
Úloha CDK12 v rozvoji nádorů
Th e role of CDK12 in tumor bio logy
Background: Physiological function of cyclin-dependent kinase 12 (CDK12) is crucial for several cellular processes, including regulation of transcription, RNA splicing, transcription termination and polyadenylation. It is well documented by now that CDK12 controls transcription of the unique set of genes involved in DNA-damage response, replication of DNA and response to cellular stress. Just recently, a key function of CDK12 in the induction of tandem duplication of specific DNA sequences within the metastatic castrate resistant prostate tumors has been documented. Therefore, it is possible to recognize CDK12 as a tumor suppressor; nevertheless, there is a growing body of evidence that CDK12 can support tumor growth under specific circumstances and thus act as a tumor oncogene. CDK12 therefore represents an alternative diagnostic approach for breast, ovarian and prostate tumors, especially when conventional treatment is not active and there is a need for more effective approaches, such as concept of synthetic lethality.
Methods: The discussed scientific papers can be reached at the PubMed and Scopus databases before 1th of April 2020.
Purpose: The aim of the review is to summarize current knowledge relevant to the function of CDK12 as a tumor suppressor or oncogene in various tumors and to discuss the use of specific CDK12 inhibitors for patient treatment. At the end of the article, we discuss the potential use of CDK12 in the treatment of specific tumors by its targeted inhibition in monotherapy or in combination with poly (ADP ribose) polymerase 1 (PARP1) and checkpoint kinase 1 (CHK1) inhibitors.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Keywords:
breast cancer – ovarian cancer – cyclin-dependent kinase 12 – CDK12 – PARP1 – CHK1
Autoři:
M. Dzimková 1; J. Procházková 1; Jaroslav Klát 2
![]() ; J. Kohoutek 1
; J. Kohoutek 1
Působiště autorů:
Oddělení chemie a toxikologie, Výzkumný ústav veterinárního lékařství, v. v. i., Brno
1; Onkogynekologické oddělení, FN Ostrava
2
Vyšlo v časopise:
Klin Onkol 2020; 33(4): 260-267
Kategorie:
Přehled
doi:
https://doi.org/10.14735/amko2020260
Souhrn
Východiska: Fyziologická aktivita cyklin-dependentní kinázy 12 (CDK12) je důležitá pro celou řadu buněčných procesů, vč. regulace transkripce, sestřihu RNA, terminaci transkripce a polyadenylaci. Je zřejmé, že CDK12 řídí transkripci specifických genů, které jsou zapojeny v odpovědi buňky na poškození DNA, v replikaci DNA a v odpovědi buňky na buněčný stres. V nedávné době byla popsána klíčová úloha CDK12 v iniciaci nádorové promoce skrze indukci tandemových duplikací specifických úseků DNA v metastazujících nádorech prostaty rezistentních na kastraci. Data z publikovaných studií ukazují na výjimečnou dualitu CDK12, kterou lze v závislosti na typu a rozsahu poškození považovat jak za nádorový supresor, tak i za onkogen. CDK12 tudíž představuje jednu z možností pro diagnostiku a léčbu nádorových onemocnění, zejména rakoviny prsu, vaječníků a prostaty, především poté, kdy konvenční léčba již přestane fungovat, a bude nutné využít např. přístupu syntetické letality pro vhodnou terapii nádorových onemocnění rezistentních ke konvenčním způsobům léčby.
Metody: Diskutovaná odborná sdělení byla získána v databázích PubMed a Scopus před 1. dubnem 2020.
Cíl: Cílem předkládané práce je shrnout dosavadní poznatky o úloze CDK12, v různých typech nádorů, a na konkrétních příkladech dokumentovat její funkci, jednak jako nádorového supresoru, ale také jako onkogenu, s potenciálním využitím nově vyvinutých inhibitorů CDK12 v léčbě nádorových onemocnění. V neposlední řadě se zabýváme využitím inhibice CDK12 v léčbě určitých typů nádorových onemocnění jak cílenou inhibicí kinázové aktivity CDK12, tak v kombinaci s inhibitory poly (ADP ribóza) polymerázy 1 (PARP1) a „checkpoint“ kinázy 1 (CHK1).
Klíčová slova:
cyklin-dependentní kináza 12 – CDK12 – karcinom prsu – karcinom vaječníků – PARP1 – CHK1
Úvod
Proces iniciace a rozvoje maligního onemocnění v současném pojetí představuje deregulaci řady fyziologických buněčných dějů, které nádor využívá k vlastnímu prospěchu. Hanahan a Weinberg v roce 2000 popsali šest takových procesů, a to:
- soběstačnost v růstových faktorech;
- rezistence na inhibitory růstu;
- deregulace a inhibice apoptózy;
- neomezené buněčné dělení (buněčný cyklus);
- podpora a udržování angiogeneze;
- tkáňová invazivita a metastazování.
Následně je rozšířili o další děje, které jsou v nádorech cíleně deregulovány, a to:
- narušení metabolické rovnováhy v buňce;
- únik nádorových buněk před specifickou imunitní odpovědí;
- nádor iniciující zánětlivé procesy;
- mutace a genomová nestabilita [1,2].
A právě narušení buněčného cyklu nejen v iniciaci, ale i v průběhu rozvoje nádorového onemocnění je zcela zásadním předpokladem pro nádorové onemocnění [3,4]. Regulace buněčného cyklu je primárně řízena aktivitou specifických enzymů, tzv. cyklin-dependentních kináz (cyclin-dependent kinase – CDK), které se ale významně podílejí i na řízení dalších buněčných procesů [5]. Z literatury je známo, že deregulace CDK a následné zvýšení jejich kinázové aktivity často přispívá ke vzniku a rozvoji onkologických onemocnění [2]. Není tedy překvapením, že CDK jsou slibným cílem studií zaměřených na popis nových léčebných postupů vhodných k léčbě nádorových onemocnění [6,7]. Kináza CDK12 patří do skupiny kináz asociovaných s transkripcí, které se podílí na různých buněčných procesech, vč. odpovědi buňky na poškození DNA (DNA damage response – DDR), replikaci DNA a sestřihu RNA [8,9]. Studie zaměřené na funkci CDK12 odhalily případy nádorů se zvýšenou hladinou CDK12, ale i případy, u kterých je prokázána ztráta funkce této kinázy, což staví CDK12 jak do role onkogenu, tak do role nádorového supresoru [10]. Předkládaný přehledový článek přináší ucelený pohled na dosavadní poznatky o úloze CDK12 v různých typech nádorů a na konkrétních příkladech dokumentuje její funkci jednak jako nádorového supresoru, ale také jako onkogenu s potenciálním využitím nově vyvinutých inhibitorů CDK12 v léčbě nádorových onemocnění.
Funkce cyklin-dependentních kináz
Cyklin-dependentní kinázy jsou významnými regulátory řady buněčných procesů [11]. Z pohledu jejich funkce se dělí na dvě podskupiny, a to CDK buněčného dělení, které přímo regulují průchod jednotlivými fázemi buněčného cyklu (CDK1, 2, 4, 6), a kinázy asociované s řízením jednotlivých fází transkripce (CDK7, 8, 9, 11, 12 a 13) [12–14]. Regulace genové transkripce pomocí CDK se odehrává skrze fosforylaci C-terminální domény (CTD) podjednotky RNA polymerázy II (RNA pol II), která je odpovědná za transkripci protein kódujících genů a nekódujících RNA (např. tzv. small nuclear RNA – sncRNA) [15]. Eukaryotická transkripce je vysoce komplexní a energeticky náročný proces. Konkrétní fáze transkripce, vč. průchodu mezi nimi, musí být pečlivě regulovány. Obecně lze konstatovat, že veškeré základní buněčné procesy závisí na bezchybné regulaci transkripce a následné koordinaci s dalšími ději v buňce [16]. A právě narušení a deregulace této křehké rovnováhy může mít za následek vznik a rozvoj nádorového onemocnění. Z provedených analýz víme, že kinázová aktivita CDK je často deregulována v nádorových buňkách; není tedy překvapením, že CDK jsou atraktivním cílem výzkumu zaměřeného na jejich využití v klinické léčbě pacientů [6].
Vlastní proces transkripce je řízen na mnoha úrovních. O časoprostorové dynamice transkripce rozhoduje nejen sekvence DNA, metylace CpG či epigenetický kód, ale především také aktivita transkripčních faktorů [16]. Transkripční faktory (TF) jsou proteiny vázající DNA sekvenci v multiproteinových komplexech. Ty TF, jejichž ztráta má za následek rozvoj nádoru, se označují jako supresory, zatímco TF, které se podílejí na vzniku nádorového onemocnění, se nazývají onkogeny. V případě, že se nádorové buňky stávají závislými na funkci daného transkripčního faktoru, hovoříme o tzv. fenoménu transkripční závislosti [17]. Je proto přirozené se domnívat, že by bylo možné tuto transkripční závislost využít v léčbě pacientů.
Objev CDK12 a její fyziologická úloha v organizmech
V roce 2001 byla poprvé popsána CDK12 (CRKRS, CRK7) jako kináza, která se pravděpodobně podílí na regulaci transkripce [18]. Avšak teprve v roce 2011 byla popsána její funkce v transkripci a současně byl potvrzen její vazebný partner, cyklin K (CycK) [19]. Nicméně s cycK asociuje také cyklin-dependentní kináza 13 (CDK13), homolog CDK12, která je výrazně méně prostudovaná a její funkce je prozatím nejasná. Z pohledu jejich funkce v buňce je důležité zmínit, že obě kinázy tvoří s CycK dva separátní komplexy, které se s velkou pravděpodobností podílí na různých buněčných procesech [19]. CDK12 je exprimována v celém savčím organismu, nicméně její hladina se liší v závislosti na konkrétní tkáni. Vysoké hladiny CDK12 byly u člověka nalezeny v ovariích, varlatech, mozku, leukocytech a v nadledvince, což naznačuje, že CDK12 může mít tkáňově specifickou úlohu v diferenciaci specifických typů buněk. Pokusy na myších demonstrovaly, že CDK12 hraje důležitou roli v udržování pluripotentního stavu embryonálních kmenových buněk [20]. V průběhu časné embryogeneze vedla úplná ztráta funkce CDK12 nebo jejího vazebného partnera CycK k embryonální letalitě v období blastocysty, a to kvůli deregulované transkripci DDR genů [19,20]. Uvědomíme-li si, že jedna ze základních charakteristik nádorových buněk je schopnost jejich dediferenciace, je možné usuzovat, že CDK12 se také podílí na udržení nediferencovaného stavu i v tomto typu buněk.
CDK12 – regulace transkripce
Jak již bylo zmíněno v úvodu, funkce CDK12 je spjata s regulací elongační fáze transkripce skrze fosforylaci CTD RNA pol II, konkrétně fosforylací serinu v pozici 2 (Ser2) [13]. Experimentální snížení hladiny CDK12 v buněčných liniích vede k významnému poklesu hladiny fosforylovaného Ser2, a tudíž k útlumu elongační fáze transkripce, což vede k výraznému oslabení transkripce genů zapojených v odpovědi buňky na poškození DNA a genů nezbytných pro iniciaci a průběh replikace DNA [8,21]. Podobně jako CDK9 může CDK12 vázat a následně fosforylovat i jiné transkripční faktory/komplexy, a regulovat tak transkripci jiným mechanizmem, než fosforylací CTD RNA pol II. Dále se domníváme, že kromě funkce v regulaci elongační fáze transkripce participuje CDK12 také v procesech sestřihu RNA a terminaci transkripce [22,23]. V prvním případě CDK12 lokalizuje do jaderných struktur (tzv. nuclear speckles), kde dochází k sestřihu RNA, a skutečně, za využití hmotnostní spektrometrie, byla potvrzena asociace CDK12 s proteiny účastnícími se sestřihu RNA [18,24,25]. CDK12 dále řídí alternativní sestřih posledního exonu v genech s velkým počtem exonů a obecně delší mRNA [26]. V případě terminace byla prokázána přímá souvislost mezi fosforylací Ser2 v CTD RNA pol II CDK12 kinázou a efektivní vazbou faktoru Ctsf77, který zabezpečuje terminaci transkripce a polyadenylaci RNA [22]. Z toho je patrné, že elongace a terminace transkripce jsou jedněmi z řídicích mechanizmů genové exprese a narušení této regulace často vede k nefyziologické změně hladin nádorových supresorů a onkogenů v buňce, a tím k iniciaci nebo rozvoji nádorového onemocnění [23,27].
Vliv ztráty funkce CDK12 na transkripci specifických genů
Klasickým a v dnešní době velmi dobře popsaným příkladem vlivu ztráty funkce CDK12 na expresi genů je kontrola transkripce DDR genů, které se, kromě jiného, podílí na regulaci homologní rekombinace (HR) a pravděpodobně i na dalších opravných mechanizmech [19,28]. Efekt CDK12 na transkripci DDR genů se odvíjí na mnoha úrovních. Snížená hladina CDK12 vede k nedostatečné iniciaci elongační fáze kvůli snížené fosforylaci Ser2 v RNA pol II [29]. Skupina prof. Sharpa dále prokázala, že ztráta CDK12 má za následek produkci aberantních transkriptů RNA, které jsou předčasně polyadenylovány, a tudíž nesou informaci o nefunkčním proteinu. A právě DDR geny se vyznačují tím, že ve svých intronech nesou polyadenylační místa vázaná na intron, která se aktivují ztrátou fyziologické funkce CDK12 [30]. Nicméně je pravdou, že buňky s umlčenou nebo katalyticky inaktivní mutantní formou CDK12 vykazují větší poškození endogenní DNA a poruchy v HR [29]. Podobný efekt byl také pozorován i po ovlivnění specifickým inhibitorem CDK12 kinázy, THZ531 [31]. Výsledná deregulace CDK12 vede nejen k buněčné smrti, ale také k senzitizaci buněk k různým látkám poškozujícím DNA, např. etoposid, mitomycin C a kamptotecin [19]. Mutantní neaktivní formy CDK12 navíc zvyšují citlivost k cis-platině a inhibitorům poly (ADP ribóza) polymerázy (PARP) 1/2 [28,32,33]. Naše skupina v nedávné době prokázala, že ztráta funkce CDK12 zvyšuje citlivost řady nádorových buněčných linií k inhibitorům enzymu „checkpoint“ kinázy 1 (CHK1) [34].
Zcela recentně byl navíc prokázán vliv inaktivace enzymatické aktivity CDK12 na transkripci genů, které řídí ustanovení preiniciačního a prereplikačního komplexu. A tyto proteiny jsou nezbytnou podmínkou pro zahájení replikace DNA v průběhu buněčného dělení. Skupina dr. Li ukázala, že CDK12 řídí společně s cyklinem K fosforylaci cyklinu E1 na serinu v pozici 366 a tím brání jeho vazbě s CDK2 během sestavování prereplikačního komplexu [35]. Naproti tomu skupina dr. Blažka prokázala, že aktivita CDK12 je nezbytná pro transkripci genů nutných k úspěšné replikaci a k přechodu G1/S v průběhu buněčného dělení [8]. A právě regulace preiniciačního a prereplikačního komplexu aktivitou CDK12 ukazuje na nové funkční propojení mezi regulací transkripce a replikací DNA.
Obecně je tedy možné konstatovat, že se CDK12 podílí na zachování genomové stability nejen skrze kontrolu transkripce DDR genů, vč. genů účastnících se HR, ale také prostřednictvím regulace transkripce genů nezbytných pro replikaci DNA. Není tedy překvapením, že právě poruchy těchto mechanizmů jsou jednou z charakteristik nádorového onemocnění [2]. Výše zmíněná pozorování tudíž vedla k hypotéze, že CDK12 se na místo ovlivnění celkové transkripce podílí na transkripci pouze specifických skupin genů, a tak se aktivně angažuje při stresových odpovědích [36,37]. Nicméně dle nedávných studií je zřejmé, že aplikace nízké dávky inhibitoru CDK12 vede ke snížení exprese genů pro DDR, BRCA1, FANCI, ERCC4 a dalších, zatímco použití vyšší dávky inhibitorů mělo za následek redukci transkripce genů asociovaných se super-enhancery v porovnání s typickými enhancery [27]. Zvýšená aktivita CDK12 by tak mohla urychlovat progresi a rezistenci nádorů a paradoxně ji stavět do role onkogenu místo předpokládané tumor supresivní role.
Mutace CDK12 v různých typech nádorů
Ztráta funkce CDK12 je častým jevem, ke kterému dochází v různých typech solidní nádorů. Z pohledu frekvence u vybraných typů nádorů byla prevalence ztráty funkce alespoň jedné alely CDK12 nalezena u nádorů močového měchýře (3,7 %), prostaty (3,4 %), jícnu a žaludku (2,1 %) a ledvin (2,1 %) [38]. Inaktivace obou alel CDK12 byla pozorována v nádorech prostaty (1,8 %), vaječníku (1,0 %) a močového měchýře (0,5 %). Specifické mutace asociované s rozvojem nádoru byly popsány pro nemalobuněčný karcinom plic [27], adenokarcinom plic [39,40], folikulární lymfom [41] a pro nádor jícnu [42]. Pro potřeby přehledového článku uvádíme výskyt mutací v nádorech prsu, vaječníku, děložního čípku a prostaty (graf 1).

High-grade serózní karcinom vaječníku
Pro 50 % všech případů high-grade serózního karcinomu vaječníku (HGSOC) je charakteristická porucha exprese genů pro HR a genomová nestabilita [43]. Nejčastěji mutovanými geny spojenými s HR jsou BRCA1, BRCA2 a také CDK12, která je jednou z rekurentních mutací v HGSOC, a to v přibližně ve 3 % případů [43]. Ve velké většině případů se jedná o primární homozygotní bodovou mutaci, převážně v kinázové doméně, což má za následek ztrátu kinázové aktivity [29]. U některých mutantních forem CDK12 je sice zachována enzymatická aktivita, ale došlo u nich k narušení vazby k cyklinu K, což má za následek sníženou expresi HR genů a nižší efektivitu oprav DNA [29]. Studie provedené na vzorcích z pacientů poukázaly na velmi zajímavý fakt, a to že nádory s mutací v BRCA1 a BRCA2 téměř výhradně nenesou mutaci pro CDK12 [29,44]. Daná pozorování tudíž podporují hypotézu, že jak BRCA, tak i CDK12 jsou součástí DDR dráhy a tento stav potvrzují i další pozorování v nádorech, ve kterých snížena hladina DDR genů asociuje s mutovanou formou CDK12.
CDK12 – deregulace u metastazujících nádorů prostaty rezistentních na kastraci (mCRPC)
Několik studií v nedávné době velmi přesvědčivě prokázalo, že genomové přestavby, které jsou hojně pozorovány v mCRPC, přímo souvisí s inaktivací funkce CDK12 [45–47]. Je zajímavé, že inaktivace obou alel genu pro CDK12 je nezbytná pro tvorbu genomových přestaveb v definovaném subtypu mCRPC nádorů, jejichž progrese nesouvisí s poškozenou DDR a s dalšími procesy, jako jsou tzv. fúze ETS (E26 transformation-specific) genů, nebo s mutacemi v genu SPOP (speckle-type POZ protein) [47]. Ztráta CDK12 dále vede ke genomovým přestavbám, které jsou charakteristické tandemovými duplikacemi, jež zahrnují oblasti enhanceru pro gen kódující androgenní receptor nebo protoonkogen MYC [46]. Tandemové duplikace vedoucí ke zvýšené expresi genů bylo možné pozorovat také v regulačních oblastech genů zapojených v řízení buněčného cyklu a v replikaci DNA [47]. Naopak tandemové duplikace vedoucí k inhibici exprese genů byly pozorovány u nádorových supresorů, TP53 a BRCA2 [45]. Daný podtyp mCRPC nádorů s mutací v genu pro CDK12 vykazoval různorodou expresi neoantigenů v důsledku genomových přetaveb a zvýšenou infiltraci nádorů T buňkami [47]. Je tedy možné usuzovat, že léčba za využití specifických inhibitorů PD-1 (programmed death-1) a PD-L1 (programmed death-ligand 1) by mohla být u mCRPC pacientů s nefunkční CDK12 velmi efektivní [48]. Obecně má tedy ztráta specifických nádorových supresorů (TP53, BRCA1, CDK12) za následek zvýšený výskyt určitého typu tandemové duplikace o určité délce [49].
Karcinom prsu
Triple-negativní nádory prsu (TNBC) obsahují spektrum mutací, které jsou podobné nádorům vaječníků, tj. nemají amplifikovaný žádný z typických receptorů – ER, HER2 nebo PR, ale vykazují mutace v DDR genech, což má za následek zvýšenou úroveň genové nestability. Mezi geny, u kterých je možné pozorovat opakující se mutace, patří zejména TP53 (80 % případů) a BRCA1 (30 % případů) [50]. V TNBC byly detekovány mutace v genu pro CDK12, a to v 1,5 % případů [50,51]. Je tedy pravděpodobné, že pacientky s TNBC s poškozeným HR kvůli ztrátě funkce CDK12 mohou být citlivé na léčbu inhibitory PARP1/2 [50].
Jiný typ nádoru prsu je charakteristický amplifikací genu/onkogenu HER2 (ERBB2, EGFR2) a označuje se jako HER2 pozitivní karcinom prsu (HER2+) [52]. Protein HER2 patří do rodiny tyrozinových kináz a je schopný stimulovat proliferaci a inhibovat apoptózu [50]. Gen pro CDK12 se nachází v blízkosti genu pro ERBB2 v genovém lokusu17q12-q21, a není proto překvapením, že v tomto typu nádoru pozorujeme až 71 % koamplifikací genu CDK12 společně s ERBB2 genem [53,54]. Z mnoha studí je zřejmé, že jak samotný HER2, tak i další koamplifikované geny mohou přispívat a skutečně přispívají k rozvoji nádoru prsu a splňují kritéria pro onkogen, a jsou tak vhodnými kandidáty pro cílenou terapii [52]. V případě genu pro CDK12 prokázaly minimálně dvě skupiny skutečně zvýšenou hladinu CDK12 na úrovni mRNA i samotného proteinu a navíc prokázaly významnou korelaci mezi zvyšující se hladinou CDK12 a gradingem nádoru [54,55]. Obdobně byla amplifikace genu CDK12 nalezena u HER2 pozitivních nádorů žaludku [56]. V tomto případě by CDK12 mohla sloužit jako vhodný prognostický marker, což potvrzují také pozorování zaměřená na validaci CDK12 coby využitelného biomarkeru pro Her2+ nádory [57,58]. V případě HER2+ karcinomů prsu se můžeme setkat i s případy, kdy amplifikace HER2 lokusu 17q12-q21 má za následek poškození čtecího rámce CDK12, a tím expresi její aberantní nefunkční formy [59]. Kromě amplifikace je dále v nádorech HER2+ možné pozorovat i bodové mutace v genu CDK12 [60]. V případě estrogen pozitivních karcinomů prsu byla nalezena příčinná souvislost mezi inhibicí aktivity CDK12 a ztrátou citlivosti nádorových buněk na léčbu tamoxifenem [61].
Obecně lze proto konstatovat, že nádory prsu se vzájemně liší mírou a typem poškození funkce CDK12, a tedy i dopadem tohoto poškození při cílené léčbě. Zatímco nádory s nízkou aktivitou nebo úplnou ztrátou funkce CDK12 vedoucí ke genomové nestabilitě jsou vhodnými kandidáty pro nasazení PARP1/2 nebo CHK1 inhibitorů, nádory s amplifikovanou nebo zvýšenou expresí CDK12 vhodným cílem pro léčbu specifickými inhibitory CDK12.
Inhibitory CDK12 a syntetická letalita
Protinádorový potenciál celé plejády inhibitorů CDK je klinicky dlouhodobě testován. Kromě širokospektrálních inhibitorů CDK, jakými jsou např. flavopiridol nebo roskovitin, byly v průběhu let intenzivně vyvíjeny také inhibitory, které cílily na aktivitu CDK zapojených nejen v regulaci buněčného cyklu, ale také v transkripci [6]. Jak bylo dokumentováno v předchozí části, CDK12 je hojně mutovaná nebo naopak vykazuje zvýšenou expresi v různých typech nádorů, tudíž specifické inhibitory pro CDK12 by mohly poskytovat atraktivní strategii v léčbě pacientů.
Jedním z potencionálních inhibitorů CDK12 je i dinaciclib, původně zařazený jako inhibitor CDK1, CDK2, CDK5 a CDK9, který má antiproliferativní účinek na různé nádorové buněčné linie [62]. V průběhu let se ukázalo, že dinaciclib je schopný inhibovat CDK12 již v daleko nižších koncentracích než CDK9. Aplikace dinaciclibu vedla k inhibici kinázové aktivity CDK12, snížení exprese HR genů a nižší fosforylaci CTD RNA pol II [28]. Kombinace PARP inhibitorů, veliparibu a inhibitoru CDK12 dinaciclibu efektivně zastavila růst nádorů u myšího modelu xenograftu [28]. Daná pozorování naznačují možnost využití CDK12 inhibitorů ke zcitlivění nádorů rezistentních k PARP1/2 inhibitorům.
Selektivním a silným inhibitorem CDK12 je nedávno vyvinutý THZ531. Mezi jeho efekty patří indukovaná apoptóza, inhibice elongační fáze transkripce, snížená exprese genů DDR a snížená exprese genů závislých na super-enhancerech [27]. Negativní efekt THZ531 na proliferaci byl pozorován u buněčné linie odvozené z akutní lymfoblastické leukemie [28]. Schopnost THZ531 zastavit buněčné dělení a indukovat senescenci nebo apoptózu byla nedávno pozorována u buněk anaplastického karcinomu štítné žlázy a hepatocelulárního karcinomu [31,63].
Proces iniciace a rozvoje nádorového onemocnění je v mnoha typech nádoru zapříčiněn zvýšenou aktivitou nádorových onkogenů a je velmi obtížné inhibovat jejich funkci. Klasickým příkladem je zvýšená exprese cMYC onkogenu u řady nádorů. Právě CDK12 je jedním z faktorů podílejících se na transkripci MYC genu [22]. Navíc testy zaměřené na plošnou identifikaci faktorů vykazujících syntetickou letalitu s onkogenem cMYC odhalily CDK12 jako jednoho z možných kandidátů pro využití k léčbě nádorů s vysokou aktivitou cMYC [64]. Dalším příkladem závislosti nádorové progrese na aktivitě silného onkogenu je fúzní protein EWS/FLI, který je exprimován v Ewingově sarkomu [65]. Ovlivnění buněk inhibitorem CDK12 aktivity THZ531 vedlo ke snížení exprese genů DDR [66]. A právě jedním z možných přístupů, jak překonat rezistenci vůči používaným léčebným terapeutikům, může být cílená inhibice CDK kináz [67].
Ztráta CDK12 a senzitivita k PARP1/2 inhibitorům
Nestabilita genomu je jednou z obecných vlastností většiny nádorových onemocnění, a proto je vhodným cílem pro potřeby současné i budoucí léčby pacientů. Pro správný účinek PARP1 inhibitorů je podmínkou oslabená nebo nefunkční HR, jež je poškozena v řadě nádorů [68]. Nádory s mutací regulátorů homologní rekombinace BRCA1 a BRCA2 jsou tudíž vhodnými kandidáty pro použití PARP inhibitorů [69,70]. Některé nádory si však dokáží v průběhu léčby vybudovat rezistenci k PARP inhibitorům, a proto je do budoucna potřeba aktivně vyvíjet nové způsoby, jak obnovit citlivost buněk na PARP1 inhibitory [71,72]. Není s podivem, že CDK12 byla odhalena jako jeden z faktorů určující citlivost k PARP inhibitorům [32]. Nádorové ovariální buňky se sníženou hladinou CDK12 byly prokazatelně citlivější k PARP inhibitoru olaparibu a cílené snížení hladiny CDK12 v dalších nádorových buněčných linií vedlo ke zvyšování citlivosti buněk k olaparibu. Navíc kombinace inhibitoru CDK12 dinaciclibu a PARP inhibitoru olaparibu mělo za následek zastavení růstu nádorových buněk in vitro, in vivo a v neposlední řadě i v modelu xenograftu derivovaného z tkáně pacientů [28].
Závěr
Fyziologická aktivita CDK12 je důležitá pro řadu buněčných procesů, vč. regulace transkripce, sestřihu RNA, terminaci transkripce a polyadenylaci. CDK12 řídí transkripci specifických genů, které jsou s vysokou aktivitou cMYC přímo zapojeny v DDR, replikaci DNA a v odpovědi na buněčný stres. Aberantní exprese CDK12 je hojně rozšířena napříč různými nádory u člověka. Data z nedávných studií ukazují na výjimečnou dualitu CDK12, kterou lze v závislosti na typu a rozsahu poškození považovat jak za nádorový supresor, tak i za onkogen. V případě HGSOC, TNBC, podtypu HER2+ nádorů prsu, adenokarcinomu plic a metastazujícího adenokarcinomu prostaty se CDK12 jeví jako nádorový supresor. Její snížená hladina v těchto nádorech negativně ovlivňuje expresi DDR genů, což má za následek poškození procesu HR a genomovou nestabilitu. Nicméně právě mutace typu „loss-of-function“ vedoucí ke ztrátě CDK12 aktivity zvyšují citlivost nádorů k PARP1/2 a PARP inhibitorům (obr. 1A).
A) CDK12 – nádorový supresor. Ztráta funkce CDK12 má za následek sníženou expresi genů zapojených v homologní rekombinaci a replikaci
DNA, vč. kontroly tandemových duplikací, což vede ke genomové nestabilitě a rozvoji karcinogeneze. Nefunkční CDK12 nebo použití
specifických inhibitorů CDK12 zvyšuje citlivost nádorových buněk k efektu PARP1/2 (PARPi) a inhibitorů CHK1 (CHK1i).
B) CDK12 – onkogen. Amplifi kace CDK12 genu má za následek zvýšenou expresi genů zapojených v genomové stabilitě a karcinogenezi.
Proto je možné uvažovat o využití specifi ckých CDK12 inhibitorů v léčbě pacientů s tímto typem nádorů.
C) CDK12 – syntetická letalita. Fyziologická hladina CDK12 udržuje expresi vybraných onkogenů (MYC, EWS/FLI), jejichž aktivita je nezbytná
pro přežití nádoru. V případě, kdy je rozvoj nádoru závislý na aktivitě konkrétního onkogenu a současně na intaktní DDR dráze,
může vést inhibice CDK12 k narušení obou drah nutných pro přežití a následně k zástavě proliferace nádorových buněk.
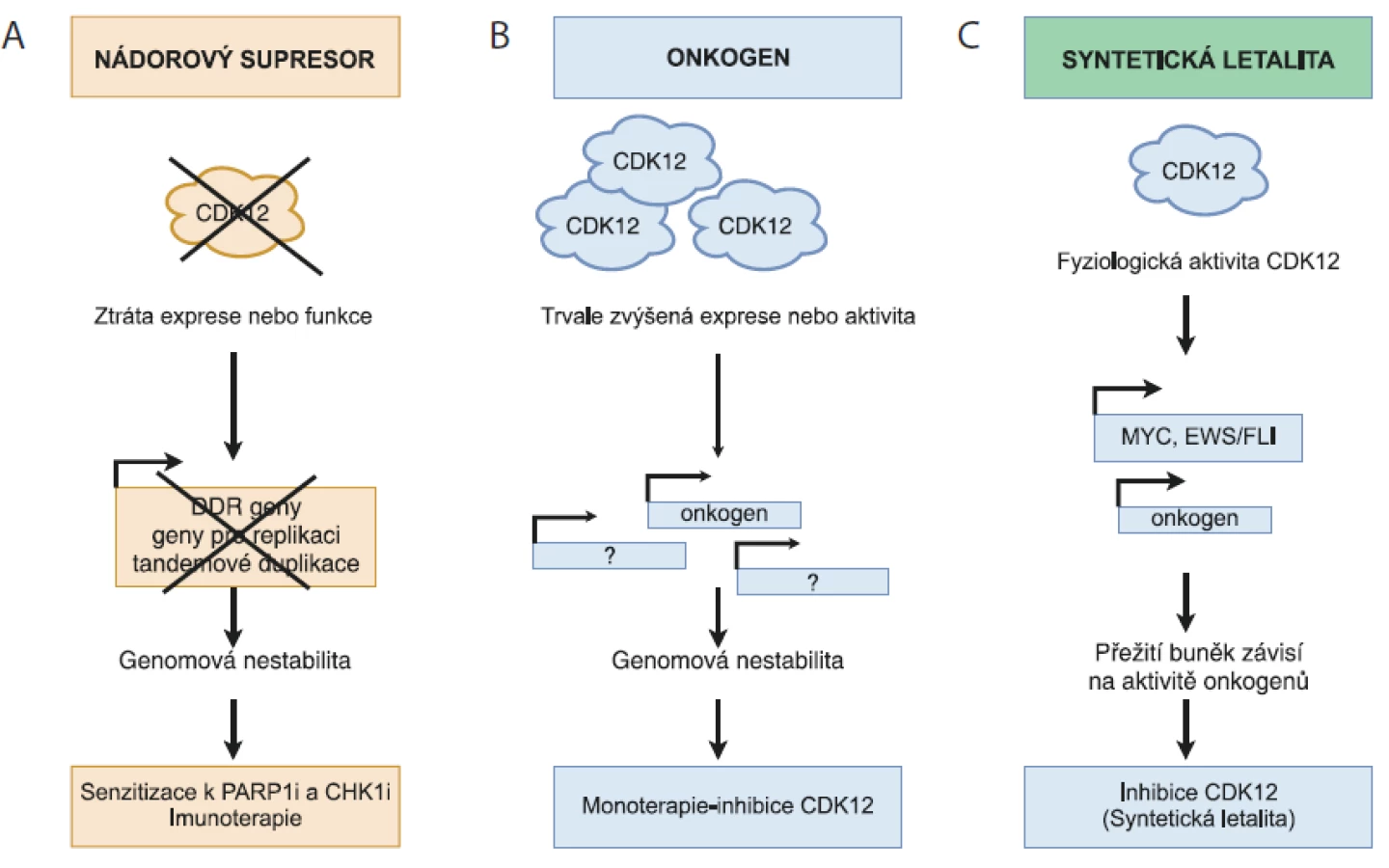
Naproti tomu v nádoru prsu subtypu HER2+ a v nádoru žaludku je možné CDK12 charakterizovat jako klasický onkogen, protože její zvýšená exprese zapříčiňuje agresivnější formu nádoru (obr. 1B). Obdobná situace nastává i v případě nádorů s vysokou aktivitou cMYC a fúzního proteinu EWS/ELI. Nicméně v tomto případě se jedná o situaci, která odpovídá konceptu transkripční závislosti, kdy jsou dané nádory závislé na konkrétním transkripčním faktoru proto, aby mohly efektivně zachovat nastavený nádorový program transkripce. V tomto kontextu by pak inhibitory transkripčních kináz mohly být vhodným způsobem, jak překonat transkripční závislost proliferujících nádorových buněk (obr. 1C).
A právě dualita CDK12 (supresor vs. onkogen) jí propůjčuje všestranné využití při personalizované léčbě pacientů. CDK12 představuje jednu z možností budoucí diagnostiky a léčby náročných nádorových onemocnění, zejména rakoviny prsu, vaječníků a prostaty především poté, kdy konvenční léčba již přestane fungovat a bude nutné využít např. přístupu syntetické letality pro vhodnou terapii nádorových onemocnění rezistentních ke konvenčním způsobům léčby.
Poděkování
Autorka by ráda poděkovala členům laboratoře za cenné připomínky při psaní přehledového článku.
Souhrnný článek vznikl za podpory programového projektu Ministerstva zdravotnictví ČR s reg. č. 16-34152A. Veškerá práva podle předpisů na ochranu duševního vlastnictví jsou vyhrazena.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Jiří Kohoutek, Ph.D.
Oddělení chemie a toxikologie
Výzkumný ústav veterinárního
lékařství, v. v. i.
Hudcova 296/70
621 00 Brno
e-mail: kohoutek76@gmail.com
Obdrženo/Submitted: 28. 3. 2020
Přijato/Accepted: 23. 4. 2020
Zdroje
1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100 (1): 57–70. doi: 10.1016/s0092-8674 (00) 81683-9.
2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144 (5): 646–674. doi: 10.1016/j.cell.2011.02.013.
3. Kent LN, Leone G. The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 2019; 19 (6): 326–338. doi: 10.1038/s41568-019-0143-7.
4. Whittaker SR, Mallinger A, Workman P et al. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther 2017; 173: 83–105. doi: 10.1016/j.pharmthera.2017.02.008.
5. Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009; 28 (33): 2925–2939. doi: 10.1038/onc.2009.170.
6. Asghar U, Witkiewicz AK, Turner NC et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov 2015; 14 (2): 130–146. doi: 10.1038/nrd4504.
7. Bruyere C, Meijer L. Targeting cyclin-dependent kinases in anti-neoplastic therapy. Curr Opin Cell Biol 2013; 25 (6): 772–779. doi: 10.1016/j.ceb.2013.08.004.
8. Chirackal Manavalan AP, Pilarova K, Kluge M et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep 2019; 20 (9): e47592. doi: 10.15252/embr.201847592.
9. Kohoutek J, Blazek D. Cyclin K goes with Cdk12 and Cdk13. Cell Div 2012; 7: 12. doi: 10.1186/1747-1028-7-12.
10. Greenleaf AL. Human CDK12 and CDK13, multi-tasking CTD kinases for the new millenium. Transcription 2019; 10 (2): 91–110. doi: 10.1080/21541264.2018.1535211
11. Malumbres M. Cyclin-dependent kinases. Genome Biol 2014; 15 (6): 122. doi: 10.1186/gb4184.
12. Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nature Reviews Molecular Cell Biology 2016; 17 (5): 280–292. doi: 10.1038/nrm.2016.27.
13. Kohoutek J. P-TEFb – the final frontier. Cell Div 2009; 4: 19. doi: 10.1186/1747-1028-4-19.
14. Malumbres M, Harlow E, Hunt T et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol 2009; 11 (11): 1275–1276. doi: 10.1038/ncb1109-1275.
15. Eick D, Geyer M. The RNA polymerase ii carboxy-terminal domain (CTD) code. Chemical Reviews 2013; 113 (11): 8456–8490. doi: 10.1021/cr400071f.
16. Jeronimo C, Bataille AR, Robert F. The writers, readers, and functions of the RNA polymerase II C-terminal domain code. Chemical Reviews 2013; 113 (11): 8491–8522. doi: 10.1021/cr4001397.
17. Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell 2017; 168 (4): 629–643. doi: 10.1016/j.cell.2016.12.013.
18. Ko TK, Kelly E, Pines S. CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J Cell Sci 2001; 114 (Pt 14): 2591–2603.
19. Blazek D, Kohoutek J, Bartholomeeusen K et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 2011; 25 (20): 2158–2172. doi: 10.1101/gad.16962311.
20. Juan HC, Lin Y, Chen HR et al. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ 2016; 23 (6): 1038–1048. doi: 10.1038/cdd.2015.157.
21. Blazek D. The cyclin K/Cdk12 complex: an emerging new player in the maintenance of genome stability. Cell Cycle 2012; 11 (6): 1049–1050. doi: 10.4161/cc.11.6.19678.
22. Davidson L, Muniz L, West S. 3‘ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev 2014; 28 (4): 342–356. doi: 10.1101/gad.231274.113.
23. Eifler TT, Shao W, Bartholomeeusen K et al. Cyclin-dependent kinase 12 increases 3‘ end processing of growth factor-induced c-FOS transcripts. Mol Cell Biol 2015; 35 (2): 468–478. doi: 10.1128/MCB.01157-14.
24. Cheng SW, Kuzyk MA, Moradian A et al. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol Cell Biol 2012; 32 (22): 4691–4704. doi: 10.1128/MCB.06267-11.
25. Liang K, Gao X, Gilmore JM et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol 2015; 35 (6): 928–938. doi: 10.1128/MCB.01426-14.
26. Tien JF, Mazloomian A, Cheng SG et al. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res 2017; 45 (11): 6698–6716. doi: 10.1093/nar/gkx187.
27. Zhang T, Kwiatkowski N, Olson CM et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol 2016; 12 (10): 876–884. doi: 10.1038/nchembio.2166.
28. Johnson SF, Cruz C, Greifenberg AK et al. CDK12 inhibition reverses de novo and acquired PARP inhibitor Rresistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep 2016; 17 (9): 2367–2381. doi: 10.1016/j.celrep.2016.10.077.
29. Ekumi KM, Paculova H, Lenasi T et al. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res 2015; 43 (5): 2575–2589. doi: 10.1093/nar/gkv101.
30. Dubbury SJ, Boutz PL, Sharp PA. CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 2018; 564 (7734): 141–145. doi: 10.1038/s41586-018-0758-y.
31. Wang C, Wang H, Lieftink C et al. CDK12 inhibition mediates DNA damage and is synergistic with sorafenib treatment in hepatocellular carcinoma. Gut 2020; 69 (4): 727-736. doi: 10.1136/gutjnl-2019-318506.
32. Bajrami I, Frankum JR, Konde A et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res 2014; 74 (1): 287–297. doi: 10.1158/0008-5472.CAN-13-2541.
33. Joshi PM, Sutor SL, Huntoon CJ et al. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly (ADP-ribose) polymerase inhibitors. J Biol Chem 2014; 289 (13): 9247–9253. doi: 10.1074/jbc.M114.551143.
34. Paculova H, Kramara J, Simeckova S et al. BRCA1 or CDK12 loss sensitizes cells to CHK1 inhibitors. Tumour Biol 2017; 39 (10) doi: 1010428317727479. doi: 10.1177/1010428317727479.
35. Lei T, Zhang P, Zhang X et al. Cyclin K regulates prereplicative complex assembly to promote mammalian cell proliferation. Nat Commun 2018; 9 (1): 1876. doi: 10.1038/s41467-018-04258-w.
36. Li X, Chatterjee N, Spirohn K e al. Cdk12 is a gene-selective RNA polymerase II kinase that regulates a subset of the transcriptome, including Nrf2 target genes. Sci Rep 2016; 6: 21455. doi: 10.1038/srep21455.
37. Quereda V, Bayle S, Vena S et al. Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell 2019; 36 (5): 545–558. doi: 10.1016/j.ccell.2019.09.004.
38. Marshall CH, Imada EL, Tang Z et al. CDK12 inactivation across solid tumors: an actionable genetic subtype. Oncoscience 2019; 6 (5-6): 312–316. doi: 10.18632/oncoscience.481.
39. Biswas R, Gao S, Cultraro CM et al. Genomic profiling of multiple sequentially acquired tumor metastatic sites from an „exceptional responder“ lung adenocarcinoma patient reveals extensive genomic heterogeneity and novel somatic variants driving treatment response. Cold Spring Harb Mol Case Stud 2016; 2 (6): a001263. doi: 10.1101/mcs.a001263.
40. Zhang X, Nguyen KD, Rudnick PA et al. Quantitative mass spectrometry to interrogate proteomic heterogeneity in metastatic lung adenocarcinoma and validate a novel somatic mutation CDK12-G879V. Mol Cell Proteomics 2019; 18 (4): 622–641. doi: 10.1074/mcp.RA118.001266.
41. Geyer JT, Subramaniyam S, Jiang Y et al. Lymphoblastic transformation of follicular lymphoma: a clinicopathologic and molecular analysis of 7 patients. Hum Pathol 2015; 46 (2): 260–271. doi: 10.1016/j.humpath.2014.10.021.
42. Riches JC, Schultz N, Ku GY et al. Genomic profiling of esophagogastric (EG) tumors in clinical practice. J Clin Oncol 2015; 33 (3_suppl): 57–57.
43. Cancer Genome Atlas Research. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474 (7353): 609–615. doi: 10.1038/nature10166.
44. Carter SL, Cibulskis, K, Helman E et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 2012; 30 (5): 413–421. doi: 10.1038/nbt.2203.
45. Quigley D A, Dang HX, Zhao SG et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 2018; 174 (3): 758–769. doi: 10.1016/j.cell.2018.06.039.
46. Viswanathan SR, Ha G, Hoff AM et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018; 174 (2): 433–447. doi: 10.1016/j.cell.2018.05.036.
47. Wu YM, Cieslik, M, Lonigro RJ et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018; 173 (7): 1770–1782. doi: 10.1016/j.cell.2018.04.034.
48. Manogue C, Cotogno P, Ledet E et al. Biomarkers for Programmed Death-1 inhibition in prostate cancer. The Oncologist 2019; 24 (4): 444–448. doi: 10.1634/theoncologist.2018-0546.
49. Menghi F, Barthel FP, Yadav V et al. The tandem duplicator phenotype is a prevalent genome-wide cancer configuration driven by distinct gene mutations. Cancer Cell 2018; 34 (2): 197–210. doi: 10.1016/j.ccell.2018.06.008.
50. Cancer Genome Atlas. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490 (7418): 61–70. doi: 10.1038/nature11412.
51. Shah SP, Roth A, Goya R et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012; 486 (7403): 395–399. doi: 10.1038/nature10933.
52. Kauraniemi P, Kallioniemi A. Activation of multiple cancer-associated genes at the ERBB2 amplicon in breast cancer. Endocr Relat Cancer 2006; 13 (1): 39–49. doi: 10.1677/erc.1.01147.
53. Mertins P, Mani DR, Ruggles KV et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016; 534 (7605): 55–62. doi: 10.1038/nature18003.
54. Sircoulomb F, Bekhouche I, Finetti P et al. Genome profiling of ERBB2-amplified breast cancers. BMC Cancer 2010; 10: 539. doi: 10.1186/1471-2407-10-539.
55. Capra M, Nuciforo PG, Confalonieri S et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res 2006; 66 (16): 8147–8154. doi: 10.1158/0008-5472.CAN-05-3489.
56. Zhou C, Feng X, Yuan F et al. Difference of molecular alterations in HER2-positive and HER2-negative gastric cancers by whole-genome sequencing analysis. Cancer Manag Res 2018; 10: 3945–3954. doi: 10.2147/CMAR.S172710.
57. Chen K, Quan J, Yang J et al. The potential markers of endocrine resistance among HR+ /HER2+ breast cancer patients. Clin Transl Oncol 2020; 22 (4): 576–584. doi: 10.1007/s12094-019-02163-2.
58. Naidoo K, Wai PT, Maguire SL et al. Evaluation of CDK12 protein expression as a potential novel biomarker for DNA damage response-targeted therapies in breast cancer. Mol Cancer Ther 2018; 17 (1): 306–315. doi: 10.1158/1535-7163.MCT-17-0760.
59. Natrajan R, Wilkerson PM, Marchio C et al. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J Pathol 2014; 232 (5): 553–565. doi: 10.1002/path.4325.
60. Chen B, Zhang G, Wei G et al. Heterogeneity of genomic profile in patients with HER2-positive breast cancer. Endocr Relat Cancer 2020; 27 (3): 153–162. doi: 10.1530/ERC-19-0414.
61. Iorns E, Martens-De Kemp SR, Lord CJ et al. Ashworth CRK7 modifies the MAPK pathway and influences the response to endocrine therapy. Carcinogenesis 2009; 30 (10): 1696–1701. doi: 10.1093/carcin/bgp187.
62. Paruch K, Dwyer MP, Alvarez C et al. Discovery of dinaciclib (SCH 727965): A potent and selective inhibitor of cyclin-dependent kinases. ACS Med Chem Lett 2010; 1 (5): 204–208. doi: 10.1021/ml100051d.
63. Geng M, Yang Y, Cao X et al. Targeting CDK12-mediated transcription regulation in anaplastic thyroid carcinoma. Biochem Biophys Res Commun 2019; 520 (3): 544–550. doi: 10.1016/j.bbrc.2019.10.052.
64. Toyoshima M, Howie HL, Imakura M et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc Natl Acad Sci U S A 2012; 109 (24): 9545–9550. doi: 10.1073/pnas.1121119109.
65. May WA, Lessnick SL, Braun BS et al. The Ewing‘s sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol 1993; 13 (12): 7393–7398. doi: 10.1128/mcb.13.12.7393.
66. Iniguez AB, Stolte B, Wang EJ et al. EWS/FLI confers tumor cell synthetic lethality to CDK12 inhibition in Ewing sarcoma. Cancer Cell 2018; 33 (2): 202–216. doi: 10.1016/j.ccell.2017.12.009.
67. Rusan M, Li K, Li Y et al. Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer Discov 2018; 8 (1): 59–73. doi: 10.1158/2159-8290.CD-17-0461.
68. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481 (7381): 287–294. doi: 10.1038/nature10760.
69. Bryant HE, Schultz N, Thomas HD et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature 2005; 434 (7035): 913–917. doi: 10.1038/nature03443.
70. Helleday T, Bryant HE, Schultz N. Poly (ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 2005; 4 (9): 1176–1178. doi: 10.4161/cc.4.9.2031.
71. Montoni A, Robu M, Pouliot E et al. Resistance to PARP-inhibitors in cancer therapy. Front Pharmacol 2013; 4: 18. doi: 10.3389/fphar.2013.00018.
72. Noordermeer SM, Van Attikum H. PARP inhibitor resistance: A tug-of-war in BRCA-mutated cells. Trends Cell Biol 2019; 29 (10): 820–834. doi: 10.1016/j.tcb.2019.07.
008.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2020 Číslo 4
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Specifika v komunikaci s pacienty s ránou – laická doporučení
- MUDr. Lenka Klimešová: Multioborová vizita může být klíčem k efektivnější perioperační léčbě chronické bolesti
Nejčtenější v tomto čísle
- Zhubné nádory krčka maternice v gravidite
- Integrovaná diagnostika difúzních gliomů
- Atypický průběh typického karcinoidu plic
- Nemoc těžkých řetězců imunoglobulinu gama