
The Yeast Complex I Equivalent NADH Dehydrogenase Rescues Mutants
Pink1 is a mitochondrial kinase involved in Parkinson's disease, and loss of Pink1 function affects mitochondrial morphology via a pathway involving Parkin and components of the mitochondrial remodeling machinery. Pink1 loss also affects the enzymatic activity of isolated Complex I of the electron transport chain (ETC); however, the primary defect in pink1 mutants is unclear. We tested the hypothesis that ETC deficiency is upstream of other pink1-associated phenotypes. We expressed Saccaromyces cerevisiae Ndi1p, an enzyme that bypasses ETC Complex I, or sea squirt Ciona intestinalis AOX, an enzyme that bypasses ETC Complex III and IV, in pink1 mutant Drosophila and find that expression of Ndi1p, but not of AOX, rescues pink1-associated defects. Likewise, loss of function of subunits that encode for Complex I–associated proteins displays many of the pink1-associated phenotypes, and these defects are rescued by Ndi1p expression. Conversely, expression of Ndi1p fails to rescue any of the parkin mutant phenotypes. Additionally, unlike pink1 mutants, fly parkin mutants do not show reduced enzymatic activity of Complex I, indicating that Ndi1p acts downstream or parallel to Pink1, but upstream or independent of Parkin. Furthermore, while increasing mitochondrial fission or decreasing mitochondrial fusion rescues mitochondrial morphological defects in pink1 mutants, these manipulations fail to significantly rescue the reduced enzymatic activity of Complex I, indicating that functional defects observed at the level of Complex I enzymatic activity in pink1 mutant mitochondria do not arise from morphological defects. Our data indicate a central role for Complex I dysfunction in pink1-associated defects, and our genetic analyses with heterologous ETC enzymes suggest that Ndi1p-dependent NADH dehydrogenase activity largely acts downstream of, or in parallel to, Pink1 but upstream of Parkin and mitochondrial remodeling.
Published in the journal:
. PLoS Genet 8(1): e32767. doi:10.1371/journal.pgen.1002456
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002456
Summary
Pink1 is a mitochondrial kinase involved in Parkinson's disease, and loss of Pink1 function affects mitochondrial morphology via a pathway involving Parkin and components of the mitochondrial remodeling machinery. Pink1 loss also affects the enzymatic activity of isolated Complex I of the electron transport chain (ETC); however, the primary defect in pink1 mutants is unclear. We tested the hypothesis that ETC deficiency is upstream of other pink1-associated phenotypes. We expressed Saccaromyces cerevisiae Ndi1p, an enzyme that bypasses ETC Complex I, or sea squirt Ciona intestinalis AOX, an enzyme that bypasses ETC Complex III and IV, in pink1 mutant Drosophila and find that expression of Ndi1p, but not of AOX, rescues pink1-associated defects. Likewise, loss of function of subunits that encode for Complex I–associated proteins displays many of the pink1-associated phenotypes, and these defects are rescued by Ndi1p expression. Conversely, expression of Ndi1p fails to rescue any of the parkin mutant phenotypes. Additionally, unlike pink1 mutants, fly parkin mutants do not show reduced enzymatic activity of Complex I, indicating that Ndi1p acts downstream or parallel to Pink1, but upstream or independent of Parkin. Furthermore, while increasing mitochondrial fission or decreasing mitochondrial fusion rescues mitochondrial morphological defects in pink1 mutants, these manipulations fail to significantly rescue the reduced enzymatic activity of Complex I, indicating that functional defects observed at the level of Complex I enzymatic activity in pink1 mutant mitochondria do not arise from morphological defects. Our data indicate a central role for Complex I dysfunction in pink1-associated defects, and our genetic analyses with heterologous ETC enzymes suggest that Ndi1p-dependent NADH dehydrogenase activity largely acts downstream of, or in parallel to, Pink1 but upstream of Parkin and mitochondrial remodeling.
Introduction
Parkinson's disease (PD (OMIM #168600)) is the most common neurodegenerative movement disorder [1]. While diverse processes including autophagy, apoptosis, oxidative stress and accumulation of protein inclusions have been implicated in the etiology of the disease, mitochondrial dysfunction appears to play a central role as well [2]–[4]. Mitochondrial toxins, such as MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) or rotenone, that block Complex I of the mitochondrial electron transport chain (ETC) cause clinical features reminiscent of PD in humans and are commonly used to create animal models of the disease [5], [6]. Furthermore, Complex I deficiency is often observed in neurons of PD patients [7] and mutations in genes causing familial forms of PD, including pink1 (PARK6, OMIM #605909, Gene ID: 65018), parkin (PARK2, OMIM #600116, Gene ID: 5071) and DJ-1 (PARK7, OMIM #606324, Gene ID: 11315) result in defects in mitochondrial morphology and/or function in model organisms [8]–[14]. Molecular genetic analyses of PD-associated genes will thus yield important insights into the mechanisms of PD.
Pink1 (CG4523 Gene ID: 31607) is a serine/threonine kinase involved in maintaining mitochondrial integrity [8]–[10] and loss-of-function mutants show hallmark mitochondrial defects including male sterility, an inability of most flies to fly as well as an inability to maintain synaptic transmission during intense stimulation, a deficit that can be rescued by supplementing synapses with ATP [15], [16]. The observation of larger clumped mitochondria in pink1 mutants suggests a model where Pink1 is involved in the clearance of dysfunctional mitochondria [17]–[21]. This is in line with experiments that show an alleviation of pink1-associated phenotypes by over-expression of Parkin (CG10523 Gene ID: 40336), an E3 ubiquitin ligase involved in mitophagy [8]–[10] and by the notion that loss-of-function pink1 trumps Parkin recruitment to mitochondria [18]. Furthermore, the morphological defects in pink1 mutants can be modulated by altering the levels of proteins involved in mitochondrial fusion or fission, and this is thought to facilitate mitophagy [17], [19], [21], [22]. However, these studies are not conclusive as functional defects in pink1 mitochondria at the level of Complex I have been observed in the absence of severe morphological alterations [15], [23] and such functional defects can eventually result in mitochondrial morphological alterations [24]–[26]. These results suggest an alternative model where functional defects in pink1 mutant mitochondria precede morphological alterations and mitophagy.
To determine if Pink1 acts to regulate ETC function we performed genetic studies with heterologous alternative enzymes that can bypass either Complex I, or Complex III and IV. Although each of the ETC complexes in flies (and humans) comprise numerous proteins (Complex I contains more than 40 subunits in humans and in flies), Saccharomyces cerevisiae Ndi1p (Gene ID: 854919) (UAS-NDI1) constitutes an alternative NADH oxidoreductase that transfers electrons from NADH to ubiquinone, delivering electrons to downstream complexes and therefore can be used to bypass electron transport in Complex I in higher order species [27], [28]. Similarly, ‘Ciona intestinalis’ alternative oxidase AOX (Gene ID: 3293227) is able to bypass the cytochrome c chain and Complexes III and IV by using electrons from ubiquinol to reduce oxygen [29]. While neither Ndi1p, nor AOX themselves transfer protons across the inner mitochondrial membrane, they may add to the proton motive force by facilitating the ubiquinone cycle, thus contributing to Complex III and IV mediated proton translocation upon expression of Ndi1p, or to Complex I mediated proton translocation upon expression of AOX. Illustrating this idea, expression of Ndi1p in Drosophila can rescue partial loss-of-function mutations in Complex I components and confers rotenone resistance [30], [31], and expression of AOX can rescue partial loss-of-function mutations in Complex III and IV components and confers cyanide resistance [32]. Hence, Ndi1p and AOX allow us to genetically dissect the ETC.
Here, we assay the role of Complex I in pink1-associated defects and find that Ndi1p can rescue pink1 phenotypes while AOX cannot. Similarly, loss-of-function of a Complex I component phenocopies many of the pink1 mutant phenotypes and these defects can also be rescued by expression of Ndi1p. In contrast, expression of Ndi1p fails to rescue parkin mutants, indicating that Ndi1p acts downstream or in parallel to Pink1 but upstream of Parkin. Further supporting this model, we do not find reduced enzymatic activity of Complex I in parkin mutants, and while modulating mitochondrial remodeling using Drp1 (CG3210 Gene ID: 33445) or Opa1 (CG8479 Gene ID: 36578) can rescue the defects in mitochondrial morphology in pink1 mutants, these manipulations do not rescue decreased enzymatic activity of Complex I observed in pink1 mutants. Thus, our studies suggest that the defects at Complex I in pink1 mutants are upstream of several of the events that lead to pink1-associated phenotypes.
Results
Yeast Ndi1p rescues pink1
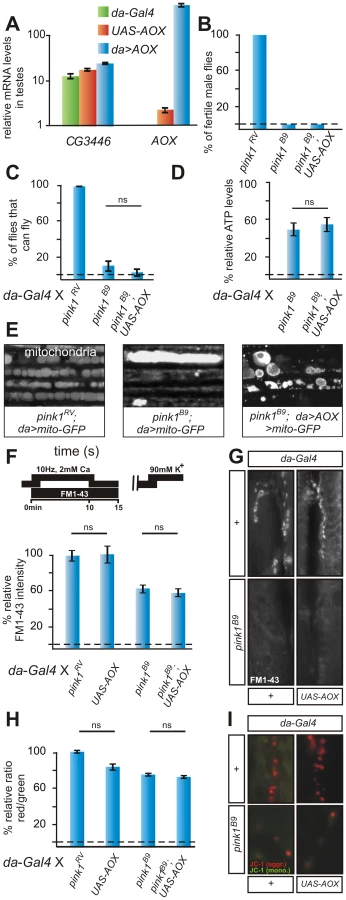
Electron transfer activity of the mitochondrial multi-protein Complex I, NADH:ubiquinone oxidoreductase (EC 1.6.5.3) can be recapitulated by a single yeast protein, Ndi1p [31], [33], [34]. To determine whether the reduced respiratory chain activity in pink1 mutants reported previously [15] are an upstream defect in the mutants, we generated transgenic flies that harbor a UAS-Sacharomyces cerevisiae NDI1 construct allowing expression under the control of GAL4. Quantitative RT-PCR using RNA from flies that harbor the UAS-NDI1 transgene and the ubiquitously expressing da-GAL4 driver indicates that NDI1 expression levels in such animals is similar to that of an endogenously expressed Complex I component CG3446 (Figure S1A). In addition, quantitative RT-PCR using RNA of UAS-NDI1 bearing flies, in the absence of GAL4 also shows low but significant expression of NDI1 RNA, particularly in the male reproductive organ (Figure 1A, blue). Ndi1p confers rotenone insensitive NADH oxidoreductase activity in flies as the enzymatic activity of isolated Complex I in the presence of rotenone, a Complex I inhibitor, remains high in mitochondria from flies expressing Ndi1p, but is dramatically reduced in rotenone-treated mitochondria from control flies (Figure S1B) [30], [31]. Ndi1p expression in Drosophila is benign as ubiquitous expression (da-GAL4) does not lead to obvious behavioral or developmental abnormalities (Figure S1C, S1D). Ndi1p expression rescues lethality associated with RNAi-induced systemic loss-of-function of CG18624 (Gene ID: 31697), an evolutionary conserved Complex I component, and also the mitochondrial defects associated with loss of CG12079 (Gene ID: 38378), another conserved Complex I component (below). These data confirm that Ndi1p is functional.

Next, we generated pink1 mutant animals that express Ndi1p. While pink1 mutant males are sterile, low expression of Ndi1p is sufficient to completely revert this defect (Figure 1B). Given that pink1 is located on the X-chromosome, homozygous pink1 females are never observed. However, in the presence of NDI1, fertile homozygous pink1 female flies are obtained (Figure S2A), whose genetic makeup we confirmed by genomic PCR (Figure S2B). Furthermore, while in pink1 mutants morphological defects in spermatid mitochondria are apparent [10], [31], expression of Ndi1p also rescues these mitochondrial defects (data not shown). Thus, expression of Ndi1p rescues pink1-associated male sterility and mitochondrial morphological defects in the germline to a level indistinguishable from wild type controls.
Drosophila flight muscles require large amounts of metabolic energy supplied by mitochondria. In adult pink1 mutant flies, mitochondrial deficits lead to muscle degeneration and a severe defect to fly. While expression of Ndi1p using da-GAL4 does not affect flight (Figure S1C), expression in pink1 mutants improves flight (Figure 1C). Although expression of Ndi1p does not restore the pink1 flight defect to control levels, it is important to note that previous experiments demonstrating rescue of Pink1 phenotypes using over-expressed Parkin showed very similar results [9].
Ndi1p expression also rescues degeneration of the indirect flight muscles in pink1 mutants as evaluated by decreased indentations in the thoraces of the flies (Figure S2C). As flight muscle degeneration correlates with an accumulation of enlarged mitochondria in pink1 mutants [8]–, we labeled the mitochondrial pool using mito-GFP (Figure 1E). In adult pink1 flight muscles, the mitochondria appear enlarged and clumped when compared to controls (Figure 1E, middle) and Ndi1p expression in pink1 mutants partially trumps this defect (Figure 1E, right). Finally, pink1 mutants that express Ndi1p have an increase in ATP levels compared to pink1 mutants not expressing Ndi1p (Figure 1D). Thus, enhanced ATP levels in pink1 mutants that express Ndi1p may dampen mitochondrial dysfunction and diminish muscle degeneration.
Previous data indicated that mitochondrial defects cause synaptic vesicle trafficking and neurotransmitter release defects at pink1 mutant synapses [15], [23]. We therefore measured neurotransmitter release at the Drosophila third instar neuromuscular junction (NMJ). Upon stimulation of the motor neuron at 1 Hz, pink1 mutants, pink1 mutants that express Ndi1p, as well as control animals that express Ndi1p, show normal neurotransmitter release as gauged by the amplitude of the excitatory junctional potential (EJP) (Figure S3A). In contrast, when stimulated at high frequency (10 Hz), neurotransmitter release in pink1 mutants gradually declines, in line with a defect to mobilize ‘reserve pool’ (RP, see below) vesicles that are only used under such ‘stressed’ conditions [16], [35]. Interestingly, pink1 mutants that express Ndi1p maintain normal levels of synaptic transmission during a 10 Hz 10 min stimulation paradigm (Figure S3B). These data indicate that synaptic transmission defects at pink1 mutant NMJs are also rescued by expression of Ndi1p. As previously shown, this neurotransmission deficit is likely caused by a lack of mobilization of the reserve pool synaptic vesicles within NMJ boutons [15]. We used the fluorescent dye FM 1–43 to label vesicles loaded in the RP [36] using the stimulation paradigm depicted in Figure 1F [37]. While pink1 mutants show a significant reduction in RP vesicle labeling, pink1 mutants that express Ndi1p display labeling of RP vesicles very similar to controls (Figure 1F, 1G). Thus, synaptic function deficits in pink1 mutants are alleviated by expression of yeast Ndi1p.
In pink1 mutant motor neurons, mitochondria are morphologically normal but show only partial mitochondrial membrane depolarization [15]. We assessed the mitochondrial membrane potential in pink1 mutants rescued with Ndi1p using the ratiometric dye JC-1. [16], [38]. Interestingly, compared to pink1 mutants, mitochondria at synaptic boutons of pink1 mutants that express Ndi1p are significantly more polarized and show more intense red JC-1 labeling (Figure 1H, 1I). Although Ndi1p itself is not involved in proton transfer over the inner mitochondrial membrane, our data indicate that expression of Ndi1p can restore a negative mitochondrial membrane potential in pink1 mutants. Potentially Ndi1p improves electron transfer from NADH to complex III/IV that in turn helps to restore the proton gradient in mitochondria at synapses.
Expression of AOX does not rescue pink1 mutant phenotypes
To further test the specificity of Ndi1p-dependent rescue of pink1-associated phenotypes, we also tested the ability of Ciona intestinalis AOX to bypass pink1 defects. Our rationale is that AOX can also transport electrons but at a different site within the ETC: AOX uses electrons from ubiquinol to reduce oxygen and thereby bypasses both Complex III and IV [29], [32]. We confirm the functionality of AOX in flies because lethality induced by expression of RNAi to cyclope (CG14028 Gene ID 46040) that encodes a Complex IV component, is rescued upon expression of AOX (data not shown) [32]. Similar to NDI1 we also find basal expression of AOX in the male reproductive organ that is much induced by the presence of da-GAL4 (Figure 2A) and ubiquitous expression of AOX also does not affect flight, muscle degeneration or the mitochondrial membrane potential (Figure S4A, S4B; see below). In contrast to Ndi1p, expression of AOX completely fails to alleviate pink1-associated phenotypes such as male fertility, flight or mitochondrial morphology (Figure 2B, 2C, 2E Figure S4C). AOX also does not revert the RP defects in pink1 mutant animals (Figure 2F, 2G) nor does it alleviate the reduced red JC-1 labeling observed in mitochondria at boutons of pink1 mutants (Figure 2H, 2I). Finally, also the reduced ATP levels, observed in pink1 mutant animals, are not rescued by AOX (Figure 2D). Thus, AOX expression does not rescue pink1 deficiency.
Downregulation of a Complex I component phenocopies many pink1 mutant phenotypes
Our work suggests that several pink1 mutant phenotypes stem from defects at the level of Complex I. To further test this hypothesis, we used RNAi mediated knock down of evolutionary conserved Complex I subunits (Figure 3A). As expected, down regulation of Complex I subunits results in significantly lower Complex I enzymatic activity (Figure 3B) and expression of NDI1 in these flies results in an increased ATP concentration compared to RNAi of Complex I subunits in the absence of NDI1 expression (Figure 3C). While expression of RNAi to some of the Complex I subunits results in developmental lethality, RNAi to other Complex I subunits yields adult flies, and we used one of those (CG11455; Gene ID: 33179) to assess flight upon Complex I knock down. Under ‘standard testing conditions’ (see methods) RNAi to this Complex I subunit does not result in a strong defect to fly (blue bar Figure 3D), however under more ‘stringent conditions’ (see methods) about half of the flies fail to fly, and this defect is rescued by expression of NDI1 (red bars Figure 3D). Thus, while reduced Complex I activity results in a defect to fly, the flight deficit upon knock down of this Complex I subunit is milder than that observed in pink1 mutants (Figure 3E).

Next, we determined mitochondrial morphology using Mito-GFP in third instar larval muscles upon knock down of a Complex I subunit. Similar to mitochondria in pink1 mutants, mitochondria in muscles of animals that express RNAi to a Complex I subunit are swollen and clumped, but the defects we observe are in general less severe than those seen in pink1B9 null mutants. Interestingly, expression of NDI1 significantly alleviates these defects (Figure 3F). To assess mitochondrial function in animals that express RNAi to a Complex I subunit with or without NDI1, we assessed the mobilization of RP vesicles and we quantified red JC-1 labeling intensity within neuromuscular mitochondria. As indicated in Figure 3G–3J, reduced Complex I activity results in reduced RP vesicle mobilization and less red JC-1 labeling in boutonic mitochondria, and expression of NDI1 can significantly rescue these defects as well. Thus, similar to pink1 mutants, loss-of-function of a component of Complex I results in reduced ATP and functional defects in synaptic mitochondria, and these defects are alleviated by NDI1p. Furthermore, we also find that reduced Complex I activity leads to morphological defects in muscular mitochondria and a defect to fly in adult flies, but these defects are in general milder than those observed in pink1 null mutants.
Expression of NDI1 does not rescue parkin mutant phenotypes
Pink1 has been suggested to act upstream of Parkin to regulate, in a linear pathway, mitophagy [8]–[10], [39], [40]. We therefore expressed Ndi1p in parkin mutant flies but we did not observe a rescue of male fertility, flight defects or muscular degeneration (Figure 4A, 4B, 4D). In line with these observations, mitochondrial morphological alterations caused by Parkin deficiency are also not rescued by expression of Ndi1p (Figure 4C). Thus, parkin mutants cannot be rescued by expression of Ndi1p.

To determine if parkin mutants also display reduced enzymatic activity of Complex I we isolated mitochondria from parkin null mutant flies and from controls and measured Complex I enzymatic activity. In contrast to pink1 mutants, the isolated enzymatic activity of Complex I in parkin mutant mitochondria is similar to controls (Figure 4E). These data are in further support of a model where Complex I defects in pink1 mutants occur upstream from the defects caused by loss of Parkin function.
Modulation of mitochondrial morphology does not rescue Complex I defects in pink1 mutants
Previous reports indicate that genetic manipulation of the mitochondrial remodeling machinery using over expression of drp1 or loss-of-function of opa1 alleviates mitochondrial morphological defects, muscle degeneration and flight deficits both in pink1 and in parkin mutant flies [17], [19] and we confirm these results (Figure 5A, 5B, data not shown). In contrast however, sterility of pink1 mutant males is not rescued by increased drp1 or decreased opa1 (Figure 5C), suggesting that manipulation of mitochondrial remodeling cannot rescue all pink1-related phenotypes. As mitochondrial morphological phenotypes may result from alterations in numerous biochemical pathways [26], we assessed directly whether drp1 or opa1 affect the enzymatic activity of Complex I in pink1 mutant flies. However, as shown in Figure 5D, the enzymatic activity of Complex I is still reduced to a level similar to that observed in pink1 mutants. These data thus indicate that the enzymatic deficiency at the level of Complex I in pink1 mutants precedes mitochondrial morphological deficits, or that they in part occur independently.

Discussion
In this work we present compelling evidence that the mitochondrial kinase Pink1 is critically required to maintain efficient Complex I enzymatic activity in mitochondria and that this function precedes mitochondrial remodeling or mitophagy (Figure 5E). While Pink1 likely acts via multiple (phospho-) targets [41], [42], our data suggests a pathway in which many of the deficiencies in pink1 can be traced back to mitochondrial dysfunction [8]–[10], [15].
Our experiments indicate that Pink1 acts at, or in parallel to, Complex I, in line with the reduced enzymatic activity of this complex in pink1 mutant mouse cells and flies [15], [23]. Expression of Ndi1p alleviates many pink1-associated phenotypes, suggesting that more efficient electron transport between NADH and ubiquinone is mediated by Ndi1p (bypassing the endogenous Complex I deficiency) [30] in pink1 mutants that boosts formation of a proton gradient by Complex III and IV. Although AOX expression also improves ETC efficiency [32], it does not rescue pink1-associated phenotypes, in contrast to AOX rescuing DJ-1β (CG1349 Gene ID: 43652) and cyclope associated phenotypes [32]. The lack of rescue is likely not due to the fact that AOX is insufficiently activated as a result of low reduced ubiquinone concentrations [43] as expression of AOX in pink1 mutants results in premature death of pink1 animals such that only few pink1 mutants that express AOX emerge as adults (data not shown). We surmise that the lower Complex I activity in pink1 mutants, which results in reduced proton transfer across the inner mitochondrial membrane [15] is further propagated by the presence of AOX, that transfers electrons to oxygen without pumping protons [29]. These data thus argue against general mitochondrial dysfunction in pink1 and a universal failing of the ETC [23] but reveals an important role for Pink1 upstream or in parallel to Complex I enzymatic activity [15].
Previous work indicates that loss of pink1 in some cell types results in mitochondrial fragmentation, a process preceding mitophagy [44]. Several lines of evidence now indicate that some of the mitochondrial morphological defects occur downstream of functional deficits in pink1 mutants. First, we show that expression of Ndi1p in pink1 mutants alleviates part of the mitochondrial morphological defects. Second, RNAi-mediated knock down of an evolutionary conserved Complex I component results in mitochondrial dysfunction but also mitochondrial swelling and clumping, indicating that functional mitochondrial defects can lead to morphological defects [24], [26], [45]. Third, mitochondrial functional defects in pink1 mutant flies and mice have been widely observed in neuronal populations where mitochondrial morphological defects are not (yet) prevalent [15], [23]. Fourth, while facilitating mitochondrial fission in pink1 mutants alleviates mitochondrial morphological defects, a deficiency at the level of the enzymatic activity of Complex I persists. Previous results in pink1 knock out cells had also indicated that mitochondrial swelling defects in pink1 mutant cells can be rescued by modulating the levels of the mitochondrial fission machinery [46], but also this manipulation failed to rescue the defect in mitochondrial membrane potential caused by loss of Pink1 [47]. Thus, our data indicate that the upstream molecular dysfunction in pink1 mutants on Complex I is a major culprit in the development of the pink1 mutant phenotypes (Figure 5E).
Our rescue experiments indicate that expression of NDI1 can significantly rescue numerous pink1 associated phenotypes, including male sterility, vesicle trafficking and mitochondrial membrane potential. Likewise, NDI1 also alleviates mitochondrial morphological defects in pink1 mutant muscles, but rescue of this morphological defect is only partial. Similarly, mitochondrial morphological defects observed in animals that express RNAi to a Complex I subunit and flight defects in such animals are in general less severe than those seen in pink1 null mutants. These results are consistent with Pink1 also acting in parallel to its role at the level of Complex1; however, we cannot exclude the possibility that the partial rescue of morphological defects in pink1 mutants upon expression of NDI1 originates from an incomplete reconstitution of ETC activity under these conditions, and that the pink1 mutant conditions at the level of Complex I may not be exactly recapitulated by knock down of the Complex I components. Furthermore, given that Complex I dysfunction results in mitochondrial morphological defects and mild flight defects that are rescued by NDI1 expression, and the observation that the enzymatic defects at the level of Complex I in pink1 mutants are not rescued upon expression of Drp1 or loss of opa1 function, the data are consistent with the pink1-associated Complex I defects to act at least in part upstream of remodeling and suggest an important and central role for Complex I in Pink1 induced mitochondrial pathology.
Our work expands on previous genetic and cell biological studies, and indicates that Pink1 can act at a different level, upstream of Parkin, to control Complex I enzymatic activity (Figure 5E). Indeed, unlike pink1 mutants, loss of parkin in flies does not cause significantly reduced enzymatic activity of Complex I seen in pink1 mutants, and in addition, parkin loss of function is not rescued by expression of Ndi1p. Our data indicate that Complex I deficiency in pink1 mutants is specific and that this defect is not a result of abnormal mitochondrial remodeling or mitophagy. This model is consistent with a role of Pink1 in controlling mitochondrial health and also does not exclude a downstream or parallel role where Pink1 can be triggered to recruit Parkin, facilitating mitophagy (Figure 5E).
Materials and Methods
Drosophila stocks and maintenance
w; UAS-mito:GFP, w; da-Gal4 and w; P{lacW}opa1-like3475/CyO (opa1S3) were obtained from Bloomington stock center (Indiana, USA); w pink1B9 and w pink1RV, parkin1 and parkinRV [48] were gifts from Jongkyeong Chung (Korea, Advanced Institute of Science and Technology) [9] and parkinΔ21 mutant flies were a gift from Graeme Mardon (Baylor College of Medicine) [11]. drp1+ genomic rescue constructs were provided by Hugo Bellen (Baylor College of Medicine) [16] and w1118; UAS-AOXF6 were a gift from Howard T. Jacobs [32] (Finland, Institute of Medical Technology, Tampere University Hospital). w1118; UAS-CG12079RNAi (w1118; P{GD5910}v13856), w1118; UAS-CG11455RNAi (w1118; P{GD4800}v12838) and cyclope (CG14028) RNAi (w1118; P{GD908}v13403) were from the Vienna Drosophila RNAi Center (VDRC) [49]. Flies were raised on standard cornmeal and molasses medium.
Generation of UAS-NDI1 transgenic flies
The coding region of Saccaromyces cerevisiae NDI1 was amplified from yeast genomic DNA (Patrick Van Dijck, VIB Leuven), with the following primers; CGG AAT TCC AAA ATG CTA TCG AAG and GGC GGC CGC CTA TAA TCC TTT A using 2 X BIO-X-ACT Short Mix (BIOLINE), cloned in the EcoR1 and Not1 of pUAST-Attb (43) and sequenced. Transgenic flies were created at GenetiVision Inc. (Houston, USA) using PhiC31 mediated transgenesis in the VK1 docking site (2R, 59D3) [50].
Quantitative RT–PCR
Testis samples from w; daGal4/+ and w; UAS-NDI1 and w; UAS-NDI1/+; da-Gal4/+ and w; UAS-AOXF6 and w; UAS-AOXF6/+; da-Gal4/+ adult flies were micro-dissected in ice-cold PBS followed by snap-freezing on dry ice. Third instar larvae from w; daGal4/+ and w; UAS-CG12079RNAi/da-Gal4 were also snap-frozen on dry ice. Q-RT-PCR was performed using standard procedures, as outlined in Text S1, and relative RNA levels were calculated according to the ΔΔCt method. Primers are listed in Table S1.
Sterility test
Single males (30) of the genotypes indicated in the figure legends were crossed to 1–3 day old w1118 virgins and a score of 1 was assigned when offspring was detected.
Flight assays
For flight assays, batches of 5 freshly eclosed male flies grown at 18°C of the genotypes indicated in the figure legends were put at room temperature for 2 days and then transferred to an empty vial (5 cm D, 10 cm H). For ‘standard testing conditions’, flies were allowed to climb above a marked line at 9 cm height, the vial was gently tapped and visually scored for flying flies over the next minute. For more ‘stringent testing conditions’ flies were allowed to climb above a marked line at 9 cm height, and flying flies were scored immediately following tapping of the vial. Flies at the bottom were removed and the remaining flies were retested. Flies that fly twice were assigned a score of 1, the others a score of 0. Student's t test was used to assess the statistical differences.
ATP measurements
Five third instar larvae or five thoraces from 2–3 days-old flies were dissected and homogenized in 50 µl of 6 M guanidine-HCl 100 mM Tris and 4 mM, EDTA, pH 7.8. These homogenates were snap-frozen in liquid nitrogen and then boiled for 3 min. Samples were then centrifuged and the supernatant was diluted (1/50) in extraction buffer, mixed with luminescent solution (ATP Determination Kit, Invitrogen) and luminescence was measured on an EnVision Multilabel Reader (Perkin Elmer). Luminiscence was normalized to protein amount (mg) (Bradford) and compared to ATP standards. n = 3. Student's t test was used to assess the statistical differences.
Microscopy
Thoraces of adult flies were viewed under an Olympus SZX12 microscope equipped with a DF PLAPO 1X PF lens and the pictures were captured with an Olympus U-CMAD3 camera.
Mitochondrial morphology in adult flight muscles of flies grown at 18°C and reared at room temperature for 2 days or in wandering third instar larvae grown at 25°C was assessed by visualizing mitochondrial tagged GFP (mito-GFP), excited using 488 nm laser light and imaged on a Zeiss LSM 510 META confocal microscope using a 63×oil NA 1.4 lens (for adult muscles) or a 40×oil NA 1.3 lens (for larvae) using a 500–530 band pass emission filter.
RP vesicles of larvae were labeled by electrically stimulating motor neurons of third instar larval fillets in HL-3 with 2 mM Ca2+ for 10 min at 10 Hz and then leaving the preparation to rest for 5 min in the presence of the dye following stimulation. This protocol labels the entire vesicle pool; the exo-endo cycling pool (ECP) and the RP. To unload the FM 1–43 from the ECP, leaving RP labeling intact, preparations were subsequently incubated for 5 min in HL-3 with 90 mM KCl and 2 mM Ca2+ (in the absence of FM 1–43) [16]. Following washing in Ca2+ free HL-3, NMJs were imaged on a Nikon FN-1 microscope with a DS-2MBWc digital camera, 40×W NA 0.8 objective and quantification of labeling intensity was performed using NIS-Elements AR 3.10.
JC-1 (Molecular Probes) labeling was performed on wandering third instar larvae of the genotypes indicated in the figure legends as described previously [15]. Red and green fluorescence was captured on a Nikon FN-1 microscope with a Hamamatsu ORCA-R2, 40W NA 0.8 objective. Quantification of red and green labeling intensity was performed using NIS-Elements AR 3.10.
Enzymatic Complex I activity measurements
Complex I activity measurements were performed as described [15]. Data represent at least 3 independent experiments where mitochondrial preparations from 50 animals were prepared for each independent experiment. The Complex I enzymatic activity was normalized to Citrate Synthase enzymatic activity.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DauerWPrzedborskiS 2003 Parkinson's disease: mechanisms and models. Neuron 39 889 909
2. MoraisVADe StrooperB 2010 Mitochondria dysfunction and neurodegenerative disorders: cause or consequence. J Alzheimers Dis 20 Suppl 2 S255 263
3. MandemakersWMoraisVADe StrooperB 2007 A cell biological perspective on mitochondrial dysfunction in Parkinson disease and other neurodegenerative diseases. J Cell Sci 120 1707 1716
4. WinklhoferKFHaassC 2009 Mitochondrial dysfunction in Parkinson's disease. Biochim Biophys Acta 1802 29 44
5. PanovADikalovSShalbuyevaNTaylorGShererT 2005 Rotenone model of Parkinson disease: multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J Biol Chem 280 42026 42035
6. ShererTBBetarbetRGreenamyreJT 2002 Environment, mitochondria, and Parkinson's disease. Neuroscientist 8 192 197
7. ParkerWDJrSwerdlowRH 1998 Mitochondrial dysfunction in idiopathic Parkinson disease. Am J Hum Genet 62 758 762
8. YangYGehrkeSImaiYHuangZOuyangY 2006 Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A 103 10793 10798
9. ParkJLeeSBLeeSKimYSongS 2006 Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441 1157 1161
10. ClarkIEDodsonMWJiangCCaoJHHuhJR 2006 Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441 1162 1166
11. PesahYPhamTBurgessHMiddlebrooksBVerstrekenP 2004 Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131 2183 2194
12. GreeneJCWhitworthAJKuoIAndrewsLAFeanyMB 2003 Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 100 4078 4083
13. MeulenerMWhitworthAJArmstrong-GoldCERizzuPHeutinkP 2005 Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr Biol 15 1572 1577
14. HaoLYGiassonBIBoniniNM 2010 DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc Natl Acad Sci U S A 107 9747 9752
15. MoraisVAVerstrekenPRoethigASmetJSnellinxA 2009 Parkinson's disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol Med 1 99 111
16. VerstrekenPLyCVVenkenKJKohTWZhouY 2005 Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47 365 378
17. DengHDodsonMWHuangHGuoM 2008 The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A 105 14503 14508
18. NarendraDPJinSMTanakaASuenDFGautierCA 2010 PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8 e1000298 doi:10.1371/journal.pbio.1000298
19. PooleACThomasREAndrewsLAMcBrideHMWhitworthAJ 2008 The PINK1/Parkin pathway regulates mitochondrial morphology. Proc Natl Acad Sci U S A 105 1638 1643
20. Vives-BauzaCZhouCHuangYCuiMde VriesRL 2010 PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107 378 383
21. YangYOuyangYYangLBealMFMcQuibbanA 2008 Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A 105 7070 7075
22. ParkJLeeGChungJ 2009 The PINK1-Parkin pathway is involved in the regulation of mitochondrial remodeling process. Biochem Biophys Res Commun 378 518 523
23. GautierCAKitadaTShenJ 2008 Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 105 11364 11369
24. ParkJKimYChoiSKohHLeeSH 2010 Drosophila Porin/VDAC affects mitochondrial morphology. PLoS ONE 5 e13151 doi:10.1371/journal.pone.0013151
25. GrahamBHLiZAlesiiEPVerstekenPLeeC 2010 Neurologic dysfunction and male infertility in Drosophila porin mutants: a new model for mitochondrial dysfunction and disease. J Biol Chem 285 11143 11153
26. IchishitaRTanakaKSugiuraYSayanoTMiharaK 2008 An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem 143 449 454
27. SeoBBKitajima-IharaTChanEKSchefflerIEMatsuno-YagiA 1998 Molecular remedy of complex I defects: rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc Natl Acad Sci U S A 95 9167 9171
28. BaiYHajekPChomynAChanESeoBB 2001 Lack of complex I activity in human cells carrying a mutation in MtDNA-encoded ND4 subunit is corrected by the Saccharomyces cerevisiae NADH-quinone oxidoreductase (NDI1) gene. J Biol Chem 276 38808 38813
29. JuszczukIMRychterAM 2003 Alternative oxidase in higher plants. Acta Biochim Pol 50 1257 1271
30. BahadoraniSChoJLoTContrerasHLawalHO 2010 Neuronal expression of a single-subunit yeast NADH-ubiquinone oxidoreductase (Ndi1) extends Drosophila lifespan. Aging Cell 9 191 202
31. SanzASoikkeliMPortero-OtinMWilsonAKemppainenE 2010 Expression of the yeast NADH dehydrogenase Ndi1 in Drosophila confers increased lifespan independently of dietary restriction. Proc Natl Acad Sci U S A 107 9105 9110
32. Fernandez-AyalaDJSanzAVartiainenSKemppainenKKBabusiakM 2009 Expression of the Ciona intestinalis alternative oxidase (AOX) in Drosophila complements defects in mitochondrial oxidative phosphorylation. Cell Metab 9 449 460
33. DeCorbyAGaskovaDSaylesLCLemireBD 2007 Expression of Ndi1p, an alternative NADH:ubiquinone oxidoreductase, increases mitochondrial membrane potential in a C. elegans model of mitochondrial disease. Biochim Biophys Acta 1767 1157 1163
34. MarellaMSeoBBNakamaru-OgisoEGreenamyreJTMatsuno-YagiA 2008 Protection by the NDI1 gene against neurodegeneration in a rotenone rat model of Parkinson's disease. PLoS ONE 3 e1433 doi:10.1371/journal.pone.0001433
35. KuromiHKidokoroY 2000 Tetanic stimulation recruits vesicles from reserve pool via a cAMP-mediated process in Drosophila synapses. Neuron 27 133 143
36. BetzWJMaoFSmithCB 1996 Imaging exocytosis and endocytosis. Curr Opin Neurobiol 6 365 371
37. VerstrekenPOhyamaTBellenHJ 2008 FM 1–43 labeling of synaptic vesicle pools at the Drosophila neuromuscular junction. Methods Mol Biol 440 349 369
38. ReersMSmithTWChenLB 1991 J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry 30 4480 4486
39. ZivianiETaoRNWhitworthAJ 2010 Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci U S A 107 5018 5023
40. PooleACThomasREYuSVincowESPallanckL 2010 The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5 e10054 doi:10.1371/journal.pone.0010054
41. Plun-FavreauHKlupschKMoisoiNGandhiSKjaerS 2007 The mitochondrial protease HtrA2 is regulated by Parkinson's disease-associated kinase PINK1. Nat Cell Biol 9 1243 1252
42. PridgeonJWOlzmannJAChinLSLiL 2007 PINK1 protects against oxidative stress by phosphorylating mitochondrial chaperone TRAP1. PLoS Biol 5 e172 doi:10.1371/journal.pbio.0050172
43. RustinPJacobsHT 2009 Respiratory chain alternative enzymes as tools to better understand and counteract respiratory chain deficiencies in human cells and animals. Physiol Plant 137 362 370
44. LutzAKExnerNFettMESchleheJSKloosK 2009 Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J Biol Chem 284 22938 22951
45. KoopmanWJVerkaartSVischHJvan der WesthuizenFHMurphyMP 2005 Inhibition of complex I of the electron transport chain causes O2-. -mediated mitochondrial outgrowth. Am J Physiol Cell Physiol 288 C1440 1450
46. ExnerNTreskeBPaquetDHolmstromKSchieslingC 2007 Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27 12413 12418
47. SandebringAThomasKJBeilinaAvan der BrugMClelandMM 2009 Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE 4 e5701 doi:10.1371/journal.pone.0005701
48. ChaGHKimSParkJLeeEKimM 2005 Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A 102 10345 10350
49. DietzlGChenDSchnorrerFSuKCBarinovaY 2007 A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448 151 156
50. VenkenKJHeYHoskinsRABellenHJ 2006 P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science 314 1747 1751
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 1
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Poly(ADP-Ribose) Polymerase 1 (PARP-1) Regulates Ribosomal Biogenesis in Nucleoli
- Microenvironmental Regulation by Fibrillin-1
- Parallel Mapping and Simultaneous Sequencing Reveals Deletions in and Associated with Discrete Inherited Disorders in a Domestic Dog Breed
- Two-Component Elements Mediate Interactions between Cytokinin and Salicylic Acid in Plant Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy