
The Coevolution of Virulence: Tolerance in Perspective
Coevolutionary interactions, such as those between host and parasite, predator and prey, or plant and pollinator, evolve subject to the genes of both interactors. It is clear, for example, that the evolution of pollination strategies can only be understood with knowledge of both the pollinator and the pollinated. Studies of the evolution of virulence, the reduction in host fitness due to infection, have nonetheless tended to focus on parasite evolution. Host-centric approaches have also been proposed—for example, under the rubric of “tolerance”, the ability of hosts to minimize virulence without necessarily minimizing parasite density. Within the tolerance framework, however, there is room for more comprehensive measures of host fitness traits, and for fuller consideration of the consequences of coevolution. For example, the evolution of tolerance can result in changed selection on parasite populations, which should provoke parasite evolution despite the fact that tolerance is not directly antagonistic to parasite fitness. As a result, consideration of the potential for parasite counter-adaptation to host tolerance—whether evolved or medially manipulated—is essential to the emergence of a cohesive theory of biotic partnerships and robust disease control strategies.
Published in the journal:
. PLoS Pathog 6(9): e32767. doi:10.1371/journal.ppat.1001006
Category:
Review
doi:
https://doi.org/10.1371/journal.ppat.1001006
Summary
Coevolutionary interactions, such as those between host and parasite, predator and prey, or plant and pollinator, evolve subject to the genes of both interactors. It is clear, for example, that the evolution of pollination strategies can only be understood with knowledge of both the pollinator and the pollinated. Studies of the evolution of virulence, the reduction in host fitness due to infection, have nonetheless tended to focus on parasite evolution. Host-centric approaches have also been proposed—for example, under the rubric of “tolerance”, the ability of hosts to minimize virulence without necessarily minimizing parasite density. Within the tolerance framework, however, there is room for more comprehensive measures of host fitness traits, and for fuller consideration of the consequences of coevolution. For example, the evolution of tolerance can result in changed selection on parasite populations, which should provoke parasite evolution despite the fact that tolerance is not directly antagonistic to parasite fitness. As a result, consideration of the potential for parasite counter-adaptation to host tolerance—whether evolved or medially manipulated—is essential to the emergence of a cohesive theory of biotic partnerships and robust disease control strategies.
Introduction: What Controls Virulence?
Evolutionary biologists define the virulence of a parasite as the reduction in host fitness caused by infection. When this reduction in host fitness is due to an increased mortality rate, the consequences for parasite evolution are clear; host death means parasite death [1]. However, virulence can also include reductions in host fecundity [2], [3], which is relevant for some parasites such as castrating parasites and obligate killers [4], [5]. In either case, typically, virulence is reasoned to increase with an increasing parasite burden. Thus, conceptually, virulence may be viewed as resulting from the density of parasites within a host (I) and the degree of damage caused by each parasite (α, the per-parasite virulence):(1)This simple equation offers considerable insight into parasite evolution. For instance, it is generally assumed that parasites can achieve higher transmission success the more numerous they are within a host (higher within-host density), but that this will in turn lead to higher virulence, which may kill hosts more rapidly and compromise transmission success [6]. The resulting idea is that the most successful parasites cause an intermediate level of virulence, which encapsulates the trade-off model of virulence evolution (reviewed in [7]).
However, virulence is ultimately a pathology of the host [8], and thus will be jointly determined by both the parasite and host. The density of parasites within hosts, I, is controlled both by intrinsic replication rates of parasites and by the rate at which hosts kill parasites (e.g., [9]), while per-parasite virulence, α, can be controlled by parasite “virulence factors” such as toxins as well as host anti-toxin molecules. Some theoretical work has acknowledged this joint control of virulence—for example, by examining how host control of I via recovery rate affects coevolution [10], [11], or how host control of α via avoidance of immunopathology can shape parasite evolution [12]. Such studies, however, are in the minority, and the virulence literature has focussed primarily on parasite-controlled traits, despite the fact that virulence is of clear relevance to host evolution.
When thinking about host evolution, it can be helpful to discuss host fitness directly, rather than virulence. A simple linear model of host fitness is:(2)which is host fitness in the absence of infection (ωo,n, the y-intercept) minus the terms that comprise virulence. Here n is the nth host genotype, and j is the jth parasite genotype. Many approaches to the study of host–parasite interactions have been less general than this. For example, much of the work on virulence has assumed constant α, or that I depends only on parasite genotype (e.g., [7]); work on host genetic control of resistance has often assumed that α is constant, but allowed I to depend on both parasite and host genotype (e.g., [13]); finally, work on the parameter αn (often referred to as tolerance) tends to assume that it is a function only of host genotype (or that ωo,n is constant [e.g., [14]]). While we appreciate that not every study needs to be holistic or coevolutionary, much might also be learned from a synthetic approach (see also [10], [15]).
In the following sections we emphasise why a full picture of the causes and consequences of virulence requires consideration of all components of Equation 2. We draw attention to the term ωo,n, which is not directly influenced by the parasite, and our discussion is weighted towards host genetic variation for tolerance (i.e., αn), both in terms of measurement and the evolutionary inferences that are possible. Despite these leanings, we aim to reinforce the notion that coevolutionary outcomes depend upon contributions from both interactors. Host-centric and parasite-centric views of virulence evolution have often been disconnected in the study of disease evolution, which has provided too narrow a perspective on the potential for interactions to generate dynamic coevolution. For example, the evolution of tolerance (i.e., selection on αn) has been thought of as having the potential to dampen coevolutionary dynamics, but this has rarely been with a full consideration of all the conditions that favour intense host or parasite counter-adaptation. Ultimately, host–parasite coevolution is about two interacting organisms gaining fitness at each other's expense, and a more holistic approach could unite a large range of perspectives on the evolutionary ecology of attack, defence, and commensalism.
Tolerance and Intercepts
A recent experimental study of virulence in a rodent defined tolerance as the slope of the regression of host fitness on I [14]. This definition is similar to that from studies on plant responses to infection or herbivory (e.g., [16], [17]) and assumes that host differences in ωo,n represent underlying differences in general vigour [14], [16], [17]. In addition to being measured as a slope, i.e., estimated across a range of parasite densities (range tolerance), tolerance has also been measured at one single parasite density (point tolerance) [18], [19], but depending on the relationship between αj,n and ωo,n, the conclusions drawn from the different measures may not be the same (Box 1, Figure 1). There are yet further definitions of tolerance, and the possibility that different measures of tolerance will not always provide the same information presents challenges for reconciling theory with data and vice versa. A key step towards suitable comparison of the different tolerance measures will be to always measure host fitness when infected alongside fitness when uninfected, ωo,n (Equation 2), although even this may be of limited value if per-parasite virulence (α) varies non-linearly with parasite density (I) (parasites get proportionally more or less benign with increasing density; Box 1, Figure 1C).
Box 1. Density Ranges and Definitions of Tolerance
The study of host-controlled α has been described as the study of tolerance: the ability of hosts to limit the damage caused by a given parasite burden, which is essentially the ability to minimize per-parasite virulence. It has been studied as a mean, i.e., where two genotypes carry the same parasite burden, but one genotype achieves higher fitness. We call this point tolerance. An empirical example of point tolerance is the striking genetic variation among strains of laboratory mice in the per-parasite virulence of Streptococcus pneumoniae infection [18]. Tolerance has also been studied as a slope to depict how quickly fitness falls as parasite density increases; more tolerant genotypes lose their fitness less quickly as densities increase (implying less sensitivity to changes in parasite numbers). We call this range tolerance. This has recently demonstrated for rodent malaria [14], using an approach in line with studies of tolerance to herbivory [16], [17].
Schematic examples of range and point tolerance are presented in Figure 1. The differences in interpretation implicit in these scenarios are important, as they would lead to different predicted evolutionary or epidemiological outcomes. These examples are meant to merely illustrate the potential confusion that may arise depending on how tolerance is measured, and we emphasise that this is not just a quibble about definitions. If we are to draw general conclusions about tolerance evolution, we need to resolve when empirical studies of point tolerance (e.g., [19]) should be freely compared with studies of range tolerance (e.g., [14]), and when either can inform theory that uses yet other definitions (e.g., [33]; see also [21]).
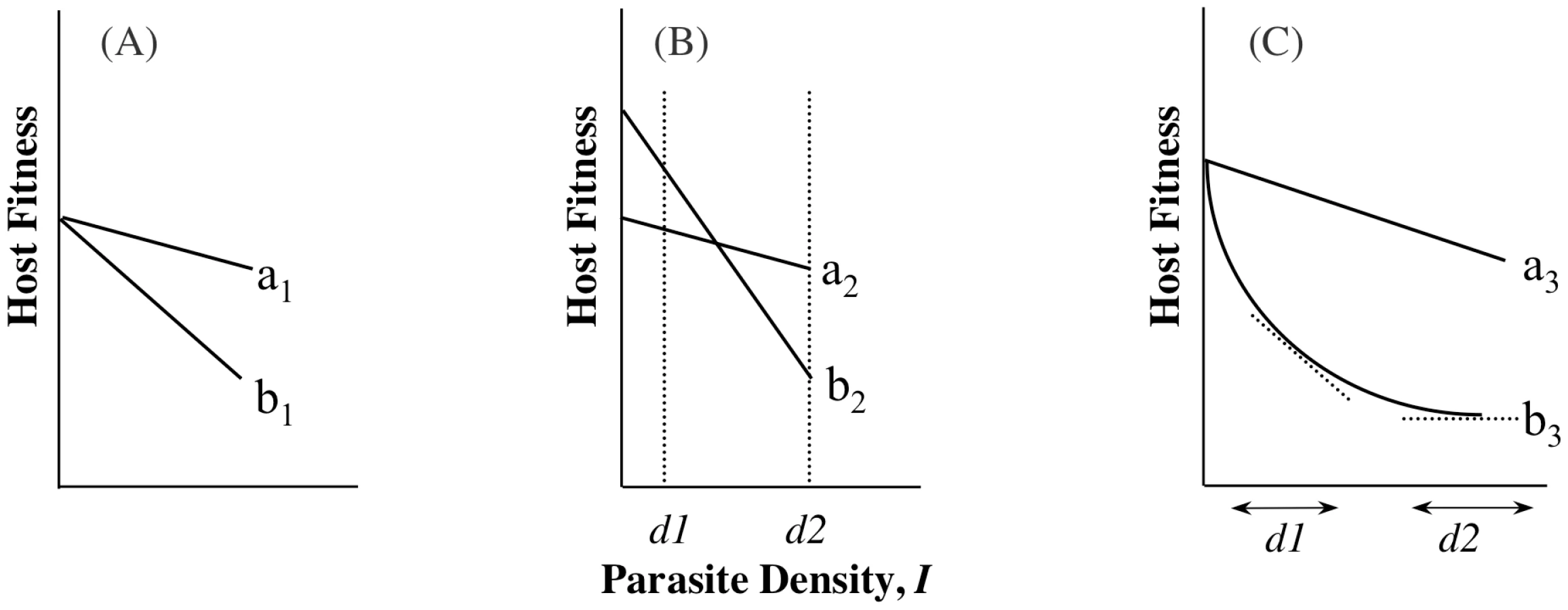
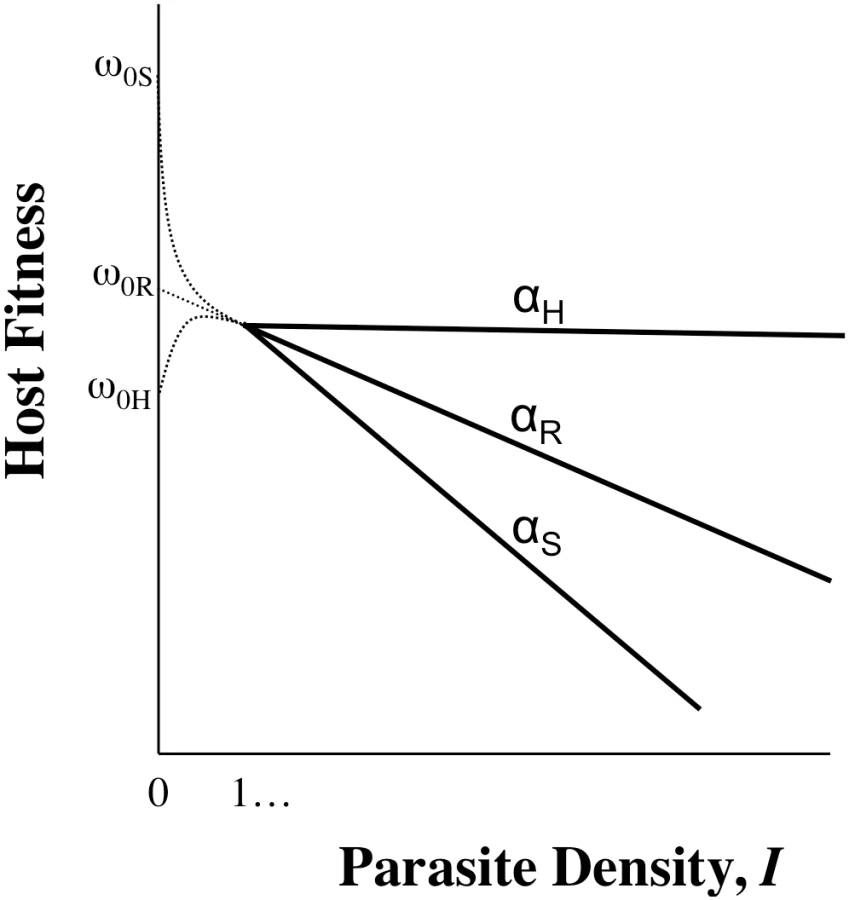
Tolerance, by definition, does not include ωo, but inference about host evolution will be restricted when ωo is not considered. First, there may be a biological relationship between tolerance and ωo due to the effects of pleiotropy, in particular if defence is traded-off with other fitness traits (Figure 2). In this case, we could not disregard variation in ωo,n as variation in general vigour that is independent of parasite-mediated selection. Moreover, variation in ωo,n, whether or not pleiotropy plays a role, will be crucial for determining evolutionary trajectories. Consider two host genotypes that differ for α but have equivalent ωo, so that one genotype has higher fitness across all values of I (compare genotypes in Figure 1A or 1C). In this case, the evolutionary outcome for hosts is certain, and α only determines the rate at which one host genotype replaces another. The more plausible and interesting scenario is where host genotypes differ for both α and ωo, but no genotype is the fittest across all parasite densities (Figure 1B). Relationships such as these can lead to the maintenance of host polymorphism, because the superiority of a genotype is entirely context dependent. Selection on tolerance traits can also maintain polymorphism when hosts recoup fitness in terms of fecundity instead of mortality [2]. In general, accurate estimates of evolutionary outcomes require that the slope of the parasitic relationship α (per-parasite virulence, or tolerance) be considered alongside the intercept ωo.
The Problem of Intimacy
As with any symbiosis, when measuring genetic variation and inferring selection on disease-related traits, it is impossible to escape the issue that infection phenotypes represent the dual contribution of host and parasite. For example, when parasite burdens are measured in the field or as an experimental outcome (as opposed to an experimentally controlled variable), how can we know if parasite burden determines host fitness, or if host fitness, itself affected by a variety of environmental factors, is mediating parasite burden? Stressed hosts, for example, may express sensitivities that lead to differences in parasite load. Here, we would be examining genetic variation in laboratory-mediated stress; it would not be clear what defence trait would be selected upon and what evolutionary response we should expect to see. In this sense, the parasitic relationship is more comparable to correlated traits than to a norm of reaction.
The difficulty of using correlations to elucidate the trait under selection is evident when trying to envision the selection process that has shaped αn and ωo,n in a population. For example, host genotypes might differ in the fat reserves that are mobilized only in periods of stress (e.g., food shortage, temperature stress, and, of course, infection). If the effect of differences in fat reserves happen to scale with stress level and stress escalates with parasite density, then hosts would differ in range tolerance. Whilst this would be tolerance of infection in a very broad sense, it might have evolved for reasons independent of infection. Conversely, differences in general vigour might arise via parasite-mediated selection. For example, a molecule that mops up pathogen-produced toxins without depressing pathogen numbers (clearly a tolerance mechanism that could affect the slope, α), might later be recruited to mop up free radicals produced during respiration in the absence of infection (raising ωo). These simple hypothetical examples reinforce the problem of separating slopes from intercepts when inferring selection: variation in ωo,n can appear as just variation in general vigour, and not subject to parasite-mediated selection (when in fact a process of parasite-mediated selection has modified ωo), whilst slopes may appear to be subject to parasite-mediated selection (when in fact slope differences arose through selection on ωo).
It is thus difficult to infer an underlying mechanism from correlational estimates of tolerance, and it is likely to be important to delve into mechanistic studies (e.g., [19]). For many, the terms “resistance” and “tolerance” carry with them some connotation of mechanism (e.g., [20], [21]), and, just as different mechanisms of resistance (e.g., when a lack of infection comes about via behavioural avoidance versus an immune reaction [22]) should predict very different trajectories of selection, different mechanisms of tolerance also predict different evolutionary trajectories. Knowing the mechanisms that confer tolerance or resistance will shed light on host evolution, and ultimately coevolution. Generally, it will be interesting to understand why the relationship between I and host fitness might become steeper or change shape: is it due to an immune system molecule that blocks toxins, for example, or prevents immunopathology [23], but only over certain parasite density ranges? Or is it simply that the environment and subsequent condition of the host mediate a change in the severity of parasitism? Indeed, it is easy to envision an additional factor added to Equation 2: the subscript e denoting the effects of the environment on the parasitic relationship. The effects of factors such as host density, resource availability, and temperature are obvious avenues of study here [24]–[26].
This problem of intimacy will often extend to the study of genetic variation: Ij,n, and αj,n are the product of two interacting genomes (Equation 2), and it is often difficult to identify which antagonist is controlling the infection phenotype due to genotype-by-genotype interactions. For example, the defence capabilities of a host genotype often depend on which parasite genotype is involved, while at the same time the impact of a particular parasite genotype depends on the host genotype it infects [9], [15], [27]–[30]. Nonetheless, previous definitions of tolerance have implicitly assumed that the sensitivity of host fitness to parasite burden (i.e., α) is entirely determined by host genotype [14]. But this sensitivity could plausibly depend on an interaction between the host and parasite genotypes as well, further calling into question simplified measures of tolerance.
Coevolution
Far from just presenting challenges for the study of parasitism, genotype-by-genotype interactions actually represent the foundation of most host–parasite coevolutionary models [9], [27], [28]. Usually this is studied in the context of resistance to infection, where the relative performance of different parasite genotypes (in terms of Ij,n) depends on which host genotype they infect. If Ij,n and thereby virulence are determined by a host–parasite genetic interaction, then this can lead to antagonistic coevolutionary dynamics and provide a mechanism for the maintenance of genetic polymorphism (specifically, negative frequency-dependent selection [e.g., [31]]). Above and in Figure 1, we highlighted how some scenarios of variation in α and ωo should also promote host polymorphism (see also [2]), and this can be extended to a full treatment of host-parasite coevolution. Here, coevolutionary dynamics will be driven by the rules of virulence optimisation. Selection for tolerance will change the selection gradient on parasites, and present them with a new optimum (or adaptive peak), towards which they will evolve. It has been suggested [14] that the evolution of tolerance dampens coevolution by not directly reducing parasite numbers, but we posit that any host evolution that knocks parasites away from their evolutionary optimum (or creates a new one) will be countered by parasite evolution.
We do not yet have a clear view on the nature of the dynamics generated by parasite evolution in response to new adaptive peaks that arise due to tolerance evolution. The tolerance theory thus far largely omits host (e.g., [32]) or parasite (e.g., [33]) counter-adaptations. Where coevolution has been permitted in optimality models, however, it has been clearly shown that parasite evolution is highly sensitive to host evolution [34]. Further investigation of host–parasite coevolution that allows host genotypes to exhibit heterogeneity in tolerance is needed, for both theoretical and practical reasons.
For instance, we do know that one step of host evolution towards greater tolerance can select for parasites with higher replication rates and populations that suffer a greater parasite burden [32]. Thus, one plausible coevolutionary scenario is that hosts that evolve ever greater tolerance can select for the evolution of parasites with higher growth rates and increased transmission [32] because the cost of virulence, in terms of killing the host prior to transmission, will be reduced. In this way, the evolution of tolerance mirrors attempts made by medical interventions (particularly vaccination) to ameliorate the pathology associated with infection, without necessarily eradicating the infection or reducing parasite densities. Such vaccination strategies can select for more virulent pathogens, presenting grave risks for those who come into contact with the more virulent parasite but are not vaccinated [35], [36]. The same is true for the spread of tolerance through populations: tolerant individuals may allow parasites to evolve greater virulence, likewise causing grave risks for intolerant or migrant individuals that become exposed to the disease [32]. Such an outcome could be important for human disease risk.
Thus, although local tolerance evolution can even lead to commensalisms [32], [33], when it is coupled with geographic structuring of populations or infrequent contact between species, tolerance evolution in one population might underlie why some zoonoses or other emerging diseases are particularly devastating to other populations. Indeed, apparent paradoxes such as “tolerance evolution might be bad” represent key lessons from viewing disease in a coevolutionary context. The risk that tolerance evolution could increase disease severity in intolerant hosts may be just scratching the surface. Equation 2 suggests a set of critical components that can be compared across host and parasite genotypes (or environments) to gain insight into host evolution, parasite evolution, or coevolution. We encourage use of such a unified, coevolutionary framework, rather than host-centric or parasite-centric alternatives, to achieve a true understanding of tolerance and to shed light on disease control strategies that will not provoke undesirable pathogen evolution. Ultimately, unification is essential if we are ever to achieve a universal theory of disease severity, or indeed a universal theory of biotic partnerships.
Zdroje
1. DayT
2002 On the evolution of virulence and the relationship between various measures of mortality. Proc R Soc Lond B Biol Sci 269 1317 1323
2. BestA
WhiteA
BootsM
2008 Maintenance of host variation in tolerance to pathogens and parasites. Proc Natl Acad Sci U S A105 20786 20791
3. JensenKN
LittleTJ
SkorpingA
EbertD
2006 Empirical support for an optimal virulence in a castrating parasite. PLoS Biol 4 e197 doi:10.1371/journal.pbio.0040197
4. EbertD
CariusH-J
LittleTJ
DecaesteckerE
2004 The evolution of virulence when parasites cause host castration and gigantism. Am Nat 164 s19 s32
5. EbertD
WeisserWW
1997 Optimal killing for obligate killers: the evolution of life histories and virulence of semelparous parasites. Proc R Soc Lond B Biol Sci 264 985 991
6. FrankSA
1996 Models of parasite virulence. Q Rev Biol 71 37 78
7. AlizonS
HurfordA
MideoN
Van BaalenM
2009 Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. J Evol Biol 22 245 259
8. EbertD
1998 Experimental evolution of parasites. Science 282 1432 1435
9. GrechK
WattK
ReadAF
2006 Host-parasite interactions for virulence and resistance in a malaria model system. J Evol Biol 19 1620 1630
10. RestifO
KoellaJC
2003 Shared control of epidemiological traits in a coevolutionary model of host-parasite interactions. Am Nat 161 827 836
11. van BaalenM
1998 Coevolution of recovery ability and virulence. Proc R Soc Lond B Biol Sci 265 317 325
12. DayT
GrahamAL
ReadAF
2007 Evolution of parasite virulence when host responses cause disease. Proc R Soc Lond B Biol Sci 274 2685 2692
13. FrankSA
1993 Evolution of host-parasite diversity. Evolution 47 1721 1732
14. RåbergL
SimD
ReadAF
2007 Disentangling genetic variation for resistance and tolerance to infectious diseases in animals. Science 318 812 814
15. de RoodeJC
AltizerS
2010 Host-parasite genetic interactions and virulence transmission relationships in natural populations of monarch butterflies. Evolution 64 502 514
16. SimmsEL
2000 Defining tolerance as a norm of reaction. Evol Ecol 14 563 570
17. TiffinP
RausherM
1999 Genetic constraints and selection acting on tolerance to herbivory in the common morning glory Ipomoea purpurea. Am Nat 154 700 716
18. GinglesNA
AlexanderJE
KadiogluA
AndrewPW
KerrA
2001 Role of genetic resistance in invasive pneumococcal infection: Identification and study of susceptibility and resistance in inbred mouse strains. Infect Immun 69 426 434
19. AyresJ
SchneiderD
2008 A signaling protease required for melanization in Drosophila affects resistance and tolerance of infections. PLoS Biol 6 e305 doi:10.1371/journal.pbio.0060305
20. BootsM
2008 Fight or learn to live with the consequences. Trends Ecol Evol 23 248 250
21. ReadAF
GrahamAL
RabergL
2009 Animal defenses against infectious agents: is damage control more important than pathogen control? PLoS Biol 5 e1000004 doi:10.1371/journal.pbio.1000004
22. DecaesteckerE
De MeesterL
EbertD
2002 In deep trouble: habitat selection constrained by multiple enemies in zooplankton. Proc Natl Acad Sci U S A 99 5481 5485
23. RabergL
GrahamAL
ReadAF
2009 Decomposing health: tolerance and resistance to parasites in animals. Phil Trans R Soc Lond B 364 37 49
24. LazzaroBP
LittleT
2009 Immunity in a variable world. Phil Trans R Soc Lond B 364 15 26
25. LivelyCM
2009 The maintenance of sex: host–parasite coevolution with density-dependent virulence. J Evol Biol 22 2086 2093
26. PagánI
Alonso-BlancoC
García-ArenalF
2009 Differential tolerance to direct and indirect density-dependent costs of viral infection in Arabidopsis thaliana. PLoS Pathog 5 e1000531 doi:10.1371/journal.ppat.1000531
27. CariusH-J
LittleTJ
EbertD
2001 Genetic variation in a host–parasite association: Potential for coevolution and frequency dependent selection. Evolution 55 1136 1145
28. Schmid-HempelP
EbertD
2003 On the evolutionary ecology of specific immune defence. Trends Ecol Evol 18 27 32
29. LambrechtsL
HalbertJ
DurandP
GouagnaLC
KoellaJC
2005 Host genotype by parasite genotype interactions underlying the resistance of anopheline mosquitoes to Plasmodium falciparum. Malar J 4 doi:10.1186
30. LambrechtsL
FellousS
KoellaJC
2006 Coevolutionary interactions between host and parasite genotypes. Trends Parasitol 22 12 16
31. HamiltonWD
1980 Sex versus non-sex versus parasite. Oikos 35 282 290
32. MillerMR
WhiteA
BootsM
2006 The evolution of parasites in response to tolerance in their hosts: the good, the bad, and apparent commensalism. Evolution 60 945 956
33. RoyBA
KirchnerJW
2000 Evolutionary dynamics of pathogen resistance and tolerance. Evolution 54 51 63
34. BestA
WhiteA
BootsM
2009 The implications of coevolutionary dynamics to host-parasite interactions. Am Nat 173 779 791
35. GandonS
MackinnonMJ
NeeS
ReadAF
2001 Imperfect vaccines and the evolution of pathogen virulence. Nature 414 751 755
36. GandonS
DayT
2008 Evidences of parasite evolution after vaccination. Vaccine 26 C4 C7
37. StearnsSC
1992 The evolution of life histories Oxford Oxford University Press
38. RolffJ
Siva-JothyMT
2003 Invertebrate ecological immunity. Science 301 472 475
39. YazdanbakhshM
KremsnerPG
van ReeR
2002 Allergy, parasites, and the hygiene hypothesis. Science 296 490 494
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 9
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- Structure of the Extracellular Portion of CD46 Provides Insights into Its Interactions with Complement Proteins and Pathogens
- The Length of Vesicular Stomatitis Virus Particles Dictates a Need for Actin Assembly during Clathrin-Dependent Endocytosis
- Inhibition of TIR Domain Signaling by TcpC: MyD88-Dependent and Independent Effects on Virulence
- Cellular Entry of Ebola Virus Involves Uptake by a Macropinocytosis-Like Mechanism and Subsequent Trafficking through Early and Late Endosomes