběrem do kapiláry pomocí systému Macroduct. Patologické hodnoty jsou ≥ 60 mmol/l, hraniční 30–59 mmol/l a normální < 30 mmol/l. Hraniční hodnoty mohou mít nemocní s atypickou formou CF, nositelé jen jedné mutace genu CFTR, dále osoby s jinými onemocněními (insuficience nadledvin, hypotyreóza, nefrotický syndrom, mentální anorexie, těžká malnutrice, celiakie, hypogamaglobulinemie a další) a rovněž osoby zdravé [19].
Molekulárně genetické vyšetření indikujeme u nemocných s odpovídajícím klinickým obrazem a opakovaně zjištěnými hraničními či patologickými hodnotami chloridů v potu, dále u pokrevních příbuzných osob s průkazem alespoň jedné mutace genu CFTR a jejich partnerů v případě plánování těhotenství (primární prevence CF), dále u podezření na CFTR-related disease (diseminované bronchiektázie, idiopatická rekurentní/chronická pankreatitida a obstruktivní azoospermie) a u dárců gamet (spermií i oocytů). Interpretace výsledku molekulárně genetického vyšetření panelu mutací genu CFTR nemusí být snadná. V případě záchytu 2 mutací je diagnóza potvrzena. V případě záchytu 1 event. i žádné mutace lze při odpovídajícím klinickém obraze a abnormálním výsledku potního testu indikovat sekvenaci celého genu CFTR. Ta může odhalit vzácné či dosud neznámé mutace [20].
Experimentálním postupem dosud zůstává vyšetření bioelektrických potenciálů nosní sliznice (in vivo) či sliznice rekta (ex vivo) k posouzení funkce CFTR proteinu. V České republice toto vyšetření dosud není běžně dostupné [21].
Na možnost přítomnosti lehčích forem CF u dospělých (obvykle pankreaticky suficientních) je třeba myslet zejména u osob s bronchiektáziemi (především v případě záchytu Pseudomonas aeruginosa, komplexu Burkholderia cepacia či netuberkulózních mykobakterií v respiračních sekretech), ABPA, idiopatickými pankreatitidami a u mužů s obstruktivní azoospermií. Dále u nejasné cirhózy jater či osteoporózy u mladých osob a u hypochloremické alkalózy (bez zvracení). U těchto nemocných je indikováno vyšetření koncentrace chloridů v potu a při hraničním či patologickém nálezu odeslání k přešetření v CF centru [22].
Diferenciální diagnostika
Vzhledem k rozmanitosti klinických projevů je diferenciální diagnostika CF velmi široká. V oblasti dolních dýchacích cest se jedná především o odlišení bronchiálního astmatu (u CF je typicky snížená hodnota vydechovaného NO), chronické obstrukční plicní nemoci a bronchiektázií jiné etiologie (imunodeficity, primární ciliární dyskineze). V oblasti gastrointestinálního traktu pak odlišení celiakie a intolerance kravského mléka [6].
Terapie
Již od 60. let minulého století jsou v léčbě CF využívány 3 základní postupy („3 pilíře“). Jednak je to respirační fyzioterapie (RFT) ke zlepšení toalety dýchacích cest, dále agresivní antibiotická léčba infekce dýchacích cest a vysokokalorická strava spolu s kvalitní substitucí pankreatickými enzymy. Mezi další postupy zásadně zlepšující přežití nemocných je nutno zařadit transplantace plic (LuTx), důsledný hygienicko-epidemiologický režim k prevenci infekce rezistentními gramnegativními bakteriemi, zavádění nových kauzálních léků cílených na jednotlivé mutace genu CFTR a především centrovou péči o nemocné, která jediná umožňuje lékařům získat dostatek zkušeností s tímto rozmanitým onemocněním. Výše uvedené a další léčebné postupy budou zmíněny v následujících oddílech věnovaných jednotlivým klinickým projevům CF.
Péče o hlavní klinické projevy cystické fibrózy
Stabilní plicní onemocnění
Péče o nemocné ve stabilní fázi plicního onemocnění zahrnuje postupy s cílem udržet průchodnost dýchacích cest, dále chronickou supresní antibiotickou a protizánětlivou léčbu u infekce Pseudomonas aeruginosa, léčbu obstrukce dýchacích cest a terapii respirační insuficience [23].
K udržení průchodnosti dýchacích cest využíváme mukoaktivní léky a RFT. Z inhalačně podávaných mukoaktivních léků se jedná o alfadornázu (Pulmozyme; dávka 1krát denně 2,5 mg – nesmí se podávat ultrazvukovými inhalátory!), hypertonický roztok soli (obvykle 5,85%; 2krát denně 4 ml – dle individuální tolerance po premedikaci bronchodilatancii) a milimolární roztok amiloridu (2krát denně 3 ml – nekombinovat s 5,85% NaCl!). Individuálně lze podat i další léky (např. ACC injekt; 2krát denně neředěný nebo 1 : 1 a Aqua pro inj). Uvedené léky podáváme výkonnými kompresorovými inhalátory, v posledních letech je k dispozici i elektronický inhalátor pracující na principu oscilující membrány (Pari eFlow), jehož výhodou je zkrácení doby inhalace a možnost provozu na baterie. RFT využívá autogenní drenáž a aktivní cyklus dechových technik (starší metody jako poklepové masáže a polohové drenáže již nepoužíváme). Instrumentální techniky využívají pozitivní výdechový přetlak buď kontinuální (PEP maska, Thera PEP), nebo oscilující (flutter, RC-Cornet, Acapella). V zahraničí jsou k dispozici i speciální vesty s vysokofrekvenčními oscilacemi aplikovanými na hrudní stěnu. RFT se standardně provádí v ranních a večerních hodinách po inhalaci 5,85% NaCl nebo amiloridu, alfadornázu nemocní inhalují v odpoledních hodinách, bez následné RFT. V zahraničí je rovněž dostupný manitol v práškové inhalační formě (Bronchitol; dávka 2krát 400 mg).
U nemocných s chronickou infekcí dýchacích cest Pseudomonas aeruginosa využíváme chronickou supresní antibiotickou a protizánětlivou léčbu. Antibiotika v tomto případě podáváme inhalačně, a to kolistin (Colomycin; 2krát denně 1–2 MIU ve 3–4 ml Aqua pro inj. dlouhodobě) nebo tobramycin (TOBI; 2krát denně 300 mg nebo TOBI Podhaler; 2krát denně 112 mg cyklicky à 28 dnů; v případě TOBI Podhaleru se jedná o práškovou inhalační formu, která zkracuje dobu inhalace). Uvedené léky podáváme dle individuální tolerance a účinnosti, lze je i kombinovat (ve 28denních pauzách TOBI podáváme kolistin). Inhalační aztreonam (Cayston; 3krát denně 75 mg cyklicky à 28 dnů) není v ČR dostupný. Vyvíjeny jsou i další inhalační formy protipseudomonádových (amikacin, ciprofloxacin) a protistafylokokových (vankomycin) antibiotik. Cyklickou léčbu nitrožilními antibiotiky (14denní kúry 3–4krát ročně) u dospělých s CF na rozdíl od dětského věku rutinně nepoužíváme. K chronické protizánětlivé léčbě využíváme azitromycin v subinhibičních dávkách (3krát týdně 250–500 mg dle tělesné hmotnosti v režimu pondělí-středa-pátek; dávka 3krát 500 mg je určena pro osoby s hmotností > 40 kg). Tato léčba je podávána na individuální bázi dle tolerance a účinnosti, navíc s ohledem na hepatopatii a přítomnost netuberkulózních mykobakterií a stafylokoků rezistentních na makrolidy. V pediatrii užívaný vysokodávkovaný ibuprofen u dospělých nepodáváme. Léčba kortikosteroidy je kontroverzní, s výjimkou CF-astmatu a ABPA není rutinně indikována. U individuálních nemocných však inhalační (budesonid 2krát 800 μg či flutikazon 2krát 500 μg) či systémové (prednison 5–20 mg obden) kortikosteroidy mohou vykazovat příznivý účinek při akceptovatelných nežádoucích účincích (stabilizace plicních funkcí u nemocných s pokročilým plicním onemocněním při chronické infekci komplexem Burkholderia cepacia).
Léčba bronchiální obstrukce je indikována u symptomatických nemocných, individuálně lze vyzkoušet inhalační betamimetika či anticholinergika (salbutamol, formoterol, salmeterol, ipratropium), teofylinové preparáty jsou nevhodné. V případě chronické respirační insuficience je indikována dlouhodobá domácí oxygenoterapie dle platných doporučení. Neinvazivní ventilační podpora je využívána nejspíše jako „most k LuTx“. Hlavním indikačním kritériem pro LuTx je u CF pokles FEV1 (usilovně vydechnutý objem za 1. s) < 30 % náležitých hodnot, neboť medián přežití u těchto nemocných činí 2 roky. Po LuTx činí medián přežití u dospělých s CF 8,9 roku.
Plicní exacerbace
Vedle navýšení mukolytik a RFT jsou hlavním léčebným prostředkem plicní exacerbace antibiotika. Dle závažnosti je exacerbace léčena ambulantně perorálními preparáty či za hospitalizace nitrožilními antibiotiky. Volba preparátu není empirická jako je tomu v běžné populaci, ale řídí se výsledky předchozích mikrobiologických vyšetření respiračních sekretů. Užívají se vyšší dávky antibiotik po delší dobu než v běžné populaci. Standardní doba léčby je 2 týdny, u nemocných s lehčím plicním postižením může být při dobré odpovědi zkrácena na 10 dnů. Těžší exacerbace vyžadují delší dobu podávání, často s využitím kombinace preparátů. Kombinované podávání antibiotik je obvyklé při pseudomonádové infekci (ambulantně inhalační antibiotika spolu s perorálně podávaným ciprofloxacinem, za hospitalizace kombinace nitrožilně aplikovaných antibiotik, obvykle betalaktamu s aminoglykosidem – s monitorováním sérových hladin) a při infekci burkholderiemi. Další léčebná opatření při plicní exacerbaci se dle stavu týkají nutriční podpory, hydratace, suplementace NaCl, oxygenoterapie, mechanické ventilační podpory a bronchoskopického odsáváním sekretů dýchacích cest [24].
Cepacia syndrom
Léčba této obávané komplikace u pacientů s infekcí komplexem Burkholderia cepacia nemá dobré výsledky. Kromě zásad uvedených v léčbě plicní exacerbace (antibiotika se podávají v 3–4kombinacích) se pokoušíme ovlivnit průběh onemocnění intravenózně podávanými imunoglobuliny, kortikosteroidy a eventuálně i dalšími imunosupresivy (cyklosporin). V případě hyperglykemie je na místě kontinuální podání inzulinu s cílem udržet normoglykemii.
Pneumotorax
Léčba se řídí obecnými pravidly pro sekundární spontánní pneumotorax (PNO). U malých PNO (max. 2 cm šíře) u asymptomatických nemocných lze observovat. Jinak provádíme hrudní drenáž. V případě jejího neúspěchu či při recidivě PNO je indikováno chirurgické ošetření obvykle torakoskopicky (s abrazí pleury apikální poloviny hemitoraxu), při neúspěchu je na místě torakotomie s parietální pleurektomií. Tyto postupy však znesnadňují následnou transplantaci plic (LuTx) [25].
Hemoptýza
Drobná hemoptýza je u dospělých s těžším bronchopulmonálním onemocněním běžnou komplikací, kterou léčíme ambulantně antibiotiky a perorálními hemostyptiky (kyselina paraaminometylbenzoová, etamsylát). Pacienty s větší či masivní hemoptýzou hospitalizujeme, a kromě uvedené léčby korigujeme event. koagulační poruchu a arteriální hypertenzi a podáváme vazoaktivní léky (terlipresin). Při neúspěchu je nutná terapeutická embolizace bronchiálních arterií. Chirurgické řešení (resekční výkony nebo podvaz bronchiálních arterií) jsou indikovány výjimečně. Nemocní s hemoptýzou nesmí při RFT užívat oscilující pomůcky (flutter) [25].
Alergická bronchopulmonální aspergilóza
U nemocných se zhoršováním respirační symptomatologie a plicních funkcí bez přítomnosti infekční exacerbace (neúspěch léčby antibiotiky) je nutno uvažovat o ABPA jako možné příčině. Senzibilizaci k Aspergillus fumigatus zjišťujeme pomocí vyšetření kožních testů (prick testy) nebo specifického IgE (senzibilizace však neznamená onemocnění!). Pro ABPA dále svědčí vysoké koncentrace celkového IgE (typicky > 500 IU/ml) a nový nález na skiagramu hrudníku (plicní infiltráty, atelektázy při hlenových zátkách). Základem léčby jsou systémové kortikoidy: prednison v iniciální dávce 0,5–2 mg/kg/den (max. 60 mg/den) po dobu 1–2 týdnů s následnou redukcí dávky; minimální doba léčby činí 2–3 měsíce za monitorování hladin celkového IgE. Recidivy ABPA vyžadují delší dobu léčby, lze zvážit přidání inhalačních kortikosteroidů a antimykotik (itrakonazol, vorikonazol, inhalačně amfotericin B) a bronchoskopické odsávání při hlenových zátkách [8].
Plicní mykobakterióza
K plicním mykobakteriózám se u nemocných s CF přistupuje dle mezinárodních doporučení [9]. V případě opakovaného záchytu v respiračních sekretech a při zhoršování plicních funkcí a/nebo nálezu na HRCT skenech plic bez jiné vysvětlitelné příčiny (zhodnocení trvá běžně i 12–15 měsíců) přistupujeme k antimikrobiální terapii. Užíváme empirické režimy dle uvedeného doporučení, při nedosažení efektu je nutná změna dle aktuálních citlivostí. Léčba je dlouhodobá, doporučuje se pokračovat alespoň 1 rok po dosažení negativizace sput. Chirurgická léčba u CF vzhledem k diseminovanému onemocnění obvykle není možná. Problémem je tolerance léků, připouští se i akceptování chronického onemocnění s intermitentní supresní léčbou (především u Mycobacterium abscessus). Nemocné s CF na přítomnost mykobakterií ve sputu pravidelně vyšetřujeme (nejméně 1krát ročně a vždy před zvažovanou protizánětlivou léčbou azitromycinem). Neřešitelné plicní mykobakteriózy v některých centrech představují kontraindikaci k LuTx.
Chronická rinosinusitida a nosní polypóza
Projevy CF v oblasti horních cest dýchacích léčíme ve spolupráci s otorinolaryngology. K terapii jsou indikováni symptomatiční nemocní, z farmakologických prostředků využíváme nosní kortikosteroidy, jindy je nutný chirurgický výkon (polypektomie, funkční endonazální chirurgie vedlejších dutin nosních/functional endoscopic sinus surgery – FESS). Sanace vedlejších dutin nosních je požadována u kandidátů LuTx.
Malnutrice a deficience vitaminů rozpustných v tucích
Léčba malnutrice je základním postupem u nemocných s CF [26]. Kalorický příjem u CF činí 110–200 % normy, a tak je nezbytná vysokokalorická strava s dostatečným obsahem tuků: 35–45 % kalorického příjmu. Doporučován je příjem potravy 5–6krát denně s „kalorickými bombami“ na noc. Další opatření ke zlepšení stavu výživy zahrnují podávání polymerních přípravků formou popíjení (sipping) nebo sondové výživy (obvykle přes noc nazogastrickou sondou či perkutánní gastrostomií), méně často i nitrožilní výživy (např. při přípravě k LuTx). Kalorický příjem má být upraven tak, aby body mass index (BMI) dosahoval cílových hodnot alespoň 22,0 (u žen), resp. 23,0 kg/m2 (u mužů). Suplementace NaCl je možná pomocí slaných pokrmů či přímo NaCl v želatinových kapslích, poměr koncentrace Na+ a kreatininu v moči se má pohybovat mezi 17–52 mmol/mmol. Vitaminy rozpustné v tucích podáváme v dávce podle sérových hladin, obvyklé dávky představují 10 000–20 000 IU vitaminu A (retinol) a 800–2 000 IU vitaminu D (cholekalciferol). Dávkování vitaminu E (α-tokoferol) se řídí poměrem vitaminu E a cholesterolu v séru (má dosahovat > 4,8 μmol/mmol); obvyklá dávka činí 100–300 mg vitaminu E denně. Vitamin K je doporučeno podávat u opakované antibiotické léčby, hepatopatií a osteoporózy v dávce 1–10 mg denně (fytomenadion). K dispozici jsou i komerční multivitaminové přípravky (např. Vitadek CF Forte ve formě hydrofilních mikrosfér).
Insuficience zevní sekrece pankreatu
Substituce kvalitními preparáty pankreatických enzymů (želatinové tobolky s acidorezistentními enterosolventními mikrotabletami: Kreon a Panzytrat) je nezbytnou podmínkou podávání vysokalorické stravy bohaté na tuky. Insuficienci zevní sekrece pankreatu verifikujeme vyšetřením elastázy 1 ve stolici (patologické hodnoty jsou < 100 μg/g, hraniční pak 100–200 μg/g). Pankreatická substituce by měla snížit ztráty tuků stolicí na < 15 % (norma < 5 %). Dávkování je individuální (cílem jsou maximálně 3 neprůjmové stolice denně) a nemá přesáhnout 10 000 j lipázy/kg tělesné hmotnosti/den, resp. 4 000 j lipázy/g tuku v potravě. Efekt lze zvýšit snížením žaludeční acidity (inhibitory protonové pumpy). Nutno upozornit na skutečnost, že pankreatickou substituci potřebují i nemocní s CF na úplné parenterální výživě (hrozí ileus z nestráveného hlenu ve střevním lumen!), alespoň v dávce 10 000 j lipázy každé 4 hod [26].
Diabetes mellitus
U dospělých nemocných s CF provádíme každoročně vyšetření orálním glukózovým tolerančním testem (oGTT) s cílem časné detekce diabetu. Nicméně, v případě porušené glukózové tolerance (nebo i normálních hodnot) podle výsledku oGTT a zhoršování plicních funkcí a/nebo stavu výživy nejasné geneze doplňujeme intravenózní glukózový toleranční test s cílem zachytit případnou inzulinopenii. V obou případech je pak indikováno zahájení inzulinoterapie. U CF nepoužíváme léčbu perorálními antidiabetiky ani diabetickou dietu, která pro tyto pacienty karenční – její indikace je hrubá chyba! Ve stravě se pouze omezují volné cukry, které je třeba nahradit tuky [11].
Metabolická kostní nemoc
Péče o kostní zdraví představuje další významnou součást léčby dospělých s CF. Vyšetření kostní denzitometrie (axiální skelet) by mělo být provedeno u všech pacientů ve věku 18 let (dříve jen při malnutrici, těžší obstrukci dýchacích cest a dlouhodobé kortikoterapii – nejdříve ve věku 8 let). Dle výsledku zahajujeme léčbu kalciem a bisfosfonáty a motivujeme nemocné k fyzické aktivitě. Pravidelně opakujeme denzitometrii k posouzení efektu (u osteopenie každé 2–4 roky, u osteoporózy každoročně). Léčba vitaminy D a K patří k rutinním postupům. Zachování kostního zdraví je zásadním opatřením u kandidátů LuTx [12].
Gastroezofageální reflux
Vyšetření a léčba gastroezofageálního refluxu se podobně jako u cholelitiázy a pankreatitid u nemocných s CF neliší od běžné populace. Významu nabývá GER u nemocných po LuTx, u nichž je ovlivnitelným rizikovým faktorem chronické dysfunkce štěpu. V těchto případech je kromě konzervativní léčby nutné provedení fundoplikace žaludku.
Syndrom obstrukce distálního střeva
Přístup k nemocným s DIOS se liší podle závažnosti stavu. Lehčí formy s nechutenstvím a pobolíváním v pravé jámě kyčelní lze řešit hydratací, zvýšením podílu vlákniny v potravě, perorálními mukolytiky (N-acetylcystein) a zvýšením dávek pankreatické substituce. Těžší formy pak vyžadují vyloučení ileu (nativní snímek břicha na hladinky) a ultrasonografické vyšetření či výpočetní tomografií k ozřejmění nálezu v pravé jámě kyčelní. Léčebně zasahujeme osmotickými laxativy (makrogoly, např. 4–8 l přípravku Fortrans, rychlostí 1 l/hod). Rozvinutý ileus je nutno řešit operačně, obvykle se provádí resekce terminálního ilea a pravostranná hemikolektomie [14].
Atypicky probíhající apendicitidy a klostridiové kolitidy
Apendicitida je u nemocných s CF méně častá než v běžné populaci a její průběh bývá atypický. V tomto případě není tendence k rozvoji difuzní peritonitidy, ale k ohraničení procesu (periapendikulární absces) s málo výraznými příznaky (subfebrilie, pobolívání v pravé jámě kyčelní). To vede často k opožděné diagnóze. Léčba je chirurgická. Klostridiová kolitida je naopak vzhledem k opakovaným antibiotickým kúrám častější, ale opět nemívá typický obraz. Vzhledem k nefunkčnímu CFTR proteinu nemusí být přítomny výrazné průjmy, pouze teploty a bolesti břicha. Nutné je opakované vyšetření stolice na toxin Clostridium difficile a při pozitivním nálezu obvyklá léčba účinnými antibiotiky (metronidazol, vankomycin).
Hepatopatie
Biliární fibróza až cirhóza jater se rozvíjí typicky v dětském věku a v dospělosti se pak setkáváme s projevy portální hypertenze, hypersplenizmu a selhání jater, které se léčí obvyklým způsobem včetně indikace transplantace jater. Z farmakologických prostředků je k dispozici kyselina ursodeoxycholová (UDCA), kterou podáváme v množství 20 mg/kg/den ve 2 dílčích dávkách. Spolu UDCA je nemocným ve špatném nutričním stavu doporučováno podávání taurinu (dostupný jako potravinový doplněk) v dávce 30–40 mg/kg/den [15].
Obstruktivní azoospermie
Informace o předpokládané neplodnosti má být pacientům s CF podána již v adolescenci (ve věku 13–14 let), k potvrzení se vyšetří spermiogram. V případě plánování rodičovství se využívají metody asistované reprodukce, nutné je genetické vyšetření partnerky na nosičství mutací genu CFTR. Odběr spermií se provádí z varlete nebo nadvarlete po předchozím ultrasonografickém vyšetření. Následně se získané spermie využijí k in vitro fertilizaci a embryotransferu.
Péče o těhotné a kojící ženy
Ženy s CF mohou otěhotnět spontánně nebo s využitím technik asistované reprodukce, opět je nutné vyšetření partnera na přítomnost mutací genu CFTR. Absolutní kontraindikací ke graviditě je plicní hypertenze, hyperkapnie a klidová hypoxemie. Mezi relativní kontraindikace patří časté infekční exacerbace, hodnota FEV1 < 50 % náležitých hodnot, infekce komplexem Burkholderia cepacia, malnutrice a diabetes mellitus. Ženy s FEV1 < 40 % náležitých hodnot mají 40% riziko úmrtí do 10 let věku dítěte. V graviditě i laktaci je nutné navýšení kalorického příjmu o 20 % (při váhovém přírůstku < 4,5 kg jsou častější nepříznivé výsledky gravidity) a je třeba ženy kontrolovat pomocí oGTT. Dále je nutné vyhnout se některým lékům, z antibiotik tetracyklinům, chinolonům, kotrimoxazolu a chloramfenikolu, u vitaminů pak vyšším dávkám vitaminu A [18].
Hygienicko-epidemiologický režim
Ve zdravotnických zařízeních i mimo ně je pro nemocné s CF zcela zásadní dodržovat pravidla hygienicko-epidemiologického režimu k zabránění přenosu infekce. Nemocní se nesmí navzájem stýkat, resp. musí od sebe dodržovat vzdálenost alespoň 1 m. Na ambulancích i lůžkových odděleních jsou od sebe separováni nemocní s odlišnou infekcí dýchacích cest (minimálně 3 skupiny: s komplexem Burkholderia cepacia, s Pseudomonas aeruginosa a nekolonizovaní). Zcela izolováni jsou pak nemocní s epidemickými kmeny (v České republice epidemický kmen ST32 Burkholderia cenocepacia) a nemocní s MRSA a s Mycobacterium abscessus. K detekci epidemických kmenů jsou využívány PCR (polymerázová řetězová reakce) metody (např. metoda MLST – multilocus sequence typing). Proti infekci z prostředí je třeba provádět dezinfekci (mytí rukou antibakteriálními mýdly, dezinfekce odpadů umyvadel a van, sterilizace inhalačních pomůcek apod). V případě prvního záchytu patogenů v respiračních sekretech je indikována eradikační léčba antibiotiky, především u Pseudomonas aeruginosa (inhalační tobramycin nebo kombinace inhalačního kolistinu s perorálním ciprofloxacinem, u symptomatických nemocných dvojkombinací nitrožilních antibiotik). Preventivní léčbu k zabránění infekci Staphylococcus aureus u dospělých nemocných s CF neprovádíme [27].
Psychosociální problematika
Nezanedbatelnou částí péče o dospělé s CF a jejich rodinu je řešení psychosociálních problémů. Těch může nastat celá řada: předávání z dětské části CF centra na dospělou, nová diagnóza CF v dospělosti, zhoršování stavu a příprava na LuTx, terminální stavy, ukončení studií a hledání vhodného zaměstnání, nebo naopak odchod do invalidního důchodu, partnerské problémy apod. Pomoc psychologa či psychiatra s medikací anxiolytiky a antidepresivy je často nezbytná [28].
Kauzální léky
Budoucnost léčby CF spočívá ve vývoji kauzálních léků. Genová léčba však dosud není a nebude v následujících 5, spíše však 10 letech dostupná. Proto jsou vyvíjeny léky specificky cílené na jednotlivé mutace genu CFTR. Od roku 2012 je ve světě (v ČR v současnosti jen omezeně) dostupný první kauzální lék na CF, a to ivakaftor (Kalydeco; dávka 2krát 150 mg), potenciátor CFTR proteinu u mutace G551D a dalších mutací III. a IV. třídy. Jeho zavedení do praxe je událostí srovnatelnou s objevem genu CFTR. V roce 2015 byl registrován další kauzální lék, a to kombinace ivakaftoru s lumakaftorem (Orkambi; dávka 2krát 400/250 mg; lumakaftor patří mezi korektory CFTR proteinu) pro homozygoty F508del. Obecně tuto skupinu léků označujeme jako modulátory CFTR proteinu, přičemž korektory zvyšují množství CFTR proteinu v buněčné membráně, kdežto potenciátory zlepšují jeho funkci [29].
Na mutaci F508del lze současně demonstrovat složitost problematiky léčby modulátory CFTR proteinu. Tato mutace je řazena do II. třídy. Pokud však po „záchraně“ pomocí korektoru (lumakaftor) dosáhne CFTR protein buněčné membrány, nabývá vlastností mutací třídy III a vyžaduje podání potenciátoru (ivakaftor). Současně se zrychluje obrat „zachráněného“ F508del-CFTR proteinu v buněčné membráně, který tím nabývá vlastnosti mutací třídy VI. Logickým krokem je zde vývoj léků schopných prodloužit dobu, po kterou je CFTR protein v buněčné membráně aktivní („stabilizátory“ CFTR proteinu).
Organizace péče o dospělé s CF
Péče o tyto nemocné je poskytována v centrech pro diagnostiku a léčbu CF. V České republice pracují tato centra při výše uvedených fakultních nemocnicích. Centrovou péči zajišťuje multidisciplinární tým. Doporučené složení multidisciplinárního týmu včetně pracovních úvazků uvádějí mezinárodní guidelines. Např. pro 100 dospělých s CF se předpokládá účast 4 lékařů (úvazky celkem 1,7), 2 specializovaných zdravotních sester a 2 fyzioterapeutů (úvazky po 2,0), nutričního terapeuta, psychologa, sociálního pracovníka, farmaceuta a sekretářky (úvazky po 0,5) a koordinátora databáze (úvazek 0,4). Nutná je návaznost na další odbornosti zajišťující péči o celé spektrum projevů CF [30]
Závěr
Prognóza nemocných s CF se za posledních 50 let dramaticky změnila a další zlepšení lze očekávat od zavádění kauzálních léků (a v budoucnu snad i genové léčby) do praxe. Tato nemoc již není doménou pediatrů a také v České republice se blíží doba, v níž bude více dospělých pacientů než pediatrických. Proto je nutné, aby se s tímto onemocněním seznámili i lékaři pečující o dospělé nemocné. Vzhledem k širokému spektru klinickým projevů stručně zmíněných v tomto článku je nezbytná široká mezioborová spolupráce koordinovaná pneumologem. Nutná je znalost hygienicko-epidemiologických opatření k zabránění přenosu infekce, nezbytnosti dechové rehabilitace (např. po operacích v oblasti hrudníku a břicha je důležitá kvalitní analgezie a přizvání respiračního fyzioterapeuta k nemocnému). Podobně je nezbytná znalost zásad antibiotické léčby (vyšší dávky po delší dobu), povědomí o nutnosti vysokokalorické stravy s adekvátní pankreatickou substitucí (včetně jejího podávání i u nemocných na plné parenterální výživě k prevenci ileu), znalost zásad léčby diabetu (neomezovat kalorický příjem a podávat pouze inzulin), povědomí o atypických průbězích apendicitid a klostridiových kolitid apod.
Především je však důležité rovněž u dospělých při diagnostické rozvaze myslet na možnost přítomnosti dosud nerozpoznané CF, a to dle zásad uvedených výše.
MUDr. Libor Fila, Ph.D.
libor.fila@fnmotol.cz
Pneumologická klinika 2. LF UK a
FN Motol,
Praha
www.fnmotol.cz
Doručeno do redakce 21. 8. 2017
Přijato po recenzi 26. 9. 2017
Zdroje
1. Cystic Fibrosis Foundation Patient Registry. 2015 Annual Data Report. Bethesda, Maryland. ©2016 Cystic Fibrosis Foundation. Dostupné z WWW:
2. Fila L, Sedlák V, Binková I et al. Dvacet let péče o dospělé nemocné s cystickou fibrózou v České republice. Vnitř Lék 2009; 55(6): 542–548.
3. Boyle MP, De Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med 2013; 1(2): 158–163. Dostupné z DOI: <http://dx.doi.org/10.1016/S2213–2600(12)70057–7>.
4. Křenková P, Piskáčková T, Holubová et al. Distribution of CFTR mutations in the Czech population: Positive impact of integrated clinical and laboratory expertise, detection of novel/de novo alleles and relevance for related/derived populations. J Cyst Fibros 2013; 12(5): 532–537. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jcf.2012.12.002>.
5. Boucher RC. New concepts of the pathogenesis of cystic fibrosis lung disease. Eur Respir J 2004; 23(1): 146–158.
6. Fila L. Cystická fibróza. In: Kolek V, Kašák V, Vašáková M et al. Pneumologie. 2. ed. Maxdorf: Praha 2014: 416–420. Dostupné z DOI: <http://dx.doi.org/978–80–7345–387–9>.
7. Yankaskas JR, Marshall BC, Sufian B et al. Cystic fibrosis adult care: consensus conference report. Chest 2004; 125(1 Suppl): S1-S39.
8. Stevens DA, Moss RB, Kurup VP et al. Participants in the Cystic Fibrosis Foundation Consensus Conference. Allergic bronchopulmonary aspergillosis in cystic fibrosis – state of the art: Cystic Fibrosis Foundation Consensus Conference. Clin Infect Dis 2003; 37(Suppl 3): S225-S264.
9. Floto RA, Olivier KN, Saiman L et al. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax 2016; 71(Suppl 1): i1-i22. Dostupné z DOI: <http://dx.doi.org/10.1136/thoraxjnl-2015–207360>.
10. Walkowiak J, Lisowska A, Blaszczyński M. The changing face of the exocrine pancreas in cystic fibrosis: pancreatic sufficiency, pancreatitis and genotype. Eur J Gastroenterol Hepatol 2008; 20(3): 157–160. Dostupné z DOI: <http://dx.doi.org/10.1097/MEG.0b013e3282f36d16>.
11. Kelly A, Moran A. Update on cystic fibrosis-related diabetes. J Cyst Fibros 2013; 12(4): 318–331. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jcf.2013.02.008>.
12. Aris RM, Merkel PA, Bachrach LK et al. Guide to bone health and disease in cystic fibrosis. J Clin Endocrinol Metab 2005; 90(3): 1888–1896.
13. Mousa HM, Woodley FW. Gastroesophageal reflux in cystic fibrosis: current understandings of mechanisms and management. Curr Gastroenterol Rep 2012; 14(3): 226–235. Dostupné z DOI: <http://dx.doi.org/10.1007/s11894–012–0261–9>.
14. Colombo C, Ellemunter H, Houwen R et al. ECFS. Guidelines for the diagnosis and management of distal intestinal obstruction syndrome in cystic fibrosis patients. J Cyst Fibros 2011; 10(Suppl 2): S24-S28. Dostupné z DOI: <http://dx.doi.org/10.1016/S1569–1993(11)60005–2>.
15. Debray D, Kelly D, Houwen R et al. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. J Cyst Fibros 2011; 10(Suppl 2): S29-S36. Dostupné z DOI: <http://dx.doi.org/10.1016/S1569–1993(11)60006–4>.
16. Davé S, Honney S, Raymond J et al. An unusual presentation of cystic fibrosis in an adult. Am J Kidney Dis 2005; 45(3): e41-e44. Dostupné z DOI: <http://dx.doi.org/10.1053/j.ajkd.2004.11.009>.
17. Wong PY. CFTR gene and male fertility. Mol Hum Reprod 1998; 4(2): 107–110.
18. Edenborough FP, Borgo G, Knoop C et al. European Cystic Fibrosis Society. Guidelines for the management of pregnancy in women with cystic fibrosis. J Cyst Fibros 2008; 7(Suppl 1): S2-S32.
19. Farrell PM, White TB, Ren CL et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J Pediatr 2017; 181(Suppl): S4-S15.e1. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jpeds.2016.09.064>.
20. Farrell PM, White TB, Derichs N et al. Cystic Fibrosis Diagnostic Challenges over 4 Decades: Historical Perspectives and Lessons Learned. J Pediatr 2017; 181(Suppl): S16-S26. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jpeds.2016.09.067>.
21. De Boeck K, Derichs N, Fajac I et al. [ECFS Diagnostic Network Working Group. EuroCareCF WP3 Group on CF diagnosis]. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros 2011; 10(Suppl 2): S53-S66. Dostupné z DOI: <http://dx.doi.org/10.1016/S1569–1993(11)60009-X>.
22. Fila L, Grandcourtová A, Valentová Bartáková L et al. Diagnostika cystické fibrózy u dospělých. Vnitř Lék 2016; 62(5): 360–364.
23. Mogayzel PJ Jr, Naureckas ET, Robinson KA et al. Pulmonary Clinical Practice Guidelines Committee. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2013; 187(7): 680–689.
24. Bhatt JM. Treatment of pulmonary exacerbations in cystic fibrosis. Eur Respir Rev 2013; 22(129): 205–216. Dostupné z DOI: <http://dx.doi.org/10.1183/09059180.00006512>.
25. Flume PA, Mogayzel PJ Jr, Robinson KA et al. [Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic Fibrosis Foundation Pulmonary Therapies Committee]. Cystic fibrosis pulmonary guidelines: pulmonary complications: hemoptysis and pneumothorax. Am J Respir Crit Care Med 2010; 182(3): 298–306. Dostupné z DOI: <http://dx.doi.org/10.1164/rccm.201002–0157CI>.
26. Turck D, Braegger CP, Colombo C et al. ESPEN-ESPGHAN-ECFS guidelines on nutrition care for infants, children, and adults with cystic fibrosis. Clin Nutr 2016; 35(3): 557–577. Dostupné z DOI: <http://dx.doi.org/10.1016/j.clnu.2016.03.004>.
27. Saiman L, Siegel JD, LiPuma JJ et al. Infection prevention and control guideline for cystic fibrosis: 2013 update. Infect Control Hosp Epidemiol 2014; 35(Suppl 1): S1-S67. Dostupné z DOI: <http://dx.doi.org/10.1086/676882>.
28. Tuchman LK, Schwartz LA, Sawicki GS et al. Cystic fibrosis and transition to adult medical care. Pediatrics 2010; 125(3): 566–573. Dostupné z DOI: <http://dx.doi.org/10.1542/peds.2009–2791>.
29. Ramsey BW, Davies J, McElvaney NG et al. VX08–770–102 Study Group. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011; 365(18): 1663–1672. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJMoa1105185>.
30. Conway S, Balfour-Lynn IM, De Rijcke K et al. European Cystic Fibrosis Society Standards of Care: Framework for the Cystic Fibrosis Centre. Cyst Fibros 2014; 13(Suppl 1): S3-S22. Dostupné z DOI: <http://dx.doi.org/10.1016/j.jcf.2014.03.009>.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2017 Číslo 11
- MINISERIÁL: Když ženám stoupá tlak...
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- Specifika v komunikaci s pacienty s ránou – laická doporučení
- Deficience vitaminu B12 navozená metforminem − aktuální poznatky a tipy pro klinickou praxi
Nejčtenější v tomto čísle
- Spirometrie – základní vyšetření funkce plic
- Neinvazivní ventilace
- Malobuněčný karcinom plic: epidemiologie, diagnostika a léčba
- Pneumonie u imunokompromitovaných