
A Kinome RNAi Screen Identified AMPK as Promoting Poxvirus Entry through the Control of Actin Dynamics
Poxviruses include medically important human pathogens, yet little is known about the specific cellular factors essential for their replication. To identify genes essential for poxvirus infection, we used high-throughput RNA interference to screen the Drosophila kinome for factors required for vaccinia infection. We identified seven genes including the three subunits of AMPK as promoting vaccinia infection. AMPK not only facilitated infection in insect cells, but also in mammalian cells. Moreover, we found that AMPK is required for macropinocytosis, a major endocytic entry pathway for vaccinia. Furthermore, we show that AMPK contributes to other virus-independent actin-dependent processes including lamellipodia formation and wound healing, independent of the known AMPK activators LKB1 and CaMKK. Therefore, AMPK plays a highly conserved role in poxvirus infection and actin dynamics independent of its role as an energy regulator.
Published in the journal:
. PLoS Pathog 6(6): e32767. doi:10.1371/journal.ppat.1000954
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000954
Summary
Poxviruses include medically important human pathogens, yet little is known about the specific cellular factors essential for their replication. To identify genes essential for poxvirus infection, we used high-throughput RNA interference to screen the Drosophila kinome for factors required for vaccinia infection. We identified seven genes including the three subunits of AMPK as promoting vaccinia infection. AMPK not only facilitated infection in insect cells, but also in mammalian cells. Moreover, we found that AMPK is required for macropinocytosis, a major endocytic entry pathway for vaccinia. Furthermore, we show that AMPK contributes to other virus-independent actin-dependent processes including lamellipodia formation and wound healing, independent of the known AMPK activators LKB1 and CaMKK. Therefore, AMPK plays a highly conserved role in poxvirus infection and actin dynamics independent of its role as an energy regulator.
Introduction
In order to successfully infect cells, viruses must remodel the cellular environment to allow for the reallocation of resources to viral production. Poxviruses are large double stranded (ds) DNA viruses that have a sophisticated lifecycle characterized by a number of temporally regulated steps. Vaccinia virus is the prototypical poxvirus, was used as the vaccine to eradicate smallpox, and has been the most thoroughly characterized [1]. To initiate infection, vaccinia first binds, enters cells, uncoats, and expresses early gene products. Next, genomic DNA replication occurs, followed by intermediate and late gene expression. Assembly, maturation, and virus release completes the cycle. Although poxviruses encode a large number of genes (>200), they remain obligate intracellular pathogens and require a multitude of activities hijacked from their host cell. While many viral factors required for various steps in the vaccinia lifecycle have been described, the specific host factor contribution is less clear.
In particular, an early step in the infection cycle involves cell penetration. This step is critical for the initial establishment of infection, and also presents a good target for anti-viral therapeutics [2]. Different families of viruses have developed diverse strategies for entering cells; some fuse at the plasma membrane, while others co-opt one of the many cellular endocytic routes [3]. Studies have demonstrated that macropinocytosis is an important endocytic route of vaccinia entry [4]. Generally, macropinocytosis is a nonselective route for bulk fluid-phase uptake and is not constitutively active, but is induced by growth factors, and also by some pathogens including vaccinia [5] [6]. This active endocytic process induces extensive actin cytoskeletal rearrangement, leading to membrane ruffling, lamellipodia formation, and the internalization of extracellular fluid and membrane. Consistent with this, vaccinia entry is dependent upon modulation of the actin cytoskeleton, and initiates macropinocytosis by inducing dramatic actin-rich microvilli protrusions followed by global myosin II-dependent blebbing, thereby promoting virus uptake [4] [7]. Induction triggers the activation of receptor tyrosine kinases (RTKs) which activate complex signaling cascades leading to the induction of these actin extensions which extend the plasma membrane allowing fluid-phase capture. This process involves signaling cascades that converge on members of the Ras superfamily of GTPases in particular, Rab5 and Rac1 [6] [8]. Rac1 contributes to a number of cellular processes that require extensive actin dynamics, and its signaling is carefully regulated by several guanine exchange factors as well as by crosstalk with other Rho family GTPases [9] [10] [11]. Again, as for growth factor dependent macropinocytosis, vaccinia-induced uptake is dependent upon Rac1 [4] [7]. Additional kinases such as p21-activated kinase (PAK1) are then activated along with actin-associated proteins that lead to large-scale actin rearrangements, lipid modifications, and ultimately macropinosome formation [4] [5]. While some specific kinase families have been implicated in macropinocytosis (e.g., protein kinase C (PKC), serine/threonine kinases, tyrosine kinases, and phosphatidylinositol kinases) [5], many of the specific factors have not been identified, and in some cases the specific role of factors such as PKC, is not well understood. Therefore, there are many additional cellular signaling factors remaining to be identified for this complicated uptake mechanism and thus, for vaccinia entry.
To take an unbiased systematic approach toward the identification of these cellular factors, we developed a system using the model organism Drosophila to perform a high-throughput RNA interference (RNAi) screen for cellular kinases and phosphatases required for early steps in vaccinia infection. The Drosophila system is particularly amenable to this approach for a number of reasons including: reduced redundancy in the genome, high conservation with mammalian systems, efficient RNAi, and previous success with this system to identify cellular factors required for viral infection [12] [13]. This Drosophila system is permissive to early steps in the vaccinia infection cycle allowing us to specifically dissect the role of cellular factors involved in the infectious entry process.
Using this system, we identified seven genes that contribute to vaccinia infection, including the three subunits of the AMP-activated kinase (AMPK) complex, the master energy sensor of the cell. Importantly, the requirement for AMPK in vaccinia infection is conserved in mammalian cells, and is specifically required for vaccinia-induced macropinocytic entry. Further characterization led to the discovery that AMPK controls a variety of virus-independent actin-dependent processes including lamellipodia formation and cell migration. Altogether, we found a new role for AMPK in actin dynamics.
Results
Vaccinia infection in Drosophila cells
Since vaccinia infection of Drosophila cells has not been reported, we first characterized the course of infection in Drosophila cells. Using a reporter virus expressing Beta-galactosidase (B-gal) under the control of an early/late promoter which is active during all stages of vaccinia infection, we found that vaccinia infection is dose-dependent with maximal expression at 48 hours post infection (hpi) (Figure S1). Next, we infected Drosophila cells using reporter viruses that express B-gal under the control of temporally regulated vaccinia promoters that are active during different phases of the virus replication cycle. We found that Drosophila cells were efficiently infected as measured by the production of B-gal from an early/late promoter (p 7.5) or by the production of E3L protein, a vaccinia gene product expressed early in infection, while there was very little expression of B-gal from either an intermediate promoter (G8R), or a late promoter (p11) (Figure 1A). Consistent with these findings, we have been unable to detect vaccinia DNA replication (data not shown) suggesting a block to infection following early protein synthesis. These findings demonstrate that while vaccinia is unable to complete all stages of the lifecycle in Drosophila cells, entry and early expression occur, providing a model system to study the host factor requirements of vaccinia entry.

Previous studies have shown that efficient vaccinia entry is dependent upon the endocytic route of macropinocytosis [4]. In order to assess whether host requirements for vaccinia entry were conserved between Drosophila and mammalian cell lines, we tested whether inhibition of macropinocytosis attenuated infection in these disparate cell types. To this end, we treated cells with several known inhibitors of macropinocytosis and vaccinia entry including; an actin inhibitor Latrunculin A, phosphoinositide-3-kinase (PI3K) inhibitor Wortmannin, Na/H antiporter inhibitor EIPA, and PKC inhibitor Rottlerin [4] [7]. We found that each of these drugs significantly inhibited vaccinia infection in both human and Drosophila cells (Figure 1B–C, quantified in Figure S2). These data show that at least early steps in the viral lifecycle are dependent upon similar pathways in insect cells allowing us to use this model to identify additional factors required for vaccinia infection in mammalian cells.
RNAi screen of Drosophila kinome
In order to systematically probe the requirements for cellular signaling factors in early vaccinia infection, we developed a quantitative assay amenable to RNAi using virally encoded B-gal expression as a measure of early infection. While a non-targeting negative control dsRNA (GFP) had no effect on the percentage of infected cells, knock-down of B-gal by RNAi reduced the percentage of B-gal expressing cells 17-fold, indicating that vaccinia infection can be quantitatively assayed using this system (Figure 2A). Moreover, dsRNA targeting the cellular gene Rab5, a small GTPase required for many endocytic processes including macropinocytosis [8] also significantly decreased vaccinia infection, validating that we can identify cell-encoded factors required for vaccinia infection using this approach (Figure 2A and 2D).

We used this assay to perform an RNAi screen against the Drosophila kinome to identify novel signaling factors that promote vaccinia infection (Schematic diagram Figure 2B). This screen consisted of approximately 440 unique genes (∼200 kinases, ∼90 phosphatases, and ∼150 regulator factors) arrayed onto 384 well plates (Table S1). Additionally, negative control wells were included containing either no dsRNA (15 wells) or dsRNA targeting GFP (28 wells), which is not expressed in this system. Lastly, 21 positive control wells with dsRNA targeting lacZ were included (Figure 2B light blue). Drosophila cells were seeded in these pre-arrayed 384 well plates, incubated for three days to allow knock down of each targeted gene, and then infected with vaccinia virus for 48 hours. For the screen, a baseline infection of 10% was within the linear range and was achieved at a multiplicity of infection (MOI) of 1.25 (Figure S1C). The plates were fixed and processed for immunofluorescence using B-gal expression as a measure of infection, and counter-stained to monitor cell number. Automated microscopy and image analysis were used to quantify the percent infection (B-gal+/Total Nuclei) that was transformed into Robust Z scores for each plate, and the Robust Z scores of the 2 replicates were plotted against each other (Figure 2B). Positive candidates were defined as having a Robust Z score of <−2 in duplicate screens (p<0.05). Using these metrics, we identified 8 genes (2%, orange and pink Figure 2B). In addition to these 8 factors, we identified 20 out of 21 (95%) of the positive control lacZ dsRNAs (Figure 2B light blue and Table S1) and none of the 43 negative controls (non-targeting dsRNA and empty wells). We also monitored the toxicity of the dsRNA treatments and found that none of the 8 genes that inhibited infection were cytotoxic (<25% decrease in cell number, Table 1). In contrast, we found that while 17 wells reduced cell number by >25% in duplicate screens, none of these genes also inhibited infection. Therefore, our screen revealed host factors required for robust infection that are not required for cell viability. Notably all 8 genes have human homologs (Table 1), including all 3 subunits of the AMP-activated kinase (AMPK) complex (SNF1A (AMPKα), SNF4Agamma (AMPKγ) and alicorn (AMPKβ)) (Figure 2B pink), a heterotrimeric complex involved in maintaining cellular energy homeostasis. RNAi resulted in ∼3-fold reduction in vaccinia infection when AMPK was depleted (Figure 2C).

Non-overlapping secondary dsRNAs were generated for each of the 8 genes and were tested to confirm the role of each of these genes in vaccinia infection. Seven of the genes validated (88%). Importantly, among the genes that validated were those encoding the three subunits of the AMPK complex (Figure 2D, Figure S3 and Table 1). Moreover, RT-PCR confirmed that AMPKα was depleted by dsRNA treatment against AMPKα (Figure S4A). Furthermore, we found that loss of AMPKα or AMPKγ also led to a defect in early viral mRNA accumulation compared to control (Figure 2E), suggesting that AMPK is required upstream of viral mRNA production in Drosophila, perhaps at the stage of entry.
AMPK promotes vaccinia infection in mammalian cells
AMPK is an important sensor of intracellular energy that is conserved in eukaryotes ranging from yeast to humans [14]. While Drosophila encodes only one copy of each AMPK subunit, mammals have multiple isoforms of each subunit encoded by several distinct genes (α1, α2, β1, β2, γ1, γ2, γ3) which can produce at least 12 possible heterotrimeric combinations [15]. The lack of redundancy in Drosophila allowed our identification of AMPK by single gene RNAi. We used this Drosophila system as a tool to identify novel host factors that contribute to vaccinia infection, but since Drosophila is not a natural host, we were interested in determining the role of AMPK in a more biologically relevant context.
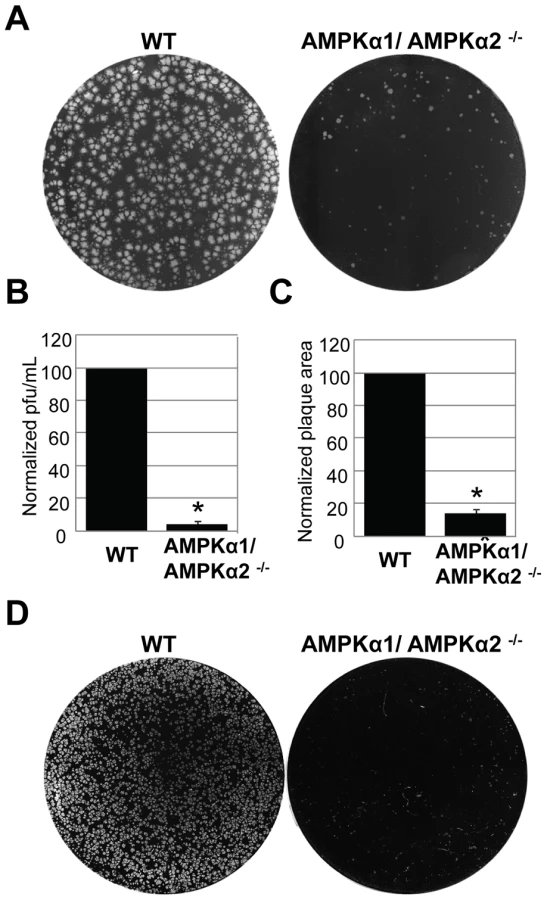
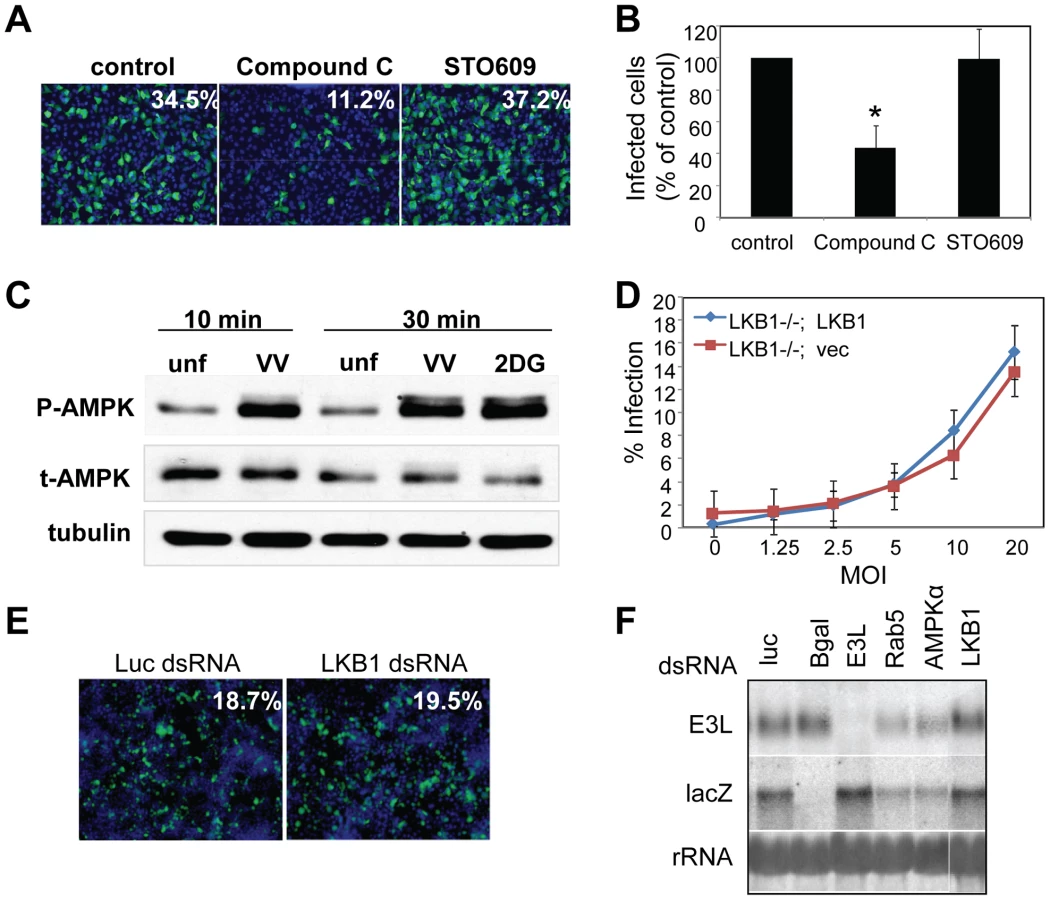
To investigate the role of AMPK in vaccinia virus infection of mammalian cells, we took advantage of mouse embryonic fibroblasts (MEFs) that are genetically altered and null for both AMPKα subunits, AMPKα1 and AMPKα2 (AMPKα1/AMPKα2 −/−) and verified the lack of these proteins by immunoblot analyses (Figure S5) [16] [17] [18]. These cell lines divide and grow indistinguishably from their sibling control AMPKα1/AMPKα2 +/+ cells (wild type) (data not shown). We challenged either the AMPKα1/AMPKα2−/− cells or their sibling control wild type cells with vaccinia virus and measured infection using a plaque assay. This revealed a 20-fold decrease in plaque number and a 15-fold decrease in plaque area in AMPKα1/AMPKα2 −/− compared to wild type cells (Figure 3A–C). The requirement for AMPK was also observed for the closely related poxvirus, cowpox virus (Figure 3D, quantified in Figure S6). These decreases in infectivity were specific for poxviruses and not simply due to a decrease in overall cell health since several unrelated RNA viruses, including Vesicular Stomatitis virus (VSV) grew as well in the AMPK deficient MEFs compared to wild type (Figure S7 and data not shown). This suggests that AMPK deficient cells are capable of supporting all stages of virus infection; including at least some forms of endocytosis and endosomal trafficking since VSV enters cells through clathrin-mediated endocytosis [19]. Therefore, we identified a specific requirement for AMPK in poxvirus infection but not for viral infection generally. In addition to plaque assays we also monitored vaccinia infection by immunofluorescence, immunoblot, and Northern blot and found that there was a significant decrease in vaccinia virus replication in AMPKα1/AMPKα2 −/− cells in each assay (Figure S8A–C). This suggests that AMPK promotes early steps of the vaccinia lifecycle in mammalian cells as well as Drosophila. Finally, to verify that the requirement for AMPK was not MEF-specific we tested whether inhibition of AMPK in the human osteosarcoma cell line U2OS attenuated vaccinia infection using two approaches. First, we pre-treated U2OS cells with the AMPK inhibitor Compound C or vehicle and challenged these cells with vaccinia [20]. Again, we found that inhibition of AMPK attenuated infection (Figure 4A, B). Next, we depleted AMPKα1, AMPKα2, or both AMPKα1 and AMPKα2 using siRNAs and observed a decrease in vaccinia infection (Figure S9A, B, Text S1). We confirmed knock-down by immunoblot (Figure S9C). Together, these data show that vaccinia infection is dependent upon AMPK for infection across disparate cell types including Drosophila, human and mouse.
Vaccinia infection activates AMPK
AMPK is activated through phosphorylation of a threonine residue on the catalytic α subunit, which can be triggered by a variety of stimuli including an increase in the cellular ratio of AMP/ATP [21] [22] [23] [24] [25] [26]. Since AMPK promotes vaccinia infection, we tested whether infection activates AMPK. We used a phospho-specific antibody against AMPKα to measure AMPK activation. Treatment with 2-deoxyglucose (2DG), a known activator of AMPK, led to an increase in AMPK phosphorylation, while little phosphorylation was detected in untreated controls (Figure 4C). Moreover, we observed an increase in phospho-AMPKα within 10 minutes of vaccinia infection (Figure 4C). This increase was not due to changes in total AMPK levels, and suggests that AMPK becomes activated very early in vaccinia infection.
Known upstream activators of AMPK are not required for vaccinia infection
Several upstream kinases have been implicated in AMPK activation under different conditions. The classic activator of AMPK is the tumor suppressor LKB1, which activates AMPK in response to energy deprivation [27] [28]. In Drosophila, LKB1 is the only described upstream kinase required for AMPK activation and lkb1 mutants phenocopy ampk mutants [29] [30]. In contrast, in mammalian systems, LKB1 is the upstream kinase in response to energy deprivation, while additional upstream kinases, such as calcium/calmodulin-dependent protein kinase kinase beta (CaMKKβ) have been implicated in AMPK activation under other conditions [31] [32]. We tested whether LKB1 was required for vaccinia infection using cells that are null for LKB1 [33] and complemented with either vector alone (LKB1−/−; Vec), or an LKB1 cDNA (LKB1−/−; LKB1) (Figure S10) and found that loss of LKB1 had no effect on vaccinia infection in mammalian cells (Figure 4D). Likewise, using RNAi to deplete LKB1 in Drosophila cells, we found that it was dispensable for infection by immunofluorescence (Figure 4E) and Northern blot (Figure 4F). RT-PCR analysis validated that LKB1 was indeed knocked down in Drosophila cells (Figure S4B). We also tested whether CaMKK, the other well-established AMPK activator in mammalian systems, was required for vaccinia infection. To this end, we pre-treated U2OS cells with the CaMKK inhibitor STO609 prior to infection, and found no effect on vaccinia infection with doses up to 5 µg/ml (Figure 4A, B). Taken together, these data show that vaccinia infection is AMPK-dependent but LKB1 and CaMKK-independent.
AMPK promotes vaccinia entry
Given that loss of AMPK led to a decrease in both viral mRNA and protein production (Figure 2, Figure S8), we tested whether AMPK was required for efficient virus entry. We monitored viral entry into wild type or AMPKα1/AMPKα2 −/− MEFs using a fluorescence-based assay. We prebound virus to the cells, and then allowed infection to proceed for one hour. Incoming virus particles were visualized using an antibody against L1R, a membrane-bound viral surface protein (Figure 5A). Deconvolution of Z stacks was used to visualize vaccinia inside of cells (Figure 5A, XZ view). Quantification revealed a ∼3-fold reduction in the number of AMPK mutant cells that internalized virus (Figure 5B). These data show that AMPK promotes infection at the stage of entry, although we have not ruled out that virus binding could also be affected by lack of AMPK.

We were also interested in whether AMPK played a role in vaccinia infection downstream of entry. First, we monitored both early and late gene expression and found that while the percentage of cells expressing an early protein (E3L) or a late protein (L1R) was reduced in AMPKα1/AMPKα2 −/− MEFs compared to wild type, 100% of the cells that expressed early genes also expressed late genes, indicating no further block to replication (Figure S11A). We also measured the infectivity of virus produced from AMPKα1/AMPKα2 −/− MEFs. We found a ∼3-fold decrease in virus titer produced from AMPKα1/AMPKα2 −/− MEFs compared to wild type (Figure S11B) which is similar to the decrease in viral entry (Figure 5B). These data suggest that while fewer AMPK deficient cells produce virus, the vaccinia released from these cells is as infectious as virus produced from wild type cells. Therefore, while AMPK is important for entry, it is dispensable for later steps in the viral lifecycle.
AMPK is required for vaccinia induced macropinocytosis
Previous studies established that a major entry route for vaccinia is macropinocytosis, which is required for and induced by vaccinia infection [4]. Since we observed a defect in viral entry in the AMPK mutant cells (Figure 5), and that inhibitors of macropinocytosis attenuated vaccinia infection (Figure 1), we tested whether AMPK plays a role in vaccinia-induced macropinocytosis. Macropinocytosis can be directly monitored by fluorescently labeled dextran uptake [34]. Neither wild type nor AMPKα1/AMPKα2 −/− MEFs efficiently endocytosed dextran under resting conditions (Figure 6A). However, upon vaccinia infection, macropinocytosis was dramatically induced in wild type MEFs as measured by an increase in dextran uptake into the cell. In contrast to the large number of dextran punctae observed in the infected wild type MEFs, AMPKα1/AMPKα2 −/− MEFs did not efficiently take up the dextran in the presence of virus (Figure 6A). We quantified the level of vaccinia-induced macropinocytosis in the wild type versus AMPK deficient cells and found an approximately five-fold reduction in the percentage of cells undergoing macropinocytosis (Figure 6B).
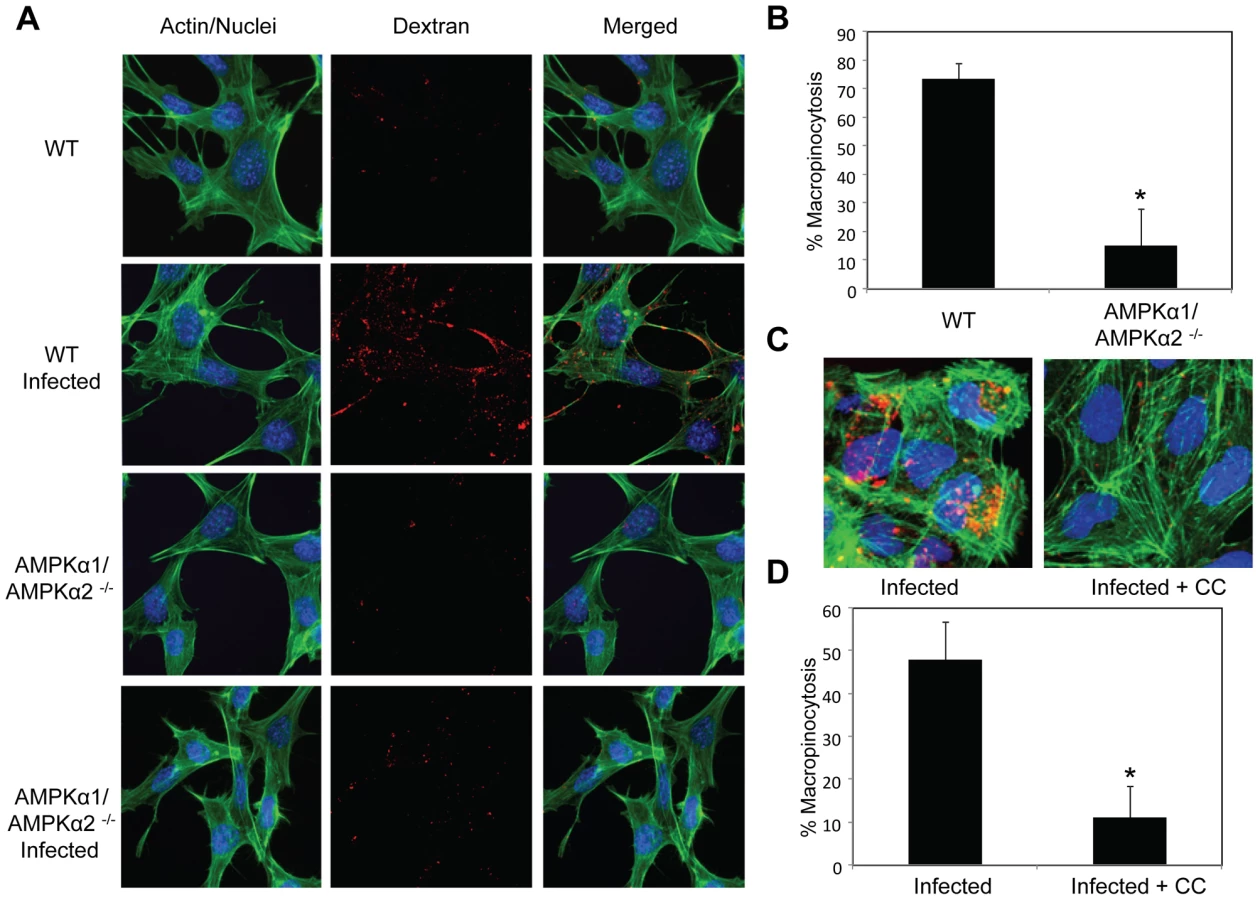
Furthermore, we found a decrease in vaccinia-induced dextran uptake in human U2OS cells pretreated with Compound C compared to vehicle control (Figure 6C). Quantification revealed an approximately five-fold decrease in the percentage of U2OS cells undergoing vaccinia-induced macropinocytosis when AMPK is inhibited (Figure 6D). Together, these data show that virus-induced macropinocytosis is dependent upon AMPK in disparate cell types and hosts.
In contrast to this dependence of macropinocytosis on AMPK, there was no defect in transferrin uptake in AMPKα1/AMPKα2 −/− MEFs (Figure S12, Text S1), indicating that receptor-mediated endocytosis is not controlled by AMPK. This is consistent with our observation that VSV infection is not attenuated in AMPKα1/AMPKα2 −/− MEFs as VSV enters cells by receptor-mediated endocytosis and not macropinocytosis (Figure S7).
AMPK promotes lamellipodia formation
One of the early steps in macropinocytosis involves extensive actin remodeling characterized by membrane ruffling and lamellipodia formation. We were interested in determining whether AMPK was required for this early step in the macropinocytic pathway, and whether the requirement for AMPK in macropinocytosis was vaccinia-dependent or if AMPK was required more generally for actin remodeling. Therefore, to test whether AMPK controlled actin-dependent remodeling independent of viral infection, we treated cells with phorbol myristic acid (PMA), which induces cells to undergo high levels of actin-mediated ruffling and lamellipodia formation; this is dependent on Rac1, a small Rho family GTPase that is also required for macropinocytosis and vaccinia infection [7] [35] [36] [37]. Dramatic lamellipodia formation, seen as thick bands of actin at the cell periphery, were observed in wild type MEFs stimulated with PMA, but abrogated in AMPK-deficient cells (Figure 7A, arrows). We also monitored PMA-induced ruffling using live cell imaging and observed a significant defect in the AMPK mutant MEFs (Videos S1, S2, Text S1).

In addition, we monitored the localization of Rac1 during PMA-induced lamellipodia formation in the wild type and AMPK deficient cells and found that both Rac1 re-localization and actin mobilization are defective in AMPK deficient cells (Figure 7B) suggesting that the defect is upstream of or parallel to Rac1 activation.
While our studies identified AMPK as a critical kinase required for vaccinia infection and actin dynamics, we found that LKB1 was dispensable for infection. This led us to test whether actin-dependent lamellipodia formation and ruffling was also LKB1-independent. We found that there was no defect in PMA-induced lamellipodia and ruffling in LKB1-deficient cells (Figure 7C, arrows). This was not unexpected since HeLa cells, a cell type routinely used for studies on actin dynamics, are mutant for LKB1, and can still undergo lamellipodia formation, and macropinocytosis [4] [38] [39] [40]. Therefore, we found that the actin remodeling activity of AMPK is independent of LKB1.
AMPK promotes cellular motility
Since extensive actin remodeling and Rac1 membrane localization are required for lamellipodia formation, macropinocytosis, and vaccinia infection [7] [35] [36] [37], we tested whether AMPK was required for another Rac1-dependent actin-dependent process namely in vitro wound healing [41]. For these studies, we created wounds in a confluent monolayer of either wild type or AMPKα1/AMPKα2−/− MEFs by scratching the surface, and monitored wound closure over time. Using this assay we found that there was a significant delay in the migration of AMPK-deficient MEFs into the wound compared to wild type cells (Figure 7D, E). While the wound was completely healed 24 hours after wounding in wild type cells, a sizable gap in the monolayer was still present in AMPK deficient cells, indicating a delay in wound healing and reduced motility. During this motile state, cells undergo a dramatic change in shape, with lamellipodia forming at the leading edge directing cell migration to close the wound. We observed the formation of lamellipodia in the WT MEFs at the edge of the wound while AMPK mutant MEFs did not polarize (Figure S13). Consistent with our observations that the role for AMPK in actin dynamics is LKB1-independent, we found that wound healing is unaffected by the loss of LKB1 (Figure S14).
Discussion
Cell penetration is a critically important step in viral infection, and one that is generally driven by cellular factors and signaling pathways. First, viruses must attach to the cell surface and bind to the viral entry receptor, which often initiates signaling within the cell. Next, viruses must fuse or penetrate the cellular membrane either at the cell surface, or in many cases, within intracellular compartments by taking advantage of the endogenous endocytic machinery. Since there is an array of different endocytic mechanisms, there is great diversity in the strategies used by viruses for entry. Therefore, studying virus entry has increased our understanding not only of viral infection, but also of the normal cellular processes of endocytosis [6].
To further dissect the cellular signaling requirements of vaccinia entry, we developed an unbiased loss-of-function screening platform in Drosophila cells, and identified seven cellular factors required for vaccinia infection. Amongst the genes were all three subunits of AMPK, implicating the entire complex in vaccinia infection. Further studies showed that in addition to its role in Drosophila, AMPK is also required in murine and human cells for poxvirus infection at the stage of entry. Moreover, AMPK becomes rapidly activated upon infection with vaccinia, suggesting that virus-induced signals converge on this complex to facilitate entry.
Studies indicate that vaccinia virus can enter cells through multiple routes via as of yet unidentified receptor(s). Studies suggest that virus particles can fuse either at the plasma membrane or from within endosomal compartments, dependent on cell type and virus strain [42] [43] [44] [45] [46]. Importantly, a major endocytic pathway for vaccinia entry has recently been described as macropinocytosis [4] [47]. Our data as well as previous reports support the idea that vaccinia uses macropinocytosis for entry, but not exclusively. We and others have shown partial inhibition of vaccinia infection using a variety of drugs that are well-established inhibitors of macropinocytosis [4] [7]. However, in no case was vaccinia entry completely dependent upon macropinocytosis for infection, demonstrating that the virus can use alternative routes for entry. Nevertheless, macropinocytosis is required for efficient entry across broad cell types suggesting that inhibition of this pathway may attenuate infection sufficiently to allow for immune-mediated clearance of the infection.
The process of macropinocytosis drives non-specific uptake of extracellular fluid, large portions of the plasma membrane, as well as large particles. Macropinosomes, unlike coated vesicles, are morphologically heterogeneous, and can vary greatly in size from 0.2–10 µm in diameter, sufficient to accommodate the large size of vaccinia virus particles (∼0.3 µm) [5] [48]. Classic induction of macropinocytosis by growth factor receptor signaling stimulates ruffles, or sheet-like extensions of the plasma membrane, formed by assembly of actin filaments beneath the plasma membrane that form cups that contract and close to form macropinosomes [5]. This process is driven by signaling events initiated at the plasma membrane and are thought to involve a number of kinase families including phosphatidylinositol 5-kinases (PI5K), PI3K, PKC, serine/threonine kinases, and receptor tyrosine kinases. In addition, as many different inducers of macropinocytosis have been identified, there are likely multiple pathways that converge on the activation of macropinocytosis, adding to the complexity of this cell biological pathway [5]. While several specific kinases, such as PAK1, and LIM kinase have well described roles in macropinocytosis [49] [50], there is much that remains unclear. Here, we have found a role for an additional kinase, AMPK, in promoting vaccinia entry through its role in macropinocytosis. We have found that AMPK deficiency attenuates vaccinia infection, concomitant with reduced entry and fluid-phase uptake, supporting an important role for AMPK in vaccinia-induced macropinocytic entry.
The process of macropinocytosis involves several steps including extensive actin-mediated membrane ruffling, cup formation, and finally cup closure, which requires the fusion of plasma membranes to close off the macropinosome, followed by fission to separate the macropinosome from the plasma membrane [5]. We discovered that AMPK is required for the formation of lamellipodia and affects the recruitment of Rac1 to the cell periphery, suggesting that the role of AMPK in macropinocytosis lies in the initial rearrangement and reorganization of the actin cytoskeleton.
While we have shown that AMPK contributes to actin remodeling during vaccinia-induced macropinocytosis, we also found that AMPK plays a role in other virus-independent remodeling processes including cell migration. During this process, forward movement is driven by the extension of a leading edge protrusion or lamellipodium, followed by contraction at the rear. This protrusive force is generated by localized polymerization of actin mediated by Rac1 [51]. In addition to its role in controlling ruffling upstream of macropinocytosis, we found AMPK also has an essential role in cellular motility and wound healing, demonstrating a broad role in Rac1-dependent actin modulation. Previously, Rac1 has been implicated in nitric oxide production and glucose uptake downstream of AMPK [52] [53]. This role for AMPK and Rac1 in glucose uptake via the translocation of the major insulin-responsive glucose transporter GLUT-4 is quite interesting because this may provide a direct link between AMPK's role in energy homeostasis and the cytoskeleton [54] [55] [56].
While the best understood role of AMPK is its role in metabolism, recent evidence suggests this kinase also has a crucial role in regulating cell structure and polarity through engagement with the actin cytoskeleton. In Drosophila, loss of AMPK leads to defects in mitotic division and epithelial cell polarity accompanied by disruption of the actin cytoskeleton [29]. In some mammalian epithelial cell lines, AMPK activation leads to polarization characterized by the formation of an actin brush-border or tight junction assembly [29] [57] [58]. Additionally, AMPK activation can induce astrocyte stellation, and actin stress fiber disassembly [59]. Finally, studies using Compound C and AMPK activators linked AMPK to macropinocytic uptake of albumin in murine macrophages [60]. Taken together with our new data, this accumulating evidence suggests a broad and conserved role for AMPK in a variety of cellular processes that require actin cytoskeletal rearrangements.
The precise signaling events that lead to these AMPK-dependent cytoskeletal changes remain unclear. Several upstream kinases have been shown to activate AMPK under different stimuli. The best studied of these is the tumor suppressor LKB1 which activates AMPK in response to changes in cellular energy levels [27] [28]. Additionally, AMPK can be activated in response to changes in intracellular calcium levels by CaMKKβ, and further evidence suggests that TGFβ-activating kinase (TAK1) may serve as a third upstream activator [31] [32] [61]. Previous studies demonstrated that LKB1 is an important mediator of cell polarity at least in part through signaling to AMPK, and has been shown to drive actin brush border formation, and the translocation of apical and basal markers during the establishment of polarity [29] [30] [57] [58] [62]. We found that at least some AMPK-dependent cytoskeletal changes are independent of LKB1 and CaMKK including lamellipodia formation, macropinocytosis and wound healing. These different actin-dependent outcomes could be controlled by the unique downstream Rho GTPase family members that may become activated by AMPK depending on the stimulus and upstream kinase (such as Rac1, which is associated with lamellipodia formation and macropinocytosis) [10] [11]. Study of the Rho family GTPases activated by AMPK under different stimuli may resolve some of these issues.
In addition to the three subunits of AMPK discovered through screening the kinome, we identified four other genes that promote vaccinia infection: Pi3K68D, Fab1, Stam, and CG9311, all of which have human homologs. Pi3K68D, Fab1, and Stam are kinases while CG9311 is the only phosphatase identified in the screen. As kinases are druggable targets, and many known factors involved in macropinocytosis are kinases, we are particularly interested in the role of these kinases in vaccinia infection. Both Pi3K68D (PIK2C2A) and Fab1 (PIP5K3/PIKfyve) have roles in metabolism of phosphatidylinositol (PtdIns), an important component of membrane trafficking, cytoskeletal rearrangements, and macropinocytosis. In particular, Pi3K68D is a member of the class II family of PI3Ks that produce PtdIns(3)P downstream of growth factor stimulation, and can modulate the activity of Rho GTPases such as Rac1 and Cdc42. Class II PI3Ks have a critical role in lamellipodia formation and in cell migration, localizing to the leading edge of migrating cells [63] [64]. Interestingly, class I PI3K have also been implicated in vaccinia infection, during virus entry, and also later stages of infection [4] [65] [66]. Fab1, the PtdIns(3)P 5-kinase that converts PtdIns(3)P into PtdIns(3,5)P2, has been implicated in fluid-phase uptake, transport, and endosomal acidification [67] [68]. The third kinase, Stam (STAM, STAM2) is activated by cytokine and growth factor stimulation, and localizes to early endosomes, where it is involved in endosomal sorting [69] [70]. Since these kinases have roles either in trafficking to or from the plasma membrane, or in cytoskeletal rearrangements, and have been implicated in processes related to macropinocytosis, it is quite possible that they also play a direct role vaccinia entry. Perhaps Pi3K68D is involved in promoting cytoskeletal rearrangements that lead to macropinocytosis, while Fab1 and Stam could be involved sequentially in later entry steps leading to membrane fusion once a macropinosome has been internalized.
Rearrangements in the actin cytoskeleton are crucial not only for vaccinia infection, but also for many essential cellular processes including: cell division, establishment and maintenance of polarity, cellular motility, and uptake of extracellular fluids, each of which must be carefully regulated. While these various processes have different outcomes for the cell, they share several important signaling components, with AMPK as a central mediator. Further characterization of AMPK as well as these additional new factors is required to determine their precise role in vaccinia infection and whether they interact with AMPK, macropinocytosis, or other actin-dependent processes. How AMPK activation in response to different signals leads to these disparate changes in the actin cytoskeleton, and how these processes fit into the larger network of AMPK-dependent pathways will drive future studies. Lastly, the development of more selective AMPK inhibitors or other inhibitors of macropinocytosis may be useful against poxviruses, and other viruses that hijack this endocytic route for their entry mechanism.
Materials and Methods
Cells, antibodies, reagents, and viruses
Drosophila DL1 cells were grown and maintained at 25°C in Schneiders Drosophila media supplemented with 10% FBS (JRH) as described [71]. MEFs, BSC-1 and U2OS cells were maintained at 37°C in DMEM supplemented with 10% FBS (Sigma) and 10 mM Hepes. HeLa S3 suspension cells were maintained in MEM supplemented with 10% FBS and 0.05% Pluronic. BSC-1 cells were maintained in MEM supplemented with 10% cosmic calf serum (Hyclone). All media were additionally supplemented with 100 µg/ml penicillin/streptomycin and 2 mM L-glutamine. LKB1−/− MEFs were complemented with MIGR (Vector) or FLAG-LKB1-MIGR (LKB1 cDNA) and maintained as above. Vaccinia strains vPRA13, vSC8, and vP30CP77, were grown in HeLa S3 suspension cells supplemented with 2.5% FBS, and tittered on BSC-1 cells as described [72] [73] [74] [75]. Cowpox Brighton Red and Vesicular Stomatitis virus (Indiana) were used. Antibodies were obtained from the following sources: anti-Bgal (Promega and Cappel), anti-E3L (gift from S. Isaacs) [76], anti-L1R (R180 gift from G. Cohen and R. Eisenberg), anti-Rac1 (Millipore),and anti-AMPK (Cell Signaling Technology). Fluorescently labeled secondary antibodies along with anti-sheep HRP were obtained from Jackson Immunochemicals or Invitrogen. All other HRP-conjugated antibodies were obtained from Amersham. AlexaFluor 488 and 594 phalloidin, FITC-conjugated dextran, and 594-conjugated Transferrin were purchased from Invitrogen. Compound C [20] and STO-609 [77] were obtained from Calbiochem. Additional chemicals were obtained from Sigma.
RNAi and infections
A mini library of dsRNAs generated against Drosophila kinase and phosphatase genes was obtained from N. Perrimon, and aliquoted onto 384 well plates at 250 ng dsRNA/384 well [78]. Secondary amplicons and control dsRNA were designed using SnapDragon and DRSC resources (www.flyrnai.org), and generated as described [79]. For 384 well assays, 16,000 DL1 cells were seeded onto 250 ng dsRNA in 10 µl serum free media. One hour later 20 µl complete media was added, and cells were incubated in a humid chamber for 3 days. For other experiments, 2,000,000 cells were seeded onto 4 µg of dsRNA/6 well in 1 mL serum free media. One hour later 2 mL complete media was added. For viral infections, vaccinia was tittered on BSC-1 cells, and MOIs added to all cell types are based on pfu/ml measured on BSC-1 cells. Media was removed and virus was added in 2% serum medium and incubated at 25°C for Drosophila cells, and 37°C for mammalian cells. Viral innocula used was adjusted to achieve ∼10% infection of Drosophila cells in the primary screen, and ∼20% infection in secondary analysis. Level of infection of mammalian cells varied depending on the assay format. Cells were processed at the indicated time point post infection.
Viral immunofluorescence
Cells were fixed and processed for immunofluorescence as previously described at 48 hours post infection for Drosophila cells and 8 hours post infection in mammalian cells [12]. Briefly, cells were fixed in 4% formaldehyde/phosphate buffered saline (PBS), washed twice in PBS/0.1% TritonX-100 (PBST), and blocked in 2% BSA/PBST. Anti-E3L and anti-B-gal primary antibodies were diluted in block, added to cells, and incubated overnight at 4°C. Cells were washed three times in PBST, and incubated in secondary antibody for one hour at room temperature. Cells were counterstained with Hoescht33342 (Sigma). Plates were imaged at 20X for Drosophila cells and 10X for mammalian cells, capturing three images per well per wavelength using an automated microscope (ImageXpress Micro), and quantification was performed using MetaXpress image analysis software. Significance was determined using a Student T-test.
Screen analysis
Image analysis was used to generate metrics from the captured images including the number of nuclei and the number of infected cells per site. The percent infection was calculated for each site, log-transformed, and the interquartile range (IQR) was used to calculate a robust Z score for each site using the following equation: log10 [(%infection-median)/(IQR*0.74)] [80]. Candidates were identified as positive if the average robust Z score of all sites in a well was <−2 in two independent replicates.
Immunoblotting, Northern blotting, and RT-PCR
For protein analysis, MEFs were prechilled to 16°C for 10 minutes and then treated with vaccinia (MOI 20) for 1 hour at 16°C to synchronize infection. Cells were incubated at 37°C for 10 or 30 minutes or treated with 10 µM 2DG for 30 minutes. Cells were then washed briefly in cold PBS and lysed in NP40 lysis buffer supplemented with protease (Boehringer) and phosphatase (Sigma) inhibitor cocktails. Samples were separated by SDS-PAGE and blotted as described [81]. HRP-conjugated secondary antibodies and Western Lightening Chemiluminescence Reagent were used for visualization.
For RNA analysis, cells were lysed in Trizol buffer, and RNA was purified and blotted as previously described with the indicated probes [12]. RT-PCR was performed using M-MLV reverse transcriptase on random primed total RNA (Invitrogen). One µL of the cDNA or a 1∶10 dilution was used for PCR amplification.
Plaque assays
Viruses were plaqued on MEF or BSC-1 cells as indicated. Confluent monolayers were treated with serial dilutions of virus for two hours, after which the cells were overlayed with agarose followed by crystal violet staining. Plaque number was determined manually, and plaque diameter was measured using MetaXpress software and used to calculate areas.
Vaccinia entry assay
MEFs plated on cover slips were chilled to 16°C for 10 minutes and then treated with vaccinia (MOI 100) at 16°C for 1 hour. Unbound virus was removed, and cells were incubated at 37°C for 1 hour, washed three times in cold PBS, and fixed. Cells were washed in ammonium chloride (50 mM) and PBST and were stained with anti-L1R and then washed and incubated with secondary antibody, Hoescht 33342, and phalloidin 488. Cover slips were mounted and imaged using a 63X objective with a Leica DMI 4000 B fluorescent microscope. Images were taken as 0.2 um Z-stacks that were deconvolved using AutoQuant X2 software using Adaptive PSF with 20 iterations. Images are displayed as max projections. To quantify, images were randomized and blindly quantified for virus entry (n>30 for each condition).
Fluid-phase dextran uptake assay
MEFs grown on glass cover slips were chilled to 16°C for 10 minutes and then treated with vaccinia (MOI 200) at 16°C for 1 hour. Unbound virus was removed, and FITC-dextran (70 kD, lysine fixable) was added at 0.5 mg/ml. Cells were incubated at 37°C for 20 minutes, washed twice in PBS, and once in pH 5.5 buffer (0.1 M sodium acetate, 0.05 M NaCl) for 5 minutes. Cells were fixed and stained with Hoescht 33342 and phalloidin 594. Cover slips were mounted and imaged using a 63X objective with a Leica DMI 4000 B fluorescent microscope. Images were randomized and blindly quantified for the percentage of cells undergoing macropinocytosis as defined by >20 punctae per cell. U2OS cells grown on glass cover slips were pretreated with 10 µM Compound C or vehicle for 1 hour and then assayed as above.
Actin ruffling assay
Cells were grown on glass cover slips and treated with vehicle or 1 µM PMA for 3 hours. For Rac1 localization experiments, cells were blocked in 8% BSA/PBST for 1 hour. Anti-Rac1 (Millipore) was added in 1% BSA/PBST overnight at 4°C. Cells were washed 3 times in PBST, and secondary antibodies were added for 1 hour at room temperature. For all experiments, cells were stained with Hoescht 33342 and phalloidin 488. Cover slips were mounted and imaged using a 63X objective with a Leica DMI 4000 B fluorescent microscope.
Transferrin uptake assay
Cells grown on glass cover slips were chilled to 16°C for 10 minutes and then treated with vaccinia (MOI 100) at 16°C for 1 hour. Unbound virus was removed, and 594-transferrin was added at 20 µg/ml. Cells were incubated at 37°C for 20 minutes, washed twice in PBS, and once in 0.1 M sodium acetate, 0.05 M NaCl, pH 5.5 buffer for 5 minutes. Cells were fixed and stained with Hoescht 33342 and phalloidin 488. Cover slips were mounted and imaged using a 63X objective with a Leica DMI 4000 B fluorescent microscope.
Wound healing assay
Cells were grown to 100% confluence overnight, then scratched with a pipet tip to wound. Several marks were made along the length of the wound, and were imaged over time, using these marks as guides. Images were analyzed for wound length at the same position over time using MetaXpress software.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MossB
2001 Poxviridae: The viruses and their replication.
HowleyDMKaPM
Fields Virology Philadelphia Lippincott, Williams, and Wilkins 2849 2884
2. QianK
Morris-NatschkeSL
LeeKH
2009 HIV entry inhibitors and their potential in HIV therapy. Med Res Rev 29 369 393
3. MarshM
HeleniusA
2006 Virus entry: open sesame. Cell 124 729 740
4. MercerJ
HeleniusA
2008 Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320 531 535
5. SwansonJA
2008 Shaping cups into phagosomes and macropinosomes. Nat Rev Mol Cell Biol 9 639 649
6. MercerJ
HeleniusA
2009 Virus entry by macropinocytosis. Nat Cell Biol 11 510 520
7. LockerJK
KuehnA
SchleichS
RutterG
HohenbergH
2000 Entry of the two infectious forms of vaccinia virus at the plasma membane is signaling-dependent for the IMV but not the EEV. Mol Biol Cell 11 2497 2511
8. LanzettiL
PalamidessiA
ArecesL
ScitaG
Di FiorePP
2004 Rab5 is a signalling GTPase involved in actin remodelling by receptor tyrosine kinases. Nature 429 309 314
9. BosJL
RehmannH
WittinghoferA
2007 GEFs and GAPs: critical elements in the control of small G proteins. Cell 129 865 877
10. HeasmanSJ
RidleyAJ
2008 Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9 690 701
11. JaffeAB
HallA
2005 Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21 247 269
12. CherryS
DoukasT
ArmknechtS
WhelanS
WangH
2005 Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev 19 445 452
13. HaoL
SakuraiA
WatanabeT
SorensenE
NidomCA
2008 Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454 890 893
14. HardieDG
ScottJW
PanDA
HudsonER
2003 Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 546 113 120
15. KahnBB
AlquierT
CarlingD
HardieDG
2005 AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1 15 25
16. JorgensenSB
ViolletB
AndreelliF
FrosigC
BirkJB
2004 Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 279 1070 1079
17. LaderouteKR
AminK
CalaoaganJM
KnappM
LeT
2006 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol 26 5336 5347
18. ViolletB
AndreelliF
JorgensenSB
PerrinC
GeloenA
2003 The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest 111 91 98
19. SunX
YauVK
BriggsBJ
WhittakerGR
2005 Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 338 53 60
20. ZhouG
MyersR
LiY
ChenY
ShenX
2001 Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108 1167 1174
21. SteinSC
WoodsA
JonesNA
DavisonMD
CarlingD
2000 The regulation of AMP-activated protein kinase by phosphorylation. Biochem J 345 Pt 3 437 443
22. DaviesSP
HelpsNR
CohenPT
HardieDG
1995 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett 377 421 425
23. HawleySA
SelbertMA
GoldsteinEG
EdelmanAM
CarlingD
1995 5′-AMP activates the AMP-activated protein kinase cascade, and Ca2+/calmodulin activates the calmodulin-dependent protein kinase I cascade, via three independent mechanisms. J Biol Chem 270 27186 27191
24. HawleySA
DavisonM
WoodsA
DaviesSP
BeriRK
1996 Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271 27879 27887
25. PanDA
HardieDG
2002 A homologue of AMP-activated protein kinase in Drosophila melanogaster is sensitive to AMP and is activated by ATP depletion. Biochem J 367 179 186
26. HardieDG
2007 AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol 8 774 785
27. AlessiDR
SakamotoK
BayascasJR
2006 LKB1-dependent signaling pathways. Annu Rev Biochem 75 137 163
28. ShawRJ
KosmatkaM
BardeesyN
HurleyRL
WittersLA
2004 The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101 3329 3335
29. LeeJH
KohH
KimM
KimY
LeeSY
2007 Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 447 1017 1020
30. MirouseV
SwickLL
KazganN
St JohnstonD
BrenmanJE
2007 LKB1 and AMPK maintain epithelial cell polarity under energetic stress. J Cell Biol 177 387 392
31. HawleySA
PanDA
MustardKJ
RossL
BainJ
2005 Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2 9 19
32. HurleyRL
AndersonKA
FranzoneJM
KempBE
MeansAR
2005 The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem 280 29060 29066
33. BardeesyN
SinhaM
HezelAF
SignorettiS
HathawayNA
2002 Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature 419 162 167
34. JonesAT
2007 Macropinocytosis: searching for an endocytic identity and role in the uptake of cell penetrating peptides. J Cell Mol Med 11 670 684
35. KellerHU
1990 Diacylglycerols and PMA are particularly effective stimulators of fluid pinocytosis in human neutrophils. J Cell Physiol 145 465 471
36. RidleyAJ
PatersonHF
JohnstonCL
DiekmannD
HallA
1992 The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 70 401 410
37. SwansonJA
1989 Phorbol esters stimulate macropinocytosis and solute flow through macrophages. J Cell Sci 94 (Pt 1) 135 142
38. SapkotaGP
DeakM
KielochA
MorriceN
GoodarziAA
2002 Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem J 368 507 516
39. KurokawaK
MatsudaM
2005 Localized RhoA activation as a requirement for the induction of membrane ruffling. Mol Biol Cell 16 4294 4303
40. WeiQ
AdelsteinRS
2002 Pitx2a expression alters actin-myosin cytoskeleton and migration of HeLa cells through Rho GTPase signaling. Mol Biol Cell 13 683 697
41. PollardTD
BorisyGG
2003 Cellular motility driven by assembly and disassembly of actin filaments. Cell 112 453 465
42. VanderplasschenA
HollinsheadM
SmithGL
1998 Intracellular and extracellular vaccinia virions enter cells by different mechanisms. J Gen Virol 79 (Pt 4) 877 887
43. TownsleyAC
WeisbergAS
WagenaarTR
MossB
2006 Vaccinia virus entry into cells via a low-pH-dependent endosomal pathway. J Virol 80 8899 8908
44. CarterGC
LawM
HollinsheadM
SmithGL
2005 Entry of the vaccinia virus intracellular mature virion and its interactions with glycosaminoglycans. J Gen Virol 86 1279 1290
45. BengaliZ
TownsleyAC
MossB
2009 Vaccinia virus strain differences in cell attachment and entry. Virology 389 132 140
46. WhitbeckJC
FooCH
Ponce de LeonM
EisenbergRJ
CohenGH
2009 Vaccinia virus exhibits cell-type-dependent entry characteristics. Virology 385 383 391
47. MossB
2006 Poxvirus entry and membrane fusion. Virology 344 48 54
48. ConditRC
MoussatcheN
TraktmanP
2006 In a nutshell: structure and assembly of the vaccinia virion. Adv Virus Res 66 31 124
49. EdwardsDC
SandersLC
BokochGM
GillGN
1999 Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1 253 259
50. LiberaliP
KakkonenE
TuracchioG
ValenteC
SpaarA
2008 The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J 27 970 981
51. RaftopoulouM
HallA
2004 Cell migration: Rho GTPases lead the way. Dev Biol 265 23 32
52. LevineYC
LiGK
MichelT
2007 Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK -> Rac1 -> Akt -> endothelial nitric-oxide synthase pathway. J Biol Chem 282 20351 20364
53. LeeYM
LeeJO
JungJH
KimJH
ParkSH
2008 Retinoic acid leads to cytoskeletal rearrangement through AMPK-Rac1 and stimulates glucose uptake through AMPK-p38 MAPK in skeletal muscle cells. J Biol Chem 283 33969 33974
54. YamaguchiS
KatahiraH
OzawaS
NakamichiY
TanakaT
2005 Activators of AMP-activated protein kinase enhance GLUT4 translocation and its glucose transport activity in 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 289 E643 649
55. HorieT
OnoK
NagaoK
NishiH
KinoshitaM
2008 Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J Cell Physiol 215 733 742
56. UedaS
KataokaT
SatohT
2008 Activation of the small GTPase Rac1 by a specific guanine-nucleotide-exchange factor suffices to induce glucose uptake into skeletal-muscle cells. Biol Cell 100 645 657
57. ZhangL
LiJ
YoungLH
CaplanMJ
2006 AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc Natl Acad Sci U S A 103 17272 17277
58. ZhengB
CantleyLC
2007 Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc Natl Acad Sci U S A 104 819 822
59. FaveroCB
MandellJW
2007 A pharmacological activator of AMP-activated protein kinase (AMPK) induces astrocyte stellation. Brain Res 1168 1 10
60. GuestCB
ChakourKS
FreundGG
2008 Macropinocytosis is decreased in diabetic mouse macrophages and is regulated by AMPK. BMC Immunol 9 42
61. MomcilovicM
HongSP
CarlsonM
2006 Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem 281 25336 25343
62. BaasAF
KuipersJ
van der WelNN
BatlleE
KoertenHK
2004 Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 116 457 466
63. DominJ
HarperL
AubynD
WheelerM
FloreyO
2005 The class II phosphoinositide 3-kinase PI3K-C2beta regulates cell migration by a PtdIns3P dependent mechanism. J Cell Physiol 205 452 462
64. MaffucciT
CookeFT
FosterFM
TraerCJ
FryMJ
2005 Class II phosphoinositide 3-kinase defines a novel signaling pathway in cell migration. J Cell Biol 169 789 799
65. SoaresJA
LeiteFG
AndradeLG
TorresAA
De SousaLP
2009 Activation of the PI3K/Akt pathway early during vaccinia and cowpox virus infections is required for both host survival and viral replication. J Virol 83 6883 6899
66. ZaborowskaI
WalshD
2009 PI3K signaling regulates rapamycin-insensitive translation initiation complex formation in vaccinia virus-infected cells. J Virol 83 3988 3992
67. IkonomovOC
SbrissaD
FotiM
CarpentierJL
ShishevaA
2003 PIKfyve controls fluid phase endocytosis but not recycling/degradation of endocytosed receptors or sorting of procathepsin D by regulating multivesicular body morphogenesis. Mol Biol Cell 14 4581 4591
68. RustenTE
RodahlLM
PattniK
EnglundC
SamakovlisC
2006 Fab1 phosphatidylinositol 3-phosphate 5-kinase controls trafficking but not silencing of endocytosed receptors. Mol Biol Cell 17 3989 4001
69. KomadaM
KitamuraN
2005 The Hrs/STAM complex in the downregulation of receptor tyrosine kinases. J Biochem 137 1 8
70. MizunoE
KawahataK
OkamotoA
KitamuraN
KomadaM
2004 Association with Hrs is required for the early endosomal localization, stability, and function of STAM. J Biochem 135 385 396
71. CherryS
PerrimonN
2004 Entry is a rate-limiting step for viral infection in a Drosophila melanogaster model of pathogenesis. Nat Immunol 5 81 87
72. AlexanderWA
MossB
FuerstTR
1992 Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J Virol 66 2934 2942
73. CochranMA
PuckettC
MossB
1985 In vitro mutagenesis of the promoter region for a vaccinia virus gene: evidence for tandem early and late regulatory signals. J Virol 54 30 37
74. ChakrabartiS
BrechlingK
MossB
1985 Vaccinia virus expression vector: coexpression of beta-galactosidase provides visual screening of recombinant virus plaques. Mol Cell Biol 5 3403 3409
75. Ramsey-EwingA
MossB
1995 Restriction of vaccinia virus replication in CHO cells occurs at the stage of viral intermediate protein synthesis. Virology 206 984 993
76. WeaverJR
ShamimM
AlexanderE
DaviesDH
FelgnerPL
2007 The identification and characterization of a monoclonal antibody to the vaccinia virus E3 protein. Virus Res 130 269 274
77. TokumitsuH
InuzukaH
IshikawaY
IkedaM
SajiI
2002 STO-609, a specific inhibitor of the Ca(2+)/calmodulin-dependent protein kinase kinase. J Biol Chem 277 15813 15818
78. MorrisonDK
MurakamiMS
CleghonV
2000 Protein kinases and phosphatases in the Drosophila genome. J Cell Biol 150 F57 62
79. ArmknechtS
BoutrosM
KigerA
NybakkenK
Mathey-PrevotB
2005 High-throughput RNA interference screens in Drosophila tissue culture cells. Methods Enzymol 392 55 73
80. ZhangXD
YangXC
ChungN
GatesA
StecE
2006 Robust statistical methods for hit selection in RNA interference high-throughput screening experiments. Pharmacogenomics 7 299 309
81. CherryS
KunteA
WangH
CoyneC
RawsonRB
2006 COPI activity coupled with fatty acid biosynthesis is required for viral replication. PLoS Pathog 2 e102
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 6
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Stillova choroba: vzácné a závažné systémové onemocnění
- Perorální antivirotika jako vysoce efektivní nástroj prevence hospitalizací kvůli COVID-19 − otázky a odpovědi pro praxi
- Choroby jater v ordinaci praktického lékaře – význam jaterních testů
- Jak souvisí postcovidový syndrom s poškozením mozku?
Nejčtenější v tomto čísle
- Requirement of NOX2 and Reactive Oxygen Species for Efficient RIG-I-Mediated Antiviral Response through Regulation of MAVS Expression
- Formation of Complexes at Plasmodesmata for Potyvirus Intercellular Movement Is Mediated by the Viral Protein P3N-PIPO
- Insight into the Mechanisms of Adenovirus Capsid Disassembly from Studies of Defensin Neutralization
- Two Novel Point Mutations in Clinical Reduce Linezolid Susceptibility and Switch on the Stringent Response to Promote Persistent Infection