
A20 () Deficiency in Myeloid Cells Protects against Influenza A Virus Infection
The innate immune response provides the first line of defense against viruses and other pathogens by responding to specific microbial molecules. Influenza A virus (IAV) produces double-stranded RNA as an intermediate during the replication life cycle, which activates the intracellular pathogen recognition receptor RIG-I and induces the production of proinflammatory cytokines and antiviral interferon. Understanding the mechanisms that regulate innate immune responses to IAV and other viruses is of key importance to develop novel therapeutic strategies. Here we used myeloid cell specific A20 knockout mice to examine the role of the ubiquitin-editing protein A20 in the response of myeloid cells to IAV infection. A20 deficient macrophages were hyperresponsive to double stranded RNA and IAV infection, as illustrated by enhanced NF-κB and IRF3 activation, concomitant with increased production of proinflammatory cytokines, chemokines and type I interferon. In vivo this was associated with an increased number of alveolar macrophages and neutrophils in the lungs of IAV infected mice. Surprisingly, myeloid cell specific A20 knockout mice are protected against lethal IAV infection. These results challenge the general belief that an excessive host proinflammatory response is associated with IAV-induced lethality, and suggest that under certain conditions inhibition of A20 might be of interest in the management of IAV infections.
Published in the journal:
. PLoS Pathog 8(3): e32767. doi:10.1371/journal.ppat.1002570
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002570
Summary
The innate immune response provides the first line of defense against viruses and other pathogens by responding to specific microbial molecules. Influenza A virus (IAV) produces double-stranded RNA as an intermediate during the replication life cycle, which activates the intracellular pathogen recognition receptor RIG-I and induces the production of proinflammatory cytokines and antiviral interferon. Understanding the mechanisms that regulate innate immune responses to IAV and other viruses is of key importance to develop novel therapeutic strategies. Here we used myeloid cell specific A20 knockout mice to examine the role of the ubiquitin-editing protein A20 in the response of myeloid cells to IAV infection. A20 deficient macrophages were hyperresponsive to double stranded RNA and IAV infection, as illustrated by enhanced NF-κB and IRF3 activation, concomitant with increased production of proinflammatory cytokines, chemokines and type I interferon. In vivo this was associated with an increased number of alveolar macrophages and neutrophils in the lungs of IAV infected mice. Surprisingly, myeloid cell specific A20 knockout mice are protected against lethal IAV infection. These results challenge the general belief that an excessive host proinflammatory response is associated with IAV-induced lethality, and suggest that under certain conditions inhibition of A20 might be of interest in the management of IAV infections.
Introduction
Viruses are a class of highly diverse pathogens which depend on the host cell for their replication. The initiation of a protective innate antiviral immune response involves the action of specialized pattern recognition receptors (PRR), which detect conserved molecular structures of the invading pathogen. Triggering of PRRs induces the production of host proinflammatory cytokines (e.g. TNF, IL-6 and IL-1) and type I interferons (interferon-α (IFN-α) and IFN-β) through activation of downstream signaling pathways that control various transcription factors such as NF-κB, AP-1, IRF3 and IRF7 [1], [2]. The presence of viral nucleic acids, such as viral RNA and DNA, viral replication intermediates and viral transcription products, can be sensed by specific intracellular PRRs [3]. Endosomal Toll-like receptors (TLRs) and cytoplasmic RNA helicase RIG-I-like receptors (RLRs) or Nod-like receptors (NLRs) detect the presence of viral single stranded (TLR7, TLR8, Nod2) or double stranded RNA (TLR3, RIG-I, MDA5). Intracellular DNA sensors that mediate antiviral immune responses to DNA viruses include TLR9, DAI [4] and the PYHIN domain containing proteins AIM2 [5], [6], [7] and IFI16 [8]. TLR mediated antiviral responses are restricted to specialized type I IFN producing plasmacytoid dendritic cells (pDC), while most other cell types, including conventional DC (cDC), macrophages and fibroblasts, depend on the cytosolic RNA and DNA sensors for the production of antiviral proteins [9].
Influenza A virus (IAV) is the etiological agent of a contagious acute respiratory disease that causes considerable mortality, which is generally believed to be due to an excessive host inflammatory response. Emergence of drug-resistant strains of influenza viruses with pandemic potential underscores the importance of developing novel antiviral strategies. In this context, understanding of the mechanisms that regulate IAV-induced immune responses is critical. IAV infection leads to the exposure in the host cell of single-stranded genomic RNA and double stranded RNA, the latter being an intermediate of viral replication. TLR3 and RIG-I have been implicated as sensors of IAV infection [10]–[12]. Both receptors contribute to the proinflammatory response to IAV, but the initiation of the innate antiviral immune response largely depends on RIG-I mediated signaling [13]. Interestingly, RIG-I deficient mice are highly susceptible to IAV [14], [15], whereas TLR3 deficient mice have a survival advantage to acute infection [16]. These results indicate that an imbalance between the beneficial and harmful effects of mediators released by immune cells is likely to contribute to the pathogenesis of influenza.
RIG-I contains a C-terminal DExD/H box helicase domain, which is required for ligand recognition, and two N-terminal CARD domains. Upon ligand binding, the CARD domains of RIG-I associate with the CARD domain of the MAVS (also termed IPS-1, VISA, Cardif) adaptor protein, which subsequently translocates to and inserts in the outer mitochondrial membrane via its C-terminal transmembrane domain [17]–[20]. Signaling downstream of MAVS requires the action of various ubiquitin modifying enzymes, which both positively and negatively regulate RIG-I mediated signal transduction [21]. K63-specific ubiquitin ligases (E3s), such as TRIM25 [22] and Riplet [23], [24], have been shown to directly promote RIG-I activation. In addition, well characterized ubiquitin ligases such as TRAF6 [25], [26] and TRAF3 [27] mediate respectively NF-κB and IRF3 activation upon RIG-I stimulation. On the other hand, deubiquitinating enzymes (DUBs), such as DUBA [28], CYLD [29], [30] and OTUB1/2 [31] have been shown to negatively regulate RLR signaling by specifically removing K63-linked polyubiquitin chains from several signaling molecules. Furthermore, various K48-specific ubiquitin ligases, such as AIP4 [32] and TRIAD3A [33] mark respectively MAVS and TRAF3 for proteasome mediated degradation, thus inhibiting further downstream signaling. Additionally, the attachment of K48-specific polyubiquitin chains to the IRF3 and IRF7 transcription factors by E3s such as RAUL [34], TRIM21 [35] and RBCK1 [36] further dampens antiviral signal transduction.
A20 is an ubiquitin-editing enzyme belonging to the OTU-domain family of DUBs. Interestingly, A20 also harbors atypical zinc finger dependent K48-specific E3 ubiquitin ligase activity. Both catalytic and noncatalytic mechanisms were previously shown to be involved in the negative regulation of proinflammatory signaling by A20 in response to multiple receptors such as TNF receptor I [37]–[39], TLR4 [40], and IL-1R [41]. The anti-inflammatory role of A20 is clearly demonstrated by the fact that A20 deficient mice die early after birth due to severe multi-organ inflammation and cachexia [37]. More recently, gene targeting of A20 in specific cell types was shown to be associated with autoimmunity and chronic inflammation [42]–[48], further illustrating that A20 is an important brake on the inflammatory response. The relevance of these findings for human disease has recently been illustrated through the identification of polymorphisms in the A20 locus that are associated with several autoimmune diseases and chronic inflammation [45]. In contrast to its well established function in the regulation of proinflammatory responses, the role of A20 in the regulation of antiviral immune responses is less well described and limited to a number of in vitro studies using overexpression or silencing in specific cell lines, indicating that A20 may regulate RIG-I- and TLR3-induced signaling to NF-κB and IRF-3 [49]–[52]. However, the precise in vivo role of A20 in the response to viral infection remains to be clarified. Using myeloid cell specific A20 knockout mice (A20myel-KO) that were recently generated in our lab and primary cells derived of these mice, we here provide evidence that A20 is a crucial negative regulator of IAV-induced proinflammatory and antiviral signaling in macrophages. Interestingly, A20myel-KO mice show enhanced survival and reduced morbidity in response to IAV lung infection compared to wild type mice. Protection against IAV in A20myel-KO mice is associated with increased cytokine and chemokine production, augmented recruitment of innate immune cells such as neutophils and alveolar macrophages, and enhanced viral clearance. These results suggest that boosting the innate immune response to IAV by targeting A20 activity in myeloid cells might have therapeutic potential.
Results
A20 inhibits RIG-I-induced NF-κB and IRF3 activation
RIG-I signaling induces the activation of NF-κB, IRF3 and IRF7 transcription factors, which promote the expression of proinflammatory cytokines and type I IFNs that restrict further viral propagation. Previous studies have shown that ectopically expressed A20 negatively regulates NF-κB and IRF3 activation upon RIG-I stimulation [50]–[52]. Similarly, we show that A20 overexpression in HEK293T cells prevents NF-κB- and IRF3-dependent luciferase reporter gene activation induced by transfection of a truncated constitutive active form of RIG-I [53], corresponding to only the two N-terminal CARD domains of RIG-I [RIG-I (2CARD)] (figure 1A, left and middle graph). We next investigated whether A20 also inhibits IRF7 activation. Unlike IRF3, IRF7 is not or weakly expressed under naïve conditions and IRF7 protein levels are rapidly upregulated upon virus-induced IRF3 activation [54], [55]. To determine the effect of A20 on IRF7 activation, we therefore transfected minor amounts of an IRF7 expression plasmid together with plasmids encoding RIG-I (2CARD), A20 and an IRF7-specific IFNα4 luciferase reporter construct. RIG-I (2CARD) expression in the absence of IRF7 co-expression showed negligible IFNα4 promoter activation (grey bar, figure 1A, right graph), whereas significant reporter gene expression was observed in the presence of IRF7. Similar to its inhibitory effect on NF-κB and IRF3 activation, A20 also prevented RIG-I-induced IRF7 activation (figure 1A, right graph). These results demonstrate the potential of A20 to inhibit RIG-I-induced NF-κB and IRF3/7 activation.
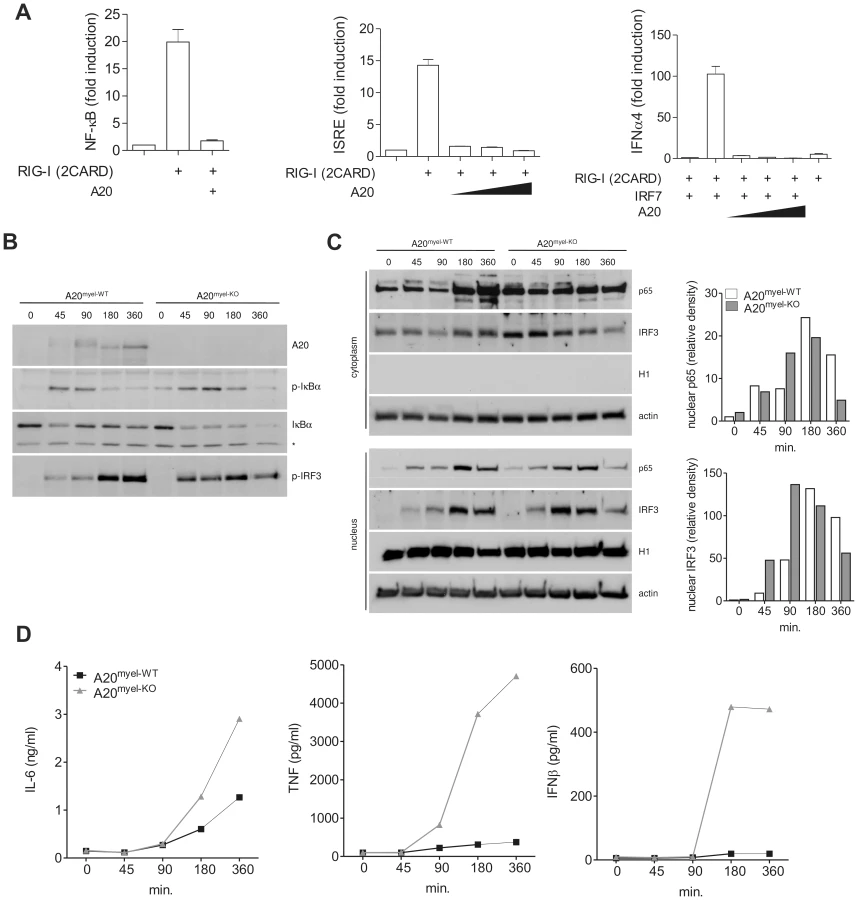
To study the effect of endogenously expressed A20 on RIG-I-induced signaling in a more immunological relevant context, we performed further experiments in A20 deficient primary macrophages. Since A20 full knockout mice die prematurely as a result of severe multi-organ inflammation [37], we generated mice carrying a conditional A20 allele in which exon IV and exon V were flanked by two loxP sites [56]. Crossing these mice with transgenic mice expressing Cre recombinase under control of the lysozyme M promoter leads to specific deletion in myeloid cells and allowed us to generate myeloid cell specific A20 knockout mice [48]. To stimulate the RIG-I receptor, we transfected A20myel-KO BMDM and wild type control cells with minimal amounts of low molecular weight (LMW) poly(I:C), which is known to preferentially bind and activate RIG-I rather than MDA5 [57]. Of note, this concentration of poly(I:C) was unable to induce significant TLR3 dependent NF-κB and IRF3 activation or cytokine production (data not shown). As expected, poly(I:C) transfection induced the rapid expression of A20 in wild type, but not in A20myel-KO BMDM (figure 1B, upper panel). At early time points, slightly slower migrating forms of A20 were observed, indicating that A20 undergoes a yet unknown modification upon poly(I:C) transfection. Compared to wild type BMBM, A20 deficient cells showed enhanced NF-κB activation as indicated by increased phosphorylation and sustained degradation of IκBα (figure 1B). Furthermore, nuclear translocation of the p65 NF-κB subunit was enhanced in poly(I:C) transfected A20myel-KO BMDM, reaching a maximum at earlier time points compared to wild type cells (figure 1C). IRF3 is known to be activated upon phosphorylation of a series of carboxyl terminal serine residues by the IKK-like kinases TBK1 and IKKε [58], leading to its dimerization and subsequent translocation to the nucleus [59]. Using immunoblotting with an antibody directed against phosphorylated Ser396, maximum IRF3 phosphorylation was detected at earlier time points in A20myel-KO BMDM compared to wild type BMDM (figure 1B). Similar to p65, IRF3 nuclear translocation reached its maximum at an earlier time point in A20 deficient BMDM compared to wild type cells (figure 1C). NF-κB controls the expression of IL-6 and TNF, and NF-κB and IRF3 control the expression of IFNβ. In line with the enhanced activation of NF-κB and IRF3 as described above, A20myel-KO BMDM secreted increased amounts of IL-6, TNF and IFNβ (figure 1D). Similar results were obtained using peritoneal macrophages (data not shown). Together, these results demonstrate that A20 plays an important role in the negative regulation of RIG-I-induced NF-κB and IRF3 activation in primary macrophages.
A20 negatively regulates IAV-induced gene expression in BMDM
To investigate the role of A20 in the IAV-induced proinflammatory and antiviral innate immune responses, we infected A20 deficient and control BMDM with IAV X-47 (H3N2). A20 mRNA levels were rapidly induced in wild type BMDM, but not in A20 deficient BMDM, upon viral infection (figure 2A). Furthermore, A20myel-KO BMDM show enhanced expression of IL-6 and IFNβ mRNA after IAV infection compared to control cells (figure 2A). In accordance with these data, cell culture supernatant collected from these cells contained higher levels of TNF and IFNβ (figure 2B).

Upon host infection with IAV, alveolar macrophages are an important source of cytokines and chemokines, attracting innate immune cells to the lung during the primary phase of infection. To test whether A20 directly controls IAV-induced gene expression in alveolar macrophages, we isolated these cells from lungs of A20myel-KO and control littermates and infected them in vitro with IAV X-47. Expression and secretion of the proinflammatory cytokines IL-6 and TNF, the type-I IFN IFNβ and IFNα4 and the chemokines MCP-1 (ccl2) and KC (cxcl1) was significantly higher in IAV infected cells lacking A20 compared to infected wild type cells (figure 2C and figure S1). Taken together these results demonstrate that A20 negatively regulates IAV-induced proinflammatory and antiviral gene expression in alveolar macrophages, consistent with the inhibitory effect of A20 seen on RIG-I-induced NF-κB and IRF3 activation.
A20 deficiency in myeloid cells protects against IAV lung infection
To determine the role of A20 expression in myeloid cells during an IAV infection in vivo, we intranasally inoculated both A20myel-KO mice and control littermates with a sublethal dose of the mouse adapted IAV X-47 (H3N2) strain and monitored morbidity in terms of weight loss. A20myel-KO mice showed reduced weight loss compared to wild type control littermates and recovered faster from the viral challenge (figure 3A). Also, total protein concentration in BAL fluid, which reflects lung damage and vascular leakage, was increased significantly at 7 and 10 days post infection in both wild type and A20myel-KO mice, and was slightly lower in A20myel-KO mice (data not shown). Next, we measured pulmonary viral titers at 4, 7 and 10 days post infection. No differences in viral titers were observed in A20myel-KO mice versus wild type mice at day 4 and 7 post infection. However, after 10 days, almost no virus could be detected in the lungs of A20myel-KO mice while abundant infectious viral particles could still be isolated from lungs of all wild type mice (figure 3B). This indicates that loss of A20 in myeloid cells does not affect early viral replication but contributes to viral clearance at later stages during infection.
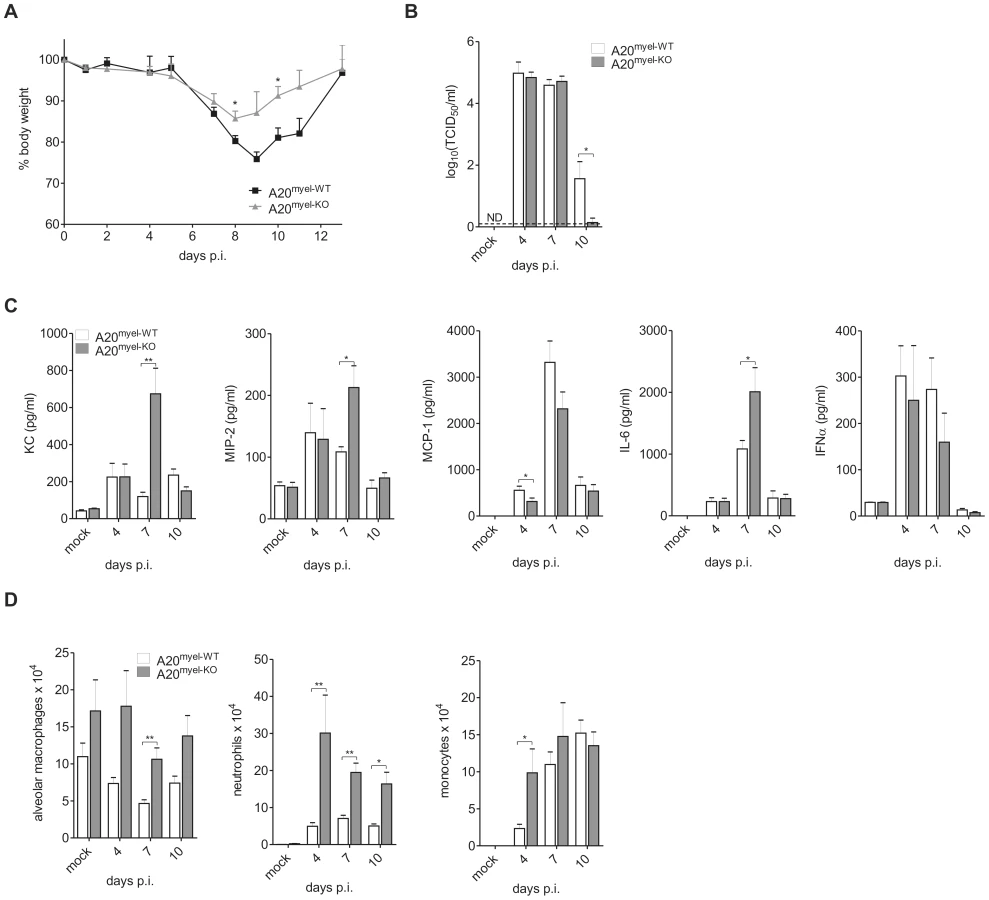
To verify if A20 deficiency in myeloid cells affects IAV-induced gene expression in the lung, we analyzed the levels of several chemokines and cytokines in the bronchoalveolar lavage (BAL) at day 4, 7 and day 10 following infection. Levels of KC and MIP-2 chemokines, as well as IL-6 were significantly higher at day 7 p.i. in BAL from IAV infected A20myel-KO mice compared to IAV infected wild type mice (figure 3C). Unlike our observations with in vitro stimulated primary macrophages we could not detect a significant increase in MCP-1 or IFNα production in the lungs of A20myel-KO animals (figure 3C). KC is the murine orthologue of IL-8 and serves together with MIP-2 as a chemoattractant for neutrophils, while MCP-1 is mainly known as a chemoattractant for monocytes, which eventually develop into macrophages upon entering the alveolar lumen [60]. Consistent with the higher KC and MIP-2 levels in A20myel-KO mice, the number of neutrophils (CD11b+ Ly6C+ Ly6G+ F4/80−) that were recruited in the bronchoalveolar spaces upon IAV infection was clearly higher throughout infection in A20myel-KO mice compared to control animals (figure 3D). Although we could detect a significant increase in monocyte (CD11b+ Ly6C+) recruitment at day 4 post infection, this was not evident at later time points after infection (figure 3D), which is consistent with the equal expression of MCP-1 in both groups of mice. The number of resident alveolar macrophages (autofluorescent+ CD11c+ F4/80+ CD11b−) was also elevated in A20myel-KO mice but did not differ significantly between IAV infected or mock infected mice (figure 3D). Elimination of IAV infected cells depends on the clonal expansion of virus specific cytotoxic CD8+ T cells (CTL) [61], [62], [63]. To test whether A20 expression in myeloid cells regulates the antiviral CTL response, total CD8+ T cells and virus specific Granzyme B (GrB) and IFNγ expressing CD8+ T cells were measured in BAL and lung parenchyma of wild type and A20myel-KO mice. A clear increase in CD8+ T cells could be detected at day 7 and 10 post infection, but no differences were observed between A20myel-KO and wild type mice (figure S2A and figure S2C). Also, the number of GrB and IFNγ CD8+ T cells as well as IFNγ expression levels in the lungs were not altered by the absence of A20 in myeloid cells (figure S2A–C).
Protection against IAV infection is also provided by the humoral immune response. To test whether loss of A20 in myeloid cells affects B cell mediated immunity, we determined hemagglutinin (HA) antibody titers in the serum of A20myel-KO and wild type littermates. However, no differences could be detected between wild type and A20myel-KO animals (figure S2D), indicating that humoral immunity is not affected by A20 expression in myeloid cells. Together, these data suggest that mechanisms other than adaptive immunity, such as an increased innate immune response, characterized by an increased influx of neutrophils and increased numbers of alveolar macrophages, contribute to the better viral clearance in A20myel-KO mice.
It is generally believed that IAV-induced mortality is due to an excessive proinflammatory response in the lung. We therefore analyzed whether the increased proinflammatory cytokine production and infiltration of proinflammatory cells in A20myel-KO mice affects mortality induced by intranasal infection with a lethal dose of IAV X-47. Surprisingly, almost all A20myel-KO mice survived (10/11), while all control mice succumbed (0/11) within 16 days after infection (figure 4A). A20myel-KO mice still showed significant weight loss and lung damage (as reflected by increased total protein concentration in the BAL; data not shown) during the course of infection but were able to recover, in contrast to wild type mice that succumbed (figure 4B). Similar to our observations with sublethal IAV infection, pulmonary KC and MIP-2 production was stronger in A20myel-KO animals compared to wild type mice following lethal IAV infection (figure 4C), which correlates with the increased numbers of neutrophils in the lungs of these mice (figure 4C). Also levels of the proinflammatory cytokines IL-6, TNF and IL-1β, which are often associated with immunopathogenesis in humans [64], were increased in the lungs of A20myel-KO mice compared to control animals (figure 4C and figure S3A). Again, MCP-1 production was not increased and even lower in A20myel-KO mice (figure 4C), and also monocyte recruitment was not different between both groups of mice. We could also not detect any differences in viral clearance or antiviral adaptive immunity at 6 h post infection (figure S3B–F). Collectively these data indicate that A20 deficiency in myeloid cells is associated with an increased innate immune response and protection against a lethal IAV infection.

Discussion
In the present study we have investigated the contribution of A20 expression in myeloid cells in the innate immune response to IAV lung infection. In the pulmonary environment, macrophages populate both lung parenchyma and the alveolar lumen where they are referred to as alveolar macrophages. Under naïve conditions, alveolar macrophages exert important immunomodulatory functions [65], [66]. However, alveolar macrophages are also crucial in the innate immune response against IAV as they are one of the first cells that encounter infectious IAV particles [67], [68]. They are an important source of proinflammatory cytokines and chemokines that attract innate immune cells to the lung during the primary phase of infection [69], and they are the primary producers of type I IFNs [70]. Alveolar macrophages are also known to phagocytose virus infected apoptotic cells and antibody coated viral particles, further contributing to viral clearance. We could show that BMDM or alveolar macrophages derived from A20myel-KO mice express higher amounts of chemokines, cytokines and type I IFNs compared to wild type mice in response to in vitro infection. Similarly, in vivo infection with IAV resulted in higher levels of primarily neutrophil attracting chemokines such as KC and MIP-2 and several proinflammatory cytokines such as IL-6, TNF and IL-1β in the lungs of A20myel-KO mice compared to wild type mice. This was associated with a selective enhancement of neutrophil recruitment to the bronchoalveolar compartment, and resulted in improved viral clearance later on during infection. Although the role of neutrophils during viral infection is still under debate, recent evidence supports a protective function of these cells during IAV infection [71], [72]. MCP-1 levels were not affected by the absence of A20 in myeloid cells, which is consistent with the notion that airway epithelial cells are the primary source of MCP-1 production during IAV infection [73]. Mice deficient for the MCP-1 receptor CCR2, which is expressed on a subset of circulating monocytes, are protected against IAV infection and display reduced signs of immunopathology [74]–[76]. During IAV infection these monocytes develop into inflammatory dendritic cells or proinflammatory macrophages [77] and are considered major contributors to IAV-induced immunopathology [78]. A20myel-KO mice were protected against a lethal IAV infection, which is rather surprising since an excessive proinflammatory cytokine response and an excessive influx of inflammatory cells is generally believed to be associated with increased lethality [64], [79]. However, the selective effect of A20 deficiency on neutrophil recruitment, without altering inflammatory monocyte (Ly6C+ CD11b+) recruitment, further support the idea that monocytes and not neutrophils are major contributors to IAV-associated immunopathology and lethality [78].
We show that A20 deficient BMDM display enhanced NF-κB and IRF3 activation in response to RIG-I stimulation by synthetic LMW double stranded RNA. RIG-I has previously been shown to play a key role in the innate immune response to IAV [13], suggesting that the increased immune response of A20myel-KO mice to IAV lung infection reflects enhanced RIG-I signaling. We propose that A20 inhibits IAV-induced proinflammatory gene expression (as shown in our manuscript for TNF, IL-6, KC, MIP-2, and IFNβ) by negatively regulating NF-κB and IRF3 activation, which are the major pathways controlling these genes. However, this does not exclude an additional effect of A20 on other signaling pathways that may contribute to proinflammatory gene expression. A20 is believed to exert its NF-κB and IRF3 inhibitory functions by modulating the ubiquitination status of different signaling proteins [80]. In this context, it was recently shown that A20 cooperates with the ubiquitin-binding proteins TAX1BP1 and ABIN1 to to disrupt the TRAF3-TBK1-IKKε complex, thereby negatively affecting K63-polyubiquitination of TBK1 and IKKε, and their ability to activity IRF3 [81], [82]. Whether similar mechanisms are involved in the regulation of RIG-I induced NF-κB activation is still unclear. So far we were unable to clearly detect ubiquitination of TBK1 and IKKε in primary macrophages, preventing us from studying the effect of A20 deficiency on their ubiquitination status. It cannot be excluded however that A20 also targets other substrates that mediate NF-κB and IRF3 activation in myeloid cells. The identification of these substrates will be the topic of future investigations in our laboratory. Multiple other deubiquitinating enzymes (DUBs), such as DUBA [28], CYLD [29], [30], OTUB1/2 [31], and A20 [49]–[52] have been shown to negatively regulate RIG-I signaling to NF-κB and IRF-3, implicating possible redundancy. However, evidence so far was limited to in vitro data and was obtained under non-physiological conditions. The clear protective phenotype of A20myel-KO mice that we here describe indicates that A20 expression in myeloid cells is a central gatekeeper of RIG-I induced signaling in response to IAV infection and that other DUBs cannot substitute for A20 deficiency under physiological conditions. If A20 has a similar non-redundant role in other cell types that are implicated in the response to IAV, such as lung epithelial cells, remains to be investigated.
Understanding and controlling the activation of innate antiviral immune responses is an important step toward novel therapies. About a fifth of world's population is infected by IAV annually, leading to high morbidity and mortality, particularly in infants, the elderly and the immunocompromised. The high mutation rate of IAV turns all attempts of vaccine and antiviral design into a never ending battle. In recent years, RNA interference, triggered by synthetic short interfering RNA (siRNA), has rapidly evolved as a potent antiviral regimen. Properly designed siRNAs have been shown to function as potent inhibitors of influenza virus replication. Although specificity and tissue delivery remain major bottlenecks in the clinical applications of RNAi in general, intranasal application of siRNA against respiratory viruses including, but not limited to influenza virus, has experienced significant success and optimism [83]. Our results suggest that not only siRNA targeting IAV components, but boosting the antiviral immune response by intranasal administration of siRNA against A20 might be a valid therapeutic approach. Also small compound inhibitors of A20 might be an interesting alternative. Finally, similar targeting of A20 might be of interest in the battle against other viral infections including RSV and SARS coronavirus.
Materials and Methods
Ethics statement
All experiments on mice were conducted according to the national (Belgian Law 14/08/1986 and 22/12/2003, Belgian Royal Decree 06/04/2010) and European (EU Directives 2010/63/EU, 86/609/EEG) animal regulations. Animal protocols were approved by the ethics committee of Ghent University (permit number LA1400091, approval ID 2010/001). All efforts were made to ameliorate suffering of animals. Mice were anesthetized by intraperitoneal (i.p.) injection of a mixture of ketamine (12 mg/kg) and xylazine (60 mg/kg).
Mice
A20fl/fl mice were generated as previously described [56]. A20fl/fl mice were crossed with LysM-Cre mice [84] (provided by I. Förster, Institute of Genetics, University of Cologne, Germany) to generate A20fl/fl LysMCre transgenes and are described in detail elsewhere [48]. Mice were housed in individually ventilated cages at the VIB Department of Molecular Biomedical Research in specific pathogen-free animal facilities. Influenza infections were performed on age- (between 7 and 9 weeks old) and sex-matched littermates. A20fl/fl LysM-Cre animals were backcrossed three times to the C57Bl/6 background. A20fl/fl mice expressing or lacking the LysM-Cre transgene were termed A20myel-KO and wild type (A20myel-WT) respectively.
Viral infection and determination of viral titers
Mouse adapted IAV X-47 (H3N2; PR8×A/Victoria/3/75) was propagated in MDCK cells. For viral inoculation, mice were anesthetized by i.p. injection with ketamine (12 mg/kg) and xylazine (60 mg/kg) and 50 µl X-47 diluted in PBS was administered intranasally. For lethal and sublethal infection, mice received respectively 2-LD50 or 0.05-LD50 X-47. To determine pulmonary viral titers, median tissue culture infectious dose (TCID50) was measured as follows: lungs were homogenized with a Polytron homogenizer (Kinematica) in PBS. Eight-fold serial dilutions of lung homogenates were incubated on MDCK cells for 5 days in DMEM supplemented with trypsin (1 µg/ml), 2 mM L-glutamine, 0.4 mM sodium pyruvate and antibiotics. For read-out, 0.5% chicken red blood cells (RBC) were added and end-point dilution of hemagglutination was monitored. TCID50 titers were then calculated according to the method of Reed and Muench [85].
Determination of HAI (hemagglutination inhibition) titers
To determine the HAI titers in infected mice, sera of these were treated with receptor-destroying enzyme (RDE/Cholera filtrate; Sigma) to remove sialic acids from serum proteins capable of aspecific inhibition of agglutination. After incubation overnight at 37°C, the RDE was inactivated by addition of 0.75% sodium citrate in PBS and heating to 56°C for 30 min. To remove sialic acid binding proteins, sera were cleared with 1/10 volume 50% chicken RBC. Titration was done by incubating a two-fold dilution series of sera with 4 HA units of X-47 virus for 1 hour at room temperature in 96-well U-bottom plates. Finally, an equal volume of 0.5% chicken RBC was added and titers were read 30 min later. Negative controls included PBS instead of immune serum (agglutination control) or PR8 instead of X-47 virus (control for agglutination effect of sera); as positive control, serum from a mouse infected twice with a sublethal dose of X-47 was used.
In vivo intracellular GrB and IFNγ staining of activated CD8+ T cells
Granzyme B (GrB) and IFNγ expressing CD8+ T cells were determined by treating the mice intranasally with 50 µg Brefeldin A (Sigma) as previously described [86]. 6 h later, BAL and lungs were isolated and single cell suspensions were prepared from the lung in the presence of 3 µg/ml Brefeldin A. Cells were stained, fixed and permeabilized (Cytofix/Cytoperm, BD Biosciences) according to the manufacturer's instructions. Activated CD8+ T cells were analyzed by flow cytometry based on CD62lo CD3+ and CD8+ expression. Live/Dead fixable aqua dead cell stain kit (Molecular Probes) was used to discriminate live from dead cells.
Cells and transfection
HEK293T and MDCK cells were grown in DMEM (Gibco) supplemented with 10% FCS, 2 mM L-glutamine, 0.4 mM sodium pyruvate and antibiotics. HEK293T cells were transfected using the calcium phosphate precipitate transfection method with specific expression vectors (pCAGGS-E-hA20 (LMBP 3778), pCAGGS-E-RIG-I-CARD (LMBP 6517), pEF-HA-IRF-7 (kindly provided by T. Taniguchi, Graduate School of Medicine and Faculty of Medicine, University of Tokyo)), NF-κB, IRF3, IRF7 reporter plasmids (respectively pConLuc (LMBP3248), pISRE-luc (LMBP4011), pGL3-IFNα4-luc (kindly provided by J. Hiscott, McGill University, Montreal, Quebec, Canada), and pACTbetagal (LMBP4341) for transfection efficiency normalization. Details of plasmids are presented along with detailed sequence maps at the BCCM-LMBP plasmid databank http://bccm.belspo.be/index.php.
For the generation of BMDM, bone marrow cells were cultured 7 days in RPMI 1640 (Gibco) supplemented with 10% FCS, 2 mM L-glutamine, 0.4 mM sodium pyruvate, antibiotics and 40 ng/ml recombinant M-CSF. BMDM were of ≥95% purity as measured by flow cytometry using F4/80 and CD11b specific antibodies. For the isolation of alveolar macrophages, the trachea was canulated and the lung was flushed 4 times with HBSS containing 1 mM EDTA. Alveolar macrophages were cultured in RPMI 1640 (Gibco) supplemented with 1% FCS, 2 mM L-glutamine, 0.4 mM sodium pyruvate and antibiotics.
Western blotting
For total lysates, cells were lysed at 4°C for 15 min in lysis buffer (200 mM NaCl, 1% NP-40, 10 mM Tris-HCl pH 7.5, 5 mM EDTA, 2 mM DTT) supplemented with protease and phosphatase inhibitors. Nuclear and cytoplasmic lysates were prepared by resuspending cells in B1 (10 mM Hepes pH 7.5, 10 mM KCl, 1 mM MgCl2, 5% glycerol, 0.5 mM EDTA and 0.1 mM EGTA supplemented with protease and phosphatase inhibitors) for 15 min at 4°C. Next, NP-40 detergent was added to a final concentration of 0.65% and cells were centrifuged at 500 g for 5 min. The nuclear fraction containing pellet was lyzed in B2 (20 mM Hepes pH 7.5, 1% NP-40, 400 mM NaCl, 10 mM KCl, 1 mM MgCl2, 20% glycerol, 0.5 mM EDTA and 0.1 mM EGTA supplemented with protease and phosphatase inhibitors) for 15 min at 4°C. The lysates were subsequently separated by SDS-PAGE and analyzed by western blotting and ECL detection (Perkin Elmer Life Sciences). Immunoblots were revealed with anti-A20, anti-IκBα, anti-p65, and anti-histon H1 (Santa Cruz), anti-IRF3 (Invitrogen), anti-phospho-IRF3 and anti-phospho-IκBα (Cell Signaling) and anti-actin (MP Biomedicals). The density of the bands was quantified (fold induction) with the ImageJ (http://rsbweb.nih.gov/ij) Gel analyzer tool. All intensities were calculated relative to the first lane ( = time 0).
Flow cytometry
Lungs were dissected and incubated with collagenase type IV (1 mg/ml; Sigma) and DNAse (100 U/ml; Roche) at 37°C for 30 min. Subsequently, samples were filtered through a 70 µm and 40 µm nylon mesh. For the preparation of BAL, trachea were canulated and airway lumen was flushed 4 times with HBSS with 1 mM EDTA. Cells were stained with monoclonal antibodies directed against MHC-II (I-A/I-E) FITC (M5/114.15.2), CD11c PerCP-Cy5.5 (N418), F4/80 APC (BM8), CD62L PE (MEL-14), Granzyme B FITC (NGZB) from eBiosciences and CD3 Molecular Complex Horizon v450 (17A2), Ly6C Horizon v450 (AL-21), Ly6G PE (1A8), CD11b APC-Cy7 (M1/70), CD8α PerCP (53-6.7), IFNγ Alexa 647 (XMG1.2) and CD16/32 (2.4G2) from BD Pharmingen. Samples were acquired on a LSRII Cytometer and analyzed using FACSDiva software (BD Biosciences). Propidium iodide was used to discriminate between live and dead cells.
Cytokine quantification
For TNF ELISA, 96-well plates were coated with TNF coating (TN3-19, eBioscience) and detection (R4-6A2, eBioscience) antibodies. IFNα and IFNβ protein levels were determined with an ELISA kit (PBL Biomedical Laboratories). For IFNγ ELISA, 96-well plates were coated with IFNγ coating (XMG1.2) and detection (R4-6A2) antibodies (eBiosciences). Detection of MCP-1, KC, TNF, IL-1β and IL-6 in BAL fluid was performed using Bioplex (BioRad) technology according to the manufacturer's instructions. Milliplex technology (Millipore) was used for the detection of MIP-2 in BAL fluid.
RNA isolation, cDNA synthesis and qPCR
Total RNA was extracted using Aurum Total RNA mini kit (BioRad) and reverse transcribed into cDNA with iScript cDNA synthesis kit (BioRad) according to the manufacturer's instructions. qPCR was performed by using SYBR Green I master mix I (Roche) in the Lightcycler 480 detection system (Roche) with the following primers: HPRT: 5′-AGTGTTGGATACAGGCCAGAC-3′ and 5′CGTGATTCAAATCCCTGAAGT-3′; IL-6: 5′-GAGGATACCACTCCCAACAGACC-3′ and 5′-AAGTGCATCATCGTTGTTCATACA-3′; IFNβ: 5′-TCAGAATGAGTGGTGGTTGC-3′ and 5′-GACCTTTCAAATGCAGTAGATTCA-3′; A20: 5′-AAACCAATGGTGATGGAAACTG-3′ and 5′-GTTGTCCCATTCGTCATTCC-3′; CCL2: 5′-TTAAAAACCTGGATCGGAACCAA-3′ and 5′-GCATTAGCTTCAGATTTACGGGT-3′; CXCL1: 5′-GAGCCTCTAACCAGTTCCAG-3′ and 5′-TGAGTGTGGCTATGACTTCG-3′ and IFNα4: 5′-TGATGAGCTACTACTGGTCAGC-3′ and 5′-GATCTCTTAGCACAAGGATGGC-3′. Primers were designed with PerlPrimer (http://perlprimer.sourceforge.net). Quantification was performed using the comparative CT method (ΔΔCT). Results are expressed relative to HPRT values.
Statistics
Results are expressed as the mean ± SEM. Statistical significance between groups was assessed using two-way ANOVA. The differences for in vivo experiments (at least 5 mice per group) were calculated using the Mann-Whitney U-test for unpaired data. Statistical significance of differences between survival rates was analyzed by comparing Kaplan-Meier curves using the log-rank test (GraphPad Prism version 5, GraphPad, San Diego, CA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. KawaiTAkiraS 2010 The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11 373 384
2. TakeuchiOAkiraS 2010 Pattern recognition receptors and inflammation. Cell 140 805 820
3. YoneyamaMFujitaT 2009 RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 227 54 65
4. TakaokaAWangZChoiMKYanaiHNegishiH 2007 DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448 501 505
5. BurckstummerTBaumannCBlumlSDixitEDurnbergerG 2009 An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 10 266 272
6. Fernandes-AlnemriTYuJWDattaPWuJAlnemriES 2009 AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458 509 513
7. HornungVAblasserACharrel-DennisMBauernfeindFHorvathG 2009 AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458 514 518
8. UnterholznerLKeatingSEBaranMHoranKAJensenSB 2010 IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol 11 997 1004
9. KatoHSatoSYoneyamaMYamamotoMUematsuS 2005 Cell type-specific involvement of RIG-I in antiviral response. Immunity 23 19 28
10. RehwinkelJTanCPGoubauDSchulzOPichlmairA 2010 RIG-I detects viral genomic RNA during negative-strand RNA virus infection. Cell 140 397 408
11. Le GofficRPothlichetJVitourDFujitaTMeursE 2007 Cutting Edge: Influenza A virus activates TLR3-dependent inflammatory and RIG-I-dependent antiviral responses in human lung epithelial cells. J Immunol 178 3368 3372
12. BaumASachidanandamRGarcia-SastreA 2010 Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci U S A 107 16303 16308
13. LooYMFornekJCrochetNBajwaGPerwitasariO 2008 Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol 82 335 345
14. KatoHTakeuchiOSatoSYoneyamaMYamamotoM 2006 Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441 101 105
15. GitlinLBarchetWGilfillanSCellaMBeutlerB 2006 Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A 103 8459 8464
16. Le GofficRBalloyVLagranderieMAlexopoulouLEscriouN 2006 Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog 2 e53
17. XuLGWangYYHanKJLiLYZhaiZ 2005 VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell 19 727 740
18. SethRBSunLEaCKChenZJ 2005 Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 122 669 682
19. MeylanECurranJHofmannKMoradpourDBinderM 2005 Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437 1167 1172
20. KawaiTTakahashiKSatoSCobanCKumarH 2005 IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 6 981 988
21. MaelfaitJBeyaertR 2012 The emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol Mol Biol Rev (in press)
22. GackMUShinYCJooCHUranoTLiangC 2007 TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 446 916 920
23. GaoDYangYKWangRPZhouXDiaoFC 2009 REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One 4 e5760
24. OshiumiHMiyashitaMInoueNOkabeMMatsumotoM 2010 The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 8 496 509
25. OganesyanGSahaSKGuoBHeJQShahangianA 2006 Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439 208 211
26. HackerHRedeckeVBlagoevBKratchmarovaIHsuLC 2006 Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439 204 207
27. KonnoHYamamotoTYamazakiKGohdaJAkiyamaT 2009 TRAF6 establishes innate immune responses by activating NF-κB and IRF7 upon sensing cytosolic viral RNA and DNA. PLoS One 4 e5674
28. KayagakiNPhungQChanSChaudhariRQuanC 2007 DUBA: a deubiquitinase that regulates type I interferon production. Science 318 1628 1632
29. ZhangMWuXLeeAJJinWChangM 2008 Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem 283 18621 18626
30. FriedmanCSO'DonnellMALegarda-AddisonDNgACardenasWB 2008 The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep 9 930 936
31. LiSZhengHMaoAPZhongBLiY 2010 Regulation of virus-triggered signaling by OTUB1- and OTUB2-mediated deubiquitination of TRAF3 and TRAF6. J Biol Chem 285 4291 4297
32. YouFSunHZhouXSunWLiangS 2009 PCBP2 mediates degradation of the adaptor MAVS via the HECT ubiquitin ligase AIP4. Nat Immunol 10 1300 1308
33. NakhaeiPMespledeTSolisMSunQZhaoT 2009 The E3 ubiquitin ligase Triad3A negatively regulates the RIG-I/MAVS signaling pathway by targeting TRAF3 for degradation. PLoS Pathog 5 e1000650
34. YuYHaywardGS 2010 The ubiquitin E3 ligase RAUL negatively regulates type i interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33 863 877
35. BollandSGarcia-SastreA 2009 Vicious circle: systemic autoreactivity in Ro52/TRIM21-deficient mice. J Exp Med 206 1647 1651
36. ZhangMTianYWangRPGaoDZhangY 2008 Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF3. Cell Res 18 1096 1104
37. LeeEGBooneDLChaiSLibbySLChienM 2000 Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 289 2350 2354
38. WertzIEO'RourkeKMZhouHEbyMAravindL 2004 De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature 430 694 699
39. SkaugBChenJDuFHeJMaAChenZJ 2011 Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell 44 559 571
40. BooneDLTurerEELeeEGAhmadRCWheelerMT 2004 The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol 5 1052 1060
41. ShembadeNMaAHarhajEW 2010 Inhibition of NF-κB signaling by A20 through disruption of ubiquitin enzyme complexes. Science 327 1135 1139
42. HammerGETurerEETaylorKEFangCJAdvinculaR 2011 Expression of A20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol 12 1184 1193
43. ChuYVahlJCKumarDHegerKBertossiA 2010 B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood 117 2227 2236
44. TavaresRMTurerEELiuCLAdvinculaRScapiniP 2010 The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity 33 181 191
45. VereeckeLBeyaertRvan LooG 2009 The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol 30 383 391
46. HovelmeyerNReissigSXuanNTAdams-QuackPLukasD 2011 A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol 41 595 601
47. KoolMvan LooGWaelputWDe PrijckSMuskensF 2011 The ubiquitin-editing protein A20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity 35 82 96
48. MatmatiMJacquesPMaelfaitJVerheugenEKoolM 2011 A20 (TNFAIP3) deficiency in myeloid cells triggers erosive polyarthritis resembling rheumatoid arthritis. Nat Genet 43 908 912
49. OnoseAHashimotoSHayashiSMaruokaSKumasawaF 2006 An inhibitory effect of A20 on NF-kappaB activation in airway epithelium upon influenza virus infection. Eur J Pharmacol 541 198 204
50. LinRYangLNakhaeiPSunQSharif-AskariE 2006 Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem 281 2095 2103
51. WangYYLiLHanKJZhaiZShuHB 2004 A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-κB and ISRE and IFN-β promoter. FEBS Lett 576 86 90
52. SaitohTYamamotoMMiyagishiMTairaKNakanishiM 2005 A20 is a negative regulator of IFN regulatory factor 3 signaling. J Immunol 174 1507 1512
53. YoneyamaMKikuchiMNatsukawaTShinobuNImaizumiT 2004 The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5 730 737
54. LinRMamaneYHiscottJ 2000 Multiple regulatory domains control IRF-7 activity in response to virus infection. J Biol Chem 275 34320 34327
55. SatoMSuemoriHHataNAsagiriMOgasawaraK 2000 Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity 13 539 548
56. VereeckeLSzeMMc GuireCRogiersBChuY 2010 Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med 207 1513 1523
57. KatoHTakeuchiOMikamo-SatohEHiraiRKawaiT 2008 Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 205 1601 1610
58. HemmiHTakeuchiOSatoSYamamotoMKaishoT 2004 The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 199 1641 1650
59. LinRHeylbroeckCPithaPMHiscottJ 1998 Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18 2986 2996
60. LandsmanLVarolCJungS 2007 Distinct differentiation potential of blood monocyte subsets in the lung. J Immunol 178 2000 2007
61. LukacherAEBracialeVLBracialeTJ 1984 In vivo effector function of influenza virus-specific cytotoxic T lymphocyte clones is highly specific. J Exp Med 160 814 826
62. BenderBSCroghanTZhangLSmallPAJr 1992 Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med 175 1143 1145
63. YoonHLeggeKLSungSSBracialeTJ 2007 Sequential activation of CD8+ T cells in the draining lymph nodes in response to pulmonary virus infection. J Immunol 179 391 399
64. La GrutaNLKedzierskaKStambasJDohertyPC 2007 A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 85 85 92
65. HoltPGOliverJBilykNMcMenaminCMcMenaminPG 1993 Downregulation of the antigen presenting cell function(s) of pulmonary dendritic cells in vivo by resident alveolar macrophages. J Exp Med 177 397 407
66. ThepenTVan RooijenNKraalG 1989 Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J Exp Med 170 499 509
67. TumpeyTMGarcia-SastreATaubenbergerJKPalesePSwayneDE 2005 Pathogenicity of influenza viruses with genes from the 1918 pandemic virus: functional roles of alveolar macrophages and neutrophils in limiting virus replication and mortality in mice. J Virol 79 14933 14944
68. TateMDPickettDLvan RooijenNBrooksAGReadingPC 2010 Critical role of airway macrophages in modulating disease severity during influenza virus infection of mice. J Virol 84 7569 7580
69. BeckerSQuayJSoukupJ 1991 Cytokine (tumor necrosis factor, IL-6, and IL-8) production by respiratory syncytial virus-infected human alveolar macrophages. J Immunol 147 4307 4312
70. KumagaiYTakeuchiOKatoHKumarHMatsuiK 2007 Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 27 240 252
71. TateMDIoannidisLJCrokerBBrownLEBrooksAG 2011 The role of neutrophils during mild and severe influenza virus infections of mice. PLoS One 6 e17618
72. TateMDBrooksAGReadingPC 2008 The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir Res 9 57
73. SmallBADresselSALawrenceCWDrakeDR3rdStolerMH 2001 CD8(+) T cell-mediated injury in vivo progresses in the absence of effector T cells. J Exp Med 194 1835 1846
74. HeroldSSteinmuellerMvon WulffenWCakarovaLPintoR 2008 Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med 205 3065 3077
75. WareingMDLyonAInglisCGiannoniFCharoI 2007 Chemokine regulation of the inflammatory response to a low-dose influenza infection in CCR2−/− mice. J Leukoc Biol 81 793 801
76. DawsonTCBeckMAKuzielWAHendersonFMaedaN 2000 Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am J Pathol 156 1951 1959
77. LinKLSuzukiYNakanoHRamsburgEGunnMD 2008 CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol 180 2562 2572
78. XuLYoonHZhaoMQLiuJRamanaCV 2004 Cutting edge: pulmonary immunopathology mediated by antigen-specific expression of TNF-alpha by antiviral CD8+ T cells. J Immunol 173 721 725
79. PeirisJSCheungCYLeungCYNichollsJM 2009 Innate immune responses to influenza A H5N1: friend or foe? Trends Immunol 30 574 584
80. VerstrepenLVerhelstKvan LooGCarpentierILeySC 2010 Expression, biological activities and mechanisms of action of A20 (TNFAIP3). Biochem Pharmacol 80 2009 2020
81. ParvatiyarKBarberGNHarhajEW 2010 TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem 285 14999 15009
82. GaoLCoopeHGrantSMaALeySC 2011 ABIN1 protein cooperates with TAX1BP1 and A20 proteins to inhibit antiviral signaling. J Biol Chem 286 36592 36602
83. BarikS 2011 Intranasal Delivery of Antiviral siRNA. Methods Mol Biol 721 333 338
84. ClausenBEBurkhardtCReithWRenkawitzRForsterI 1999 Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8 265 277
85. ReedLJMuenchH 1938 A simple method of estimating fifty percent endpoints. Am J Hyg 27 493 497
86. HuffordMMKimTSSunJBracialeTJ 2011 Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J Exp Med 208 167 180
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2012 Číslo 3
- Stillova choroba: vzácné a závažné systémové onemocnění
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- A Foot in the Door for Dermatophyte Research
- An Entomopathogenic Nematode by Any Other Name
- New Insights into spp.: A Potential Link with Irritable Bowel Syndrome
- Short ORF-Dependent Ribosome Shunting Operates in an RNA Picorna-Like Virus and a DNA Pararetrovirus that Cause Rice Tungro Disease