
Systémová sklerodermie v roce 2017
Systemic sclerosis in 2017
Systemic sclerosis is classed as a diffuse (systemic) disease of connective tissue. It is a heterogeneous disease significantly shortening life expectancy. Its etiology is unknown. Pathogenetic interplay is assumed to involve a triad of pathological autoimmune inflammation, vasculopathy and fibrosis. Clinical manifestations can be classed based on the preponderant pathogenetic process. Vasculopathy is manifested by secondary Raynaud’s phenomenon with abnormal findings on the nailfold capillaroscopy, skin telangiectasias, gastric antral vascular ectasia, life threatening scleroderma renal crisis, digital ulcerations and prognostically severe pulmonary arterial hypertension. The treatment of vascular manifestations uses medicines with vasodilation effect. The manifestation of inflammation is accentuated by pleurisy, pericarditis, myositis, synovitis/arthritis and alveolitis. Finally, the manifestation of fibrosis predominates in association with dermatosclerosis, interstitial lung disease and fibrotic impairment of the gastrointestinal tract. Medicines with immunomodulatory or immunosupressive effects are used to affect the inflammation and fibrosis. Despite the aforementioned, there is still no universally effective treatment available. The pharmacological therapy of this disease is organ specific and symptomatic.
Key words:
capillaroscopy – digital ulcers – interstitial lung disease – pulmonary arterial hypertension – scleroderma renal crisis – systemic sclerosis
Autoři:
Tomáš Soukup 1; Tomáš Veleta 2,3
Působiště autorů:
Subkatedra revmatologie II. interní gastroenterologické kliniky LF UK a FN Hradec Králové
1; Oddělení urgentní medicíny FN Hradec Králové
2; Katedra interních oborů LF UK a FN Hradec Králové
3
Vyšlo v časopise:
Vnitř Lék 2018; 64(2): 146-154
Kategorie:
Přehledné referáty
Souhrn
Systémová sklerodermie patří do skupiny difuzních (systémových) onemocnění pojiva. Jedná se o heterogenní nemoc významně zkracující život. Etiologie je neznámá. V patogenetické souhře se předpokládá triáda patologického autoimunitního zánětu, vaskulopatie a fibrózy. Klinické projevy je možné rozdělit podle převahy patogenetického děje. Vaskulopatie se prezentuje sekundárním Raynaudovým fenoménem s abnormálními nálezy na kapilárách nehtového lůžka, kožními teleangiektaziemi, vaskulárními ektaziemi v antru žaludku, život ohrožující sklerodermickou renální krizí, digitálními ulceracemi a prognosticky závažnou plicní arteriální hypertenzí. Léčba vaskulárních projevů se zaměřuje na léky s vazodilatačním účinkem. Manifestace zánětu je akcentována u pleuritidy, perikarditidy, myozitidy, synovitidy/artritidy a alveolitidy. Konečně manifestace fibrózy dominuje při postižení kůže dermosklerózou, při intersticiálním plicním postižení a fibrotickém postižení stěny trávicí trubice. Na ovlivnění zánětu a fibrózy jsou používána léčiva s imunomodulačním nebo imunosupresivním účinkem. Přes výše uvedené dosud neexistuje univerzální účinná léčba. Farmakologická terapie tohoto onemocnění je orgánově specifická a symptomatická.
Klíčová slova:
digitální ulcerace – intersticiální plicní postižení – kapilaroskopie – plicní arteriální hypertenze – sklerodermická renální krize – systémová sklerodermie
Úvod
Termín skleroderma je vytvořen spojením řeckých slov skleros (tuhý) a derma (kůže). Jedná se o popis na první pohled zřetelného příznaku, ztluštělé a ztuhlé kůže. První zmínka týkající se tuhé kůže, na které nelze vytvořit řasu, pochází již od Hippokrata asi 400 let př. n. l. Přesvědčivý popis kožních změn ve smyslu sklerodermy popsal až neapolský lékař Carlo Curzio v roce 1753 [1].
Systémová sklerodermie (SSc) patří do skupiny difuzních (systémových) onemocnění pojiva. Patogeneze této skupiny nemocí je založena na patologické autoimunitě vznikající za přítomnosti autoprotilátek namířených proti jaderným strukturám, tzv. antinukleárních protilátek (ANA). Od počátku svého rozvoje SSc poškozuje malé cévy a v některých lokalizacích změny dosahují až na cévy středního průsvitu. Patogenetický děj vede posléze k fibroproduktivním změnám ve tkáních. Organickému postižení cév často předchází vazomotorická neuróza nazývaná podle autora prvního popisu Maurice Raynauda z roku 1862 Raynaudovým fenoménem [2]. V asociaci s SSc je Raynaudův fenomén označován jako sekundární. Klinickému obrazu dominuje tuhnutí kůže postupující typicky od akrálních částí prstů. To většinou odlišuje SSc od jiných nemocí s tuhou kůží. Z vnitřních orgánů jsou nejčastěji postiženy trávicí trubice, plíce, plicní oběh a ledviny.
Epidemiologie
Významně častěji postihuje SSc ženy, 3–5 : 1 v poměru proti mužům. Prevalence činí 4–253 případů a roční incidence 3–19 nových případů na milion obyvatel. Z údajů jasně vyplývá geografická variabilita výskytu nemoci. Nemoc začíná obvykle ve středním věku [3–5].
Klinické formy systémové sklerodermie
Rozlišení klinických forem SSc se používá v praxi, neboť má opodstatnění v rozdílné klinické manifestaci. Umožňuje predikovat prognózu a frekvenci jednotlivých klinických projevů vyvinutých v přirozeném průběhu nemoci [6].
Kožně difuzní systémová sklerodermie
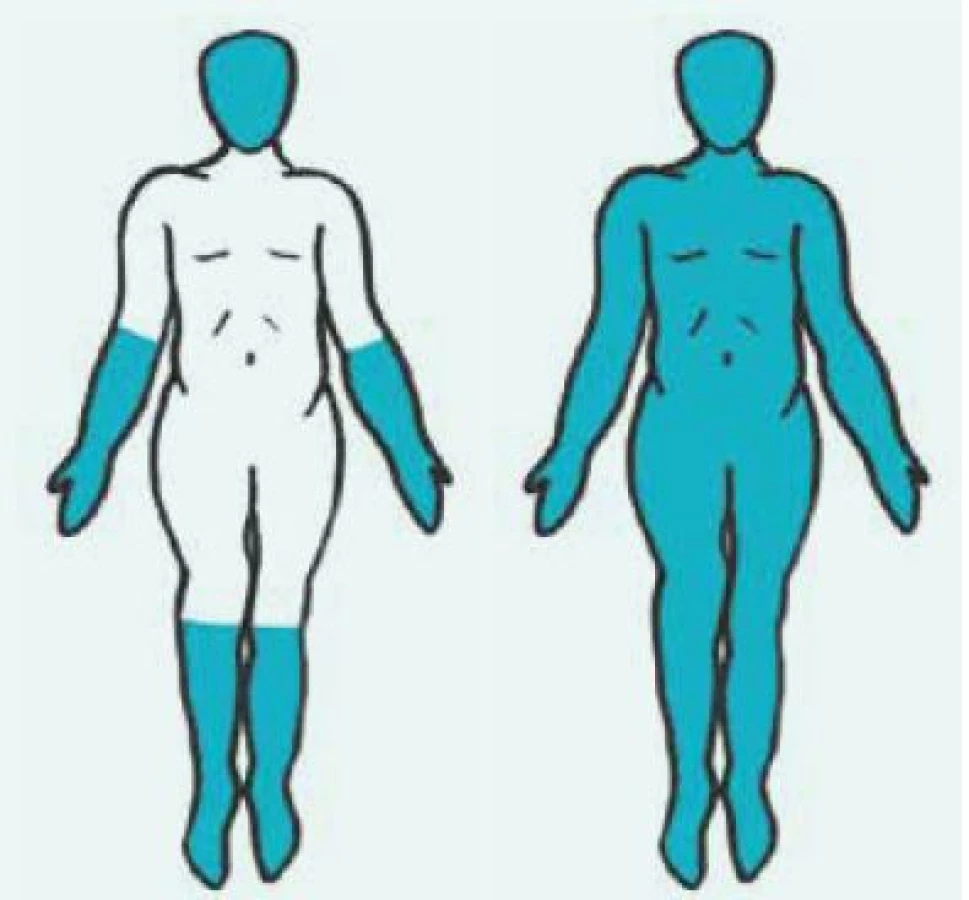
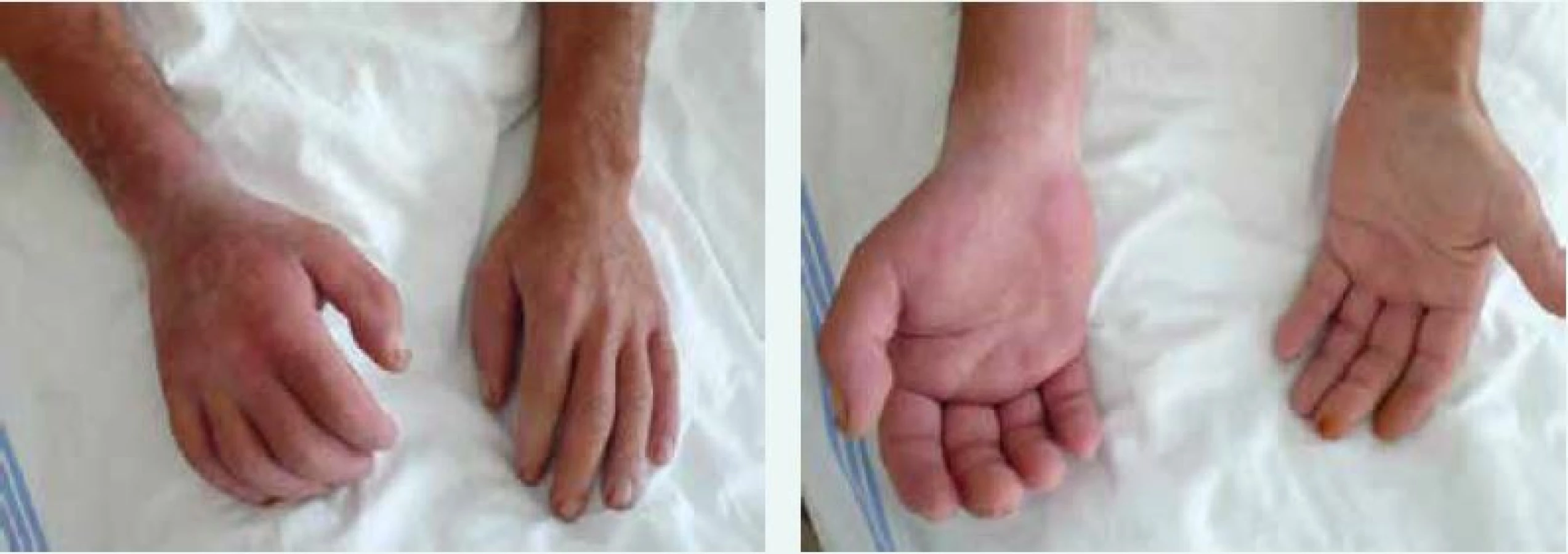
Kožně difuzní systémová sklerodermie (dSSc) se obvykle manifestuje rozvojem Raynaudova fenoménu v 1. roce nemoci. Následují tuhé otoky kůže a podkoží na prstech, končetinách, obličeji, trupu a alespoň části paží nebo stehen (obr. 1 vpravo). Dostavují se projevy pohybového aparátu, zejména myalgie, artralgie/artritidy a tenosynovitidy. Typické jsou hmatné šlachové třecí zvuky (friction rubs). Postižení vnitřních orgánů je příznačné a rozvíjí se během prvních 5 let nemoci. Zahrnuje postižení plic fibrózou, gastrointestinálního traktu, ledvin a srdce [7].
Kožně limitovaná systémová sklerodermie
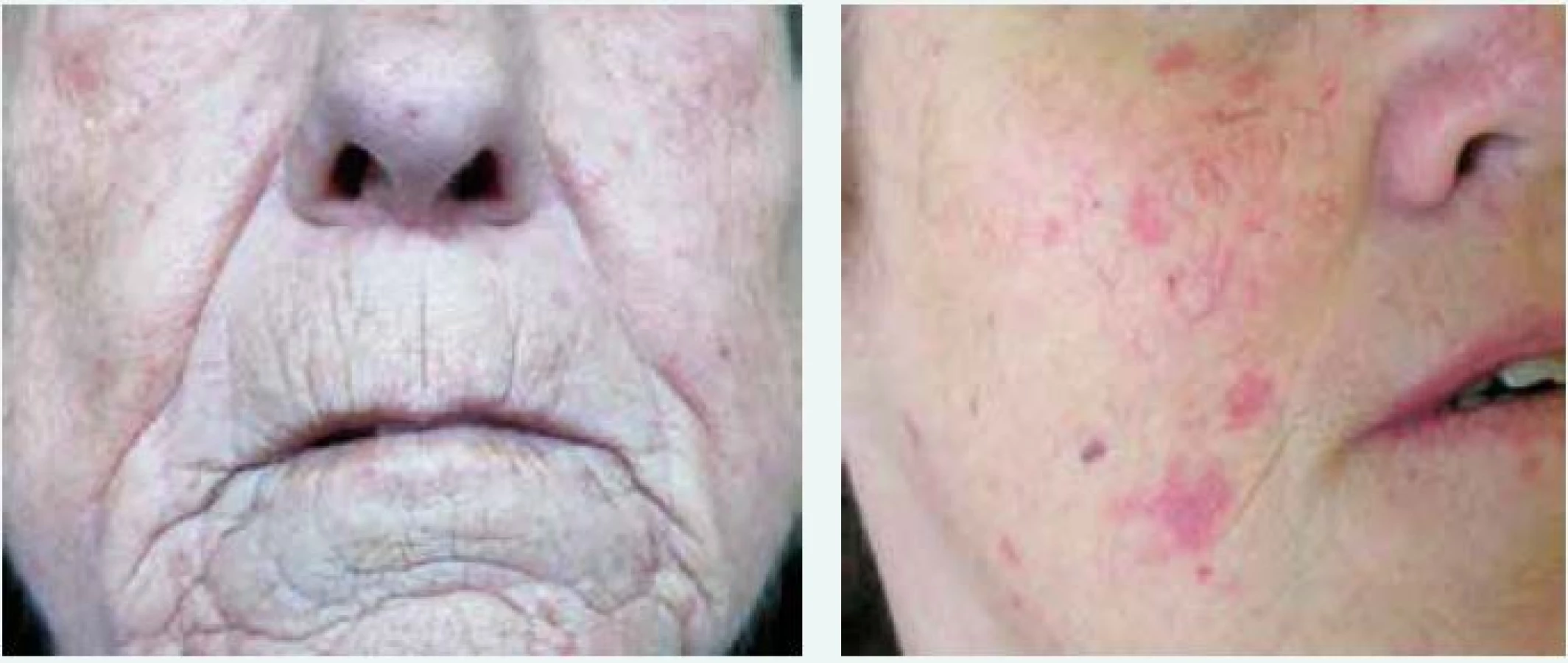
Kožně limitovaná systémová sklerodermie (lSSc) vzniká plíživěji, obvykle u osob s víceletým průběhem Raynaudova fenoménu. Kožní změny jsou zejména na periferních částech končetin distálně od kolenních a loketních kloubů (akroskleróza). Zahrnují postižení prstů (sklerodaktylie). V oblasti vrcholu prstu mohou být malé jizvičky (pitting scars) až digitální ulcerace. Změny pozorujeme v obličeji, ale nepostihují trup (obr. 1 vlevo). Změny v obličeji zahrnují hlavně radiální jizvy kolem úst, fenomén mizejících rtů a cévní ektazie (teleangiektazie) (obr. 2). U části nemocných se vyvine CREST syndrom charakterizovaný kalcinózou/C (jde o patologické ukládání vápníku do kloubů a měkkých tkání zejména v okolí kloubů v podobě fosforečnanových a uhličitanových solí), Raynaudovým fenoménem/R, ezofageální hypomotilitou/E, sklerodaktylií/S a teleangiektaziemi/T. Z útrobních manifestací je nejčastěji postižen jícen a plicní intesticium a bývá přítomna plicní arteriální hypertenze [7].
Systémová sklerodermie/skleróza bez sklerodermie
Systémová sklerodermie/skleróza bez sklerodermie je raritní skupinou SSc bez kožních změn a Raynaudova fenoménu, ale s útrobním postižením, kapilaroskopickými nálezy a přítomností protilátek příznačných pro SSc [8].
Systémová sklerodermie jako součást překryvného syndromu
Systémová sklerodermie je součást překryvného syndromu (overlap syndrome) s jiným onemocněním ze skupiny difuzních nemocí pojiva. Tendence ke sdružení nemocí je obecnou charakteristikou difuzních nemocí pojiva.
Nediferencované difuzní onemocnění pojiva
Klinické projevy SSc jsou součástí nediferencovaného difuzního onemocnění pojiva nebo smíšeného onemocnění pojiva, které patří do skupiny tzv. sklerodermii příbuzných nemocí (scleroderma related diseases).
Ke stanovení diagnózy SSc lze v současnosti použít klasifikační kritéria Americké revmatologické společnosti a Evropské ligy proti revmatismu (American College for Rheumatolgy – ACR; The European League Against Rheumatism – EULAR) z roku 2013 [9]. Výhodou těchto relativně nových kritérií je širší hodnocení různých klinických projevů, a tedy možnost časnějšího záchytu nemocných s ne zcela plně vyjádřenými příznaky, jak je tomu např. u méně vyjádřených kožních změn pacientů s lSSc. Nicméně vzhledem k vysokému váženému skóre pro kožní postižení stále přetrvává riziko poddiagnostikování této podskupiny při nevýrazných kožních změnách (tab. 1)
![ACR/EULAR klasifikační kritéria systémové sklerodermie. Upraveno podle [9]](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image/184b94a0cdb63718c14bf8aaa28287eb.jpg)
Prognóza
Mortalita SSc je oproti celkové populaci prokazatelně zvýšená. Podle prací z Evropy je 10leté přežívání ve Španělsku 75 % [10], v Itálii je to 70 % [11]. Studie Steenové et al ukázala, že plicní fibróza a plicní arteriální hypertenze jsou nyní oproti dříve dominující sklerodermické renální krizi dvěma hlavními příčinami smrti v příčinné souvislosti s SSc [12]. Zlepšení prognózy u renální krize spočívá v prevenci a zlepšení intenzivní péče. K prognosticky nepříznivým faktorům patří mužské pohlaví, dSSc, věk nad 65 let, postižení viscerálních orgánů (plíce, srdce, gastrointestinálního traktu), pokles renálních funkcí, anémie a trombocytopenie.
Etiopatogeneze
Přestože máme u SSc řadu potvrzených informací o abnormalitách imunitního systému, endotelu a funkci fibroblastů, doposud nebyl nalezen prvotní impulz ke spuštění patogenetických drah. Etiologie SSc je tedy neznámá. Nebylo tedy zjištěno, že by byl jednotlivý samotný gen nebo spouštěč (trigger) z prostředí sám o sobě odpovědný za rozvoj SSc. Na druhou stranu genetické faktory jasně ovlivňují náchylnost k nemoci. Genetická dispozice je založena na kombinaci polymorfizmů ve více genech. V tomto kontextu je velmi zajímavý vysoký výskyt SSc u amerického domorodého kmene indiánů Chotcaw s 20násobným rizikem rozvoje SSc oproti celkové populaci [13]. Vedle genetických faktorů hrají roli vnější faktory (polyvinylchlorid, křemík, organická rozpouštědla aj) a imunologické děje [14]. Patogenetický koncept SSc předpokládá triádu:
- autoimunitní zánět přičítaný humorálním a buněčným abnormalitám – kromě ANA nespecifických autoprotilátek nacházíme specifické ANA autoprotilátky: u dSSc je to většinou protilátka proti DNA topoizomeráze I (antiScl-70, výskyt ve 40–50 % případů dSSc), u lSSc jsou to anticentromerové protilátky namířené proti vřeténkům dělících se buněk (ACA, výskyt u 40–50 % případů), u překryvného syndromu s polymyozitidou nalézáme anti-PM/Scl [15]
- difuzní vaskulopatii s poškozením endotelu, následuje postižení všech 3 částí cévní stěny (intimy, medie a adventicie) zejména fibrózou a nakonec obliterující vaskulopatií s následnou hypoxií [14]
- progresivní fibrózu v perivaskulárním prostoru a v intersticiu [14,16] způsobenou excesivní syntézou extracelulární matrix
Zánětlivé a autoimunitní procesy aktivují fibroblasty k proliferaci a tvorbě složek extracelulární matrix, především kolagenu typu I a III [15]. Všechny 3 klíčové procesy jsou vzájemně propojeny, což odlišuje SSc od jiných difuzních onemocnění pojiva a jiných fibrotizujících onemocnění. Zjednodušeně řečeno, vaskulopatie a zánět předcházejí rozvoji fibrózy a fibróza s vaskulopatií následně udržují zánětlivou aktivitu, a tím uzavírají bludný kruh [16].
Diferenciální diagnostika
O´Leavy et al v roce 1930 odlišili generalizovanou systémovou formu sklerodermie (tedy SSc) od lokalizované formy sklerodermie spojené s lokalizovaným asymetrickým postižením kůže nazývaným morfea, lineární sklerodermie. Lokalizovanou sklerodermii lze snadno odlišit, nevykazuje akrální změny kůže a útrobní postižení [3]. Skleroderma se též vyskytuje u různých nemocí bez etiopatogenetického a klinického vztahu k SSc (tab. 2).
Klinický obraz
SSc je značně heterogenní nemoc. Onemocnění je doprovázeno celkovými projevy. K časným příznakům řadíme nechutenství, hubnutí, slabost a únavu, později se objevuje reaktivní deprese.
Kožní postižení
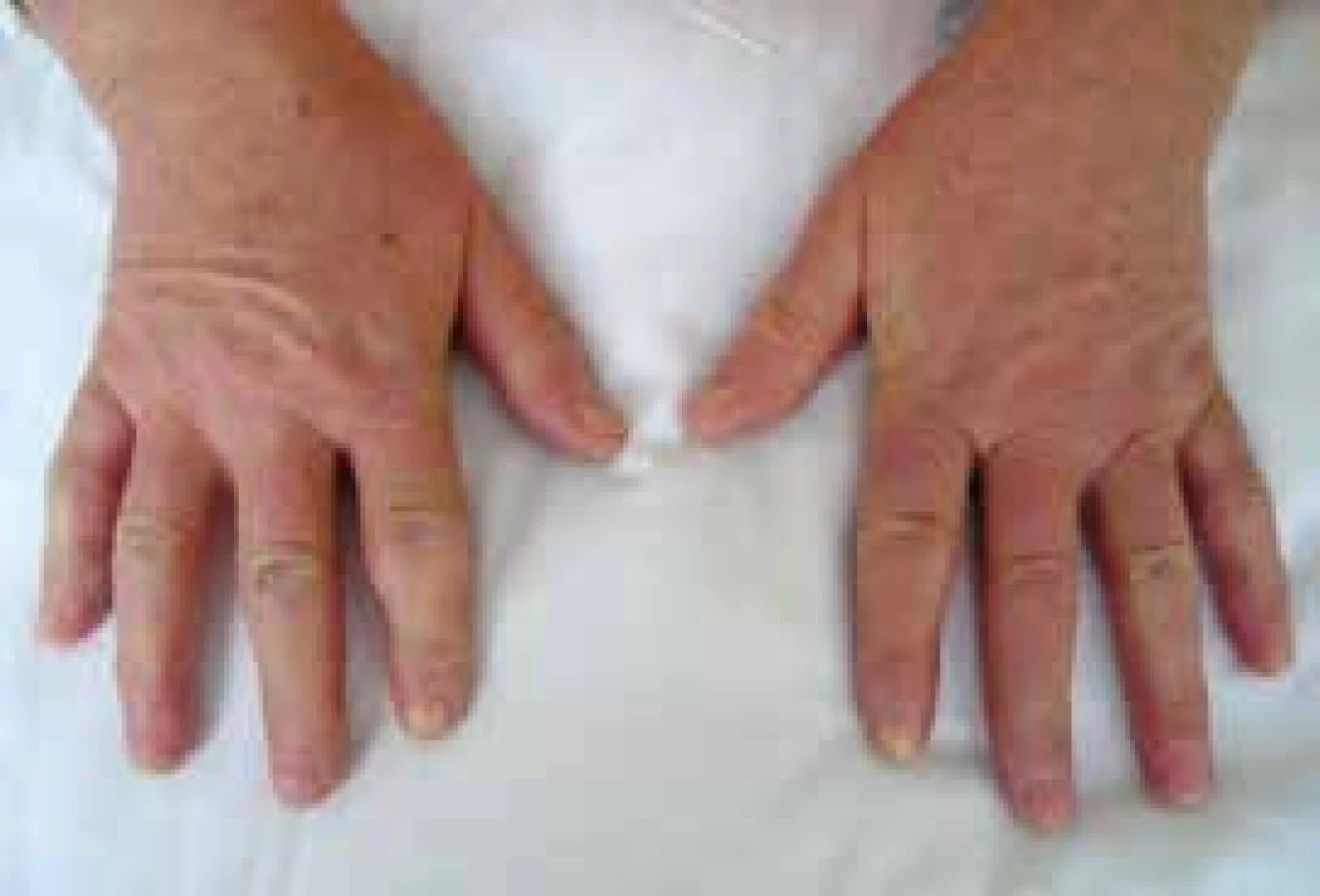
Postižení kůže je charakteristickou manifestací SSc. Rozsah a závažnost kožního postižení se mění v průběhu času. Dosahuje vrcholu v průběhu 2–5 let od začátku onemocnění, pak následuje stabilizace a pozvolného zmírnění procesu. Tento fakt vedl často k mystifikaci v hodnocení účinnosti léčby v rámci klinických hodnocení. Kožní postižení se v první fázi projevuje otokem (typické jsou otoky prstů – tzv. puffy fingers), následně indurací a ztrátou pružnosti kůže (obr. 3 a 4). V časné fázi dSSc spojené s rychlou progresí kožních změn je riziko promptního rozvoje orgánového postižení a cílem imunosupresivní léčby je snaha o zpomalení progrese těchto změn a následných komplikací. Ze všech klinických projevů SSc má však kožní postižení v dosud prováděných randomizovaných placebem kontrolovaných studiích historicky nejmenší odpověď na protizánětlivou nebo imunosupresivní léčbu. Tíha léčby spočívá stále na metotrexátu [17].
Postižení oběhového systému
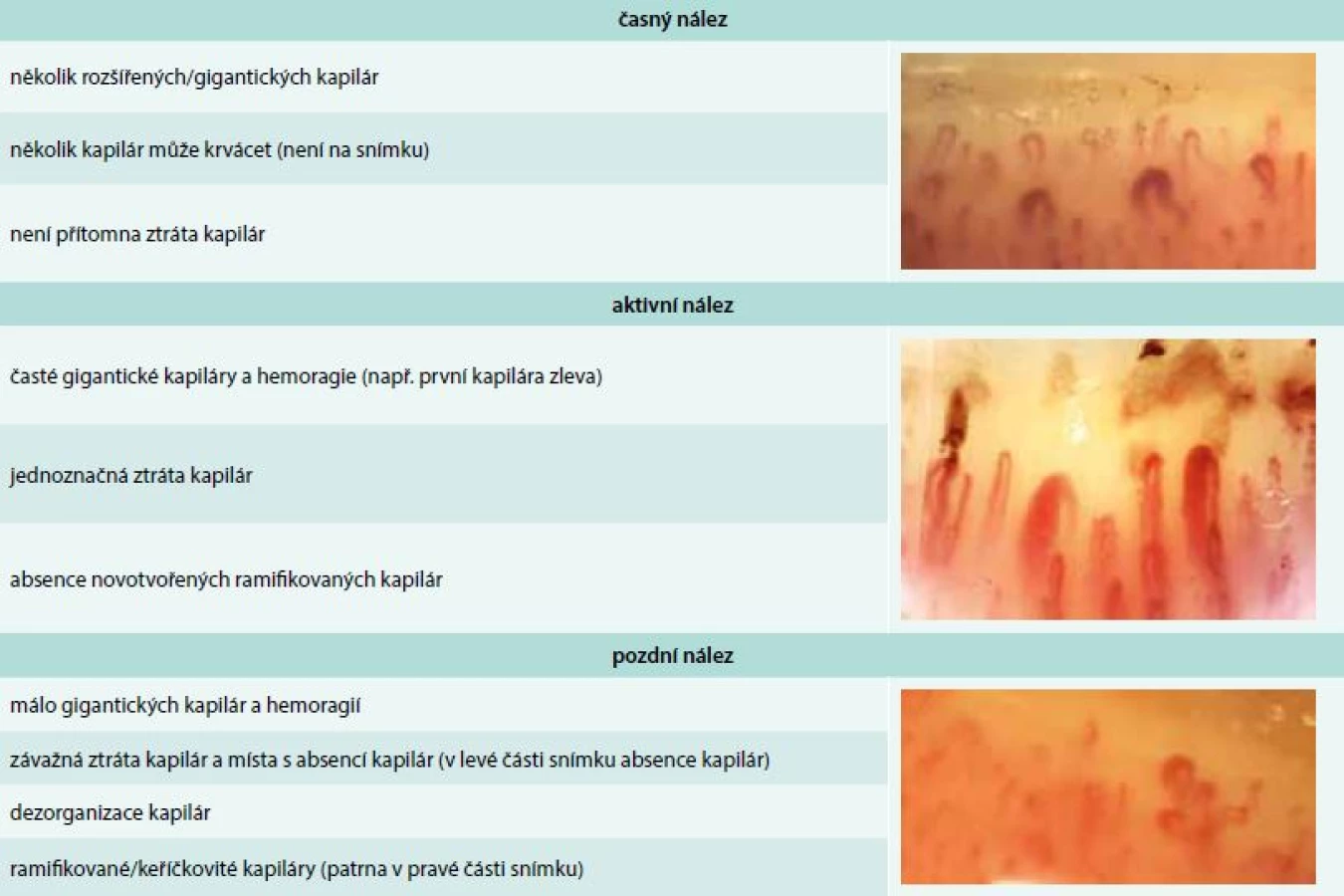
Vaskulární poškození je nejčasnějším a pravděpodobně i primárním procesem v patogenezi SSc a je histopatologicky prokazatelné před rozvojem fibrózy a objevením prvního klinického příznaku, kterým obvykle bývá Raynaudův fenomén. Ten je charakterizován epizodickými barevnými změnami aker od bílé přes fialovou, vyvolanými chladem nebo stresem. Záchvat může být ukončen reaktivním hyperemickým zčervenáním kůže. Raynaudův fenomén je jednou z časných manifestací SSc, objevuje se asi u 95 % nemocných [18]. Postižení se během času stupňuje ve strukturální změny cév. Postižení četných cévních lůžek je velmi dobře patrné při kapilaroskopii nehtových valů. Již ve fázi Raynaudova fenoménu lze při kapilaroskopii nehtových valů zjistit typické změny, které předznamenávají rozvoj cévního postižení při SSc – snížený počet kapilár na 1 mm pole, porucha uspořádání a dilatace kapilár, mikrohemoragie z kapilár, následné avaskulární zóny a novotvorba kapilár (tab. 3) [19].
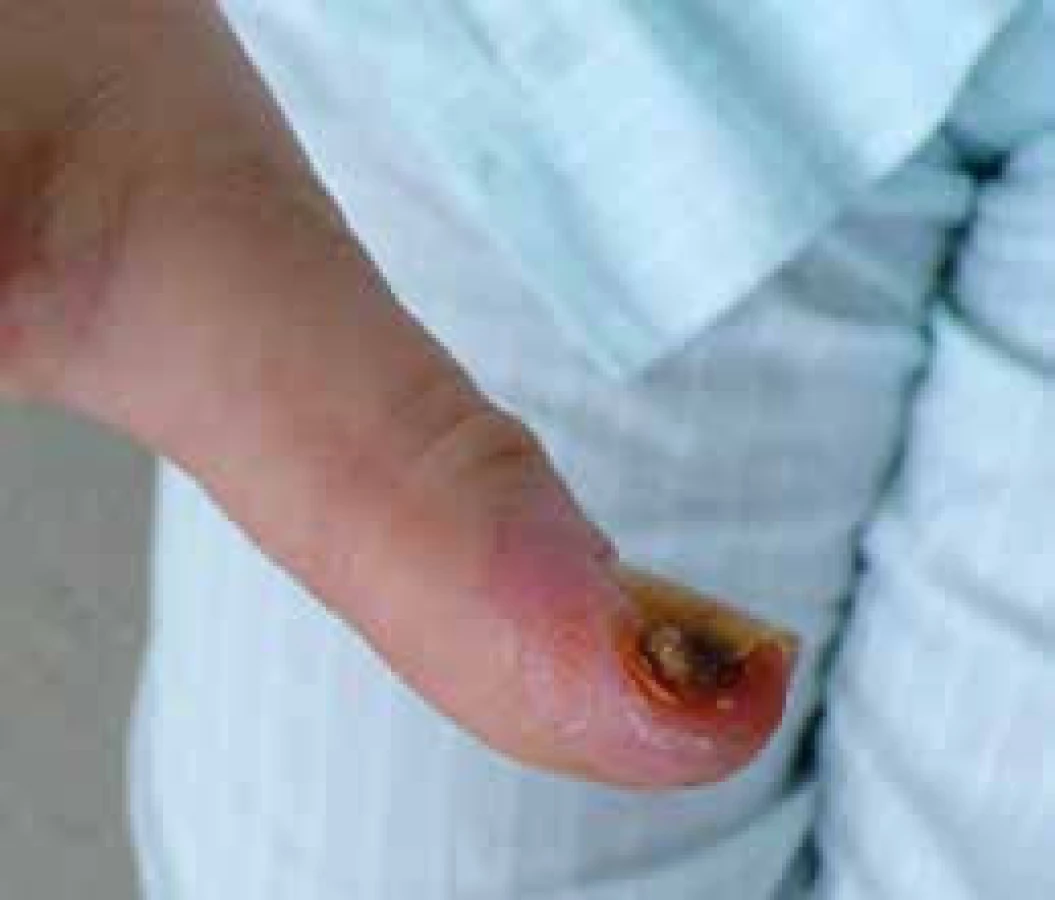
Progresivní strukturální změny v malých cévách akrálních částí končetin vedou k rozvoji ischemických změn v podobě digitálních ulcerací spojených s nekrózou a gangrénou prstů (obr. 5) až k obrazu kritické digitální ischemie. Arteriografické studie odhalily okluze a stenózy digitálních arterií, ulnární arterie a povrchového palmárního oblouku. Digitální ulcerace jsou často provázeny infekcí, ztrátou tkání a v některých případech vedou k invalidizaci nemocného. Za 1 rok dochází ke ztrátě akrálních částí u 1–2 % SSc pacientů [20]. Léčba je problematická a dlouhodobá (viz níže).
Postižení cév stejného charakteru jako u digitálních arterií se manifestuje v řečišti plicní arterie u plicní arteriální hypertenze. Výskyt známek plicní hypertenze u pacientů se SSc je relativně častý, vyskytuje se asi u 12 % pacientů [21]. K hlavním příčinám zvýšeného tlaku v plicnici patří plicní arteriální hypertenze spojená se přímým postižením plicní arterie vaskulopatií a dále plicní hypertenze spojená s postižením plicního intersticia nebo levého srdce. K rizikovým faktorům rozvoje této manifestace patří lSSc, trvání choroby je 10–15 let a začátek onemocnění ve vyšším věku. Ve srovnání s idiopatickou plicní hypertenzí mívá plicní arteriální hypertenze spojená s SSc závažnější prognózou [22].
Pozdní diagnóza plicní arteriální hypertenze nepříznivě ovlivňuje účinnost léčby a následně prognózu nemocných. Jediným způsobem, jak dosáhnout diagnózy v časnějším stadiu onemocnění, je pravidelný screening. Zásadním vyšetřením v odhalení plicní hypertenze je transtorakální echokardiografie [23]. Echokardiografický screening plicní hypertenze je indikován u nemocných s SSc každoročně. S cílem zvýšit výtěžnost screeningu a snížit zbytečné indikace srdečních katetrizací se doporučuje kombinovat echokardiografii a vyšetření sérových koncentrací natriuretických peptidů a difuzní plicní kapacity pro oxid uhelnatý [24,25]. Definitivní diagnostika plicní (arteriální) hypertenze, včetně katetrizační verifikace, patří do rukou expertních center. Léčit by se měli již pacienti s hraničními hodnotami tlaku v plicnici.
Léčba vaskulárních projevů se zaměřuje na vazodilatační léky. Obecně lze shrnout, že mechanizmus účinku léků je zaměřen na vazoaktivní patogenetické mechanizmy [26]. Ty jsou u SSc spojeny se ztrátou vazodilatačních mediátorů prostacyklinu, oxidu dusnatého (v léčbě jsou používány inhibitory 5-fosfodiesterázy, resp. prostanoidy) a abnormální odpovědí na vazokostriktivní mediátor endotelin-1 (použití blokátorů endotelinových receptorů je opodstatněno kvalitními klinickými hodnoceními RAPIDS 1–2, DUAL 1–2, BREATHE 1–2) [27–29]. Léčba plicní hypertenze spadá do působení specializovaných center [30]. Do jisté míry je efektivní léčba blokátory kalciových kanálů v indikaci tzv. nefixované plicní hypertenze a Raynaudova fenoménu, oxygenoterapie, antikoagulační léčba. Perspektivní se zdá být léčba riociguatem, agonistou solubilní guanylátcyklázy s vazodilatačním a antifibrotickým potenciálem [31].
Plicní postižení
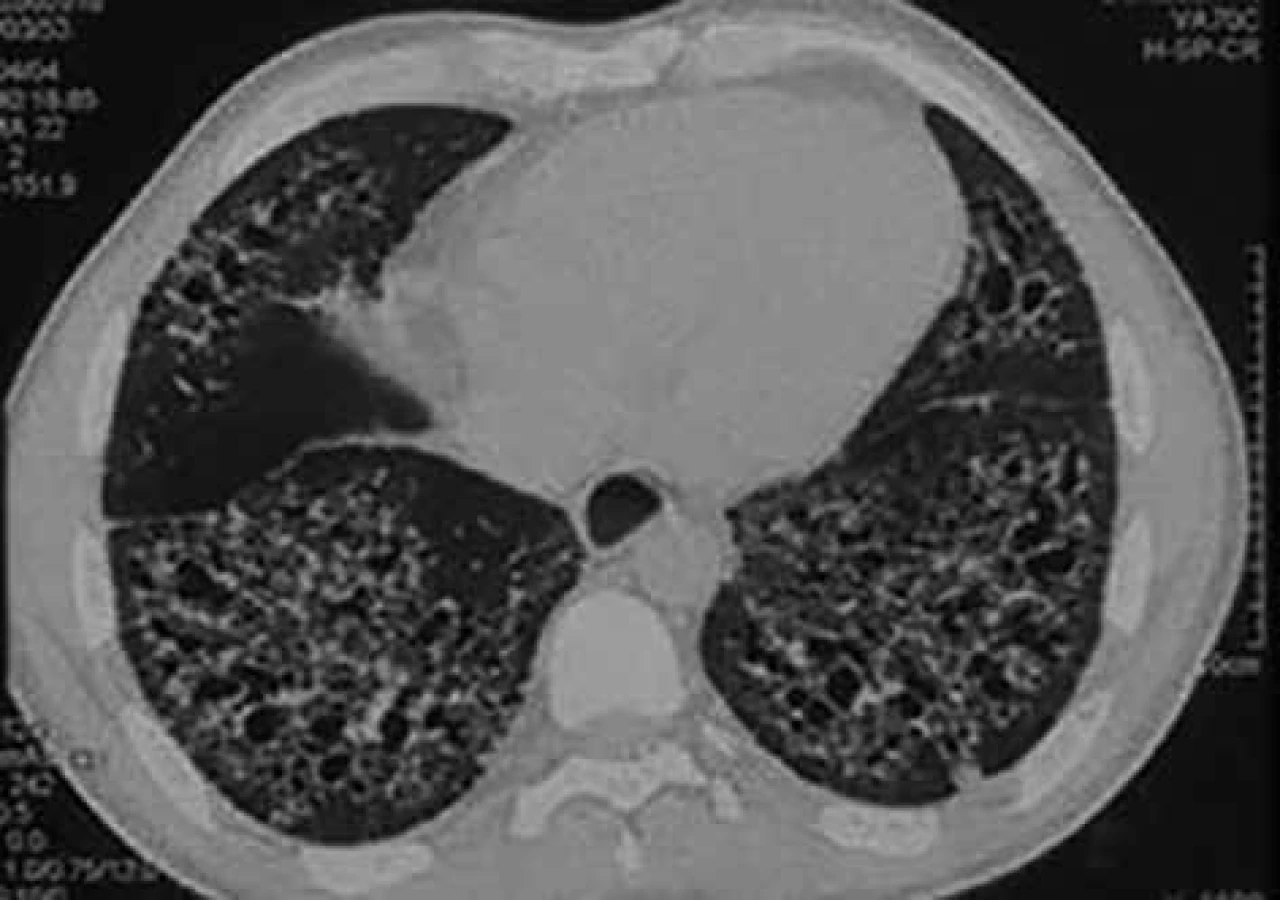
Plicní postižení při SSc obvykle spadá do skupiny intersticiálních plicních procesů. Intersticiální plicní postižení postihuje 53 % nemocných s dSSc a 35 % s lSSc. Deset let přežívá 29–69 % nemocných. Hlavními symptomy jsou dušnost a suchý kašel [32]. Diagnosticky je důležité funkční vyšetření plic. Nález odpovídající restrikční ventilační poruše však nemusí přesně odrážet postižení plicního parenchymu, proto je nutné vyšetření difuzní plicní kapacity pro oxid uhelnatý. Pokles tohoto parametru (tzv. transfer faktoru) může provázet jak intersticiální plicní postižení, tak postižení plicní cirkulace. Přesto je doporučena každoroční depistáž. V případě změn plicních funkcí je nezbytné doplnění vyšetření hrudníku vysoce rozlišovací výpočetní tomografií. Obraz mléčných opacit v plicním parenchymu při výpočetní tomografii hrudníku upozorňuje na možné časné postižení plicního intersticia, ale tento nález není zcela specifický a může být způsoben jinými procesy, např. městnáním v malém oběhu [32]. Často spolupřítomný obraz plicní fibrózy (obr. 6) u nemocných se SSc bývá natolik charakteristický, že není důvod indikovat plicní biopsii [33].
U pacientů s fibrotizujícím plicním postižením nelze již vzniklé postižení odvrátit, cílem je proces zpomalit. Léčba je obvykle zahajována u pacientů s rozsáhlým postižením nebo progresivní chorobou. Jako iniciální léčba se nejčastěji používá intravenózní cyklofosfamid. Obvyklá dávka je 0,5–2 g/m2 každý měsíc po dobu 6–18 měsíců nebo 6 měsíců a pak kvartální aplikace celkem 2 roky. Alternativně lze zvážit mykofenolát mofetil nebo rituximab. Navazujeme pokračovací léčbou ve formě azatioprinu nebo mykofenolát mofetilu [17]. Dvě klinická hodnocení hodnotila účinek autologní transplantace kmenových buněk. Prokázala stabilizaci plicních funkcí, také zlepšení kožních změn a vyšší četnost přežití a bez relapsů [34,35]. Tato metoda je doporučena k provedení na specializovaném pracovišti u rychle progredujícího onemocnění s rizikem orgánového selhání [17].
Muskuloskeletální manifestace
Vyskytuje se přibližně u 75 % nemocných. Bývají přítomny artralgie, artritidy doprovázené ranní ztuhlostí, postižení šlach s třecími šelesty šlachových pochev v oblasti prstů, loktů a ramen. Svalové postižení doprovázejí myalgie a slabost proximálních svalových skupin. Při léčbě lehkých forem artritidy většinou postačuje imunosupresivní léčba indikovaná pro jiné projevy nemoci, jako je např. kožní postižení. Závažnější artritida nebo myozitida by měla být léčena jako u nemocí s artritidou bez SSc. V úvahu připadá například indikace metotrexátu [31].
Sklerodermická renální krize
Vzácným, ale velmi závažným projevem postižení při SSc, je sklerodermická renální krize. V současnosti je tradována incidence okolo 2,5 % nemocných se SSc. Podstatou je porucha mikrocirkulace v ledvinách při vaskulopatii a zvýšená aktivita renin-angiotenzinové osy [36]. Krize je typicky spojena s náhle vzniklou arteriální hypertenzí a rychle progredujícím renálním selháním s oligurií nebo anurií. Klinicky bývá přidružena hypertenzní encefalopatie, akutní srdeční selhání, mikroangiopatická hemolytická anémie a trombocytopenie. Jde o vážný, život ohrožující stav [12]. V kontextu renální krize je třeba zmínit zásadní vliv dvou lékových skupin, negativní roli systémových glukokortikoidů s rizikem pro rozvoj krize, a naopak pozitivní efekt inhibitorů angiotenzin konvertujícího enzymu (ACEi) v léčbě renální krize. Analýza literárních dat v současnosti neopodstatňuje podávání ACEi v prevenci krize. Dokonce jsou dostupná data ukazující zvýšené riziko nutné dialýzy nebo i úmrtí spojené s léčbou ACEi před rozvojem prvních příznaků krize [37,38].
Pro dobrou prognózu nemocného je zásadní včasné nasazení ACEi a okamžité předání do péče odborného centra. Jestliže se diagnóza sklerodermické renální krize nepotvrdí, vhodnými antihypertenzivy jsou blokátory kalciových kanálů, jak je to doporučeno u jiných manifestací SSc (Raynaudova fenoménu, plicní arteriální hypertenze) nebo centrálně působící antihypertenziva a diuretika [39,40]. Pacienti se sklerodermickou renální krizí, kteří nevykazují žádné známky renální funkční regenerace navzdory včasné kontrole krevního tlaku, jsou kandidáty pro léčbu metodami náhrady funkce ledvin (dialýza, transplantace).
Gastrointestinální postižení
Gastrointestinální postižení je nejčastější orgánovou manifestací SSc. Více než 90 % pacientů se SSc má postižení gastrointestinálního traktu [41]. Postižení se může objevit v kterékoli jeho části. Časně dochází k vaskulárním lézím sliznice a submukózy, neurálnímu postižení, později k atrofii hladkého svalstva a fibróze. Následkem je dysmotilita trávicí trubice. Podle lokalizace vede k gastroezofageálnímu refluxu, dokonce i k extraezofageálnímu refluxu, poruše evakuace žaludku, syndromu bakteriálního přerůstání v tenkém střevě (small intestine bacterial overgrowth – SIBO) s malabsorpcí, pseudoobstrukcí, pomalým průchodem skrz kolon a anorektální dysfunkcí (např. fekální inkontinencí) [7,42].
Celková léčba SSc nevede ke zlepšení gastrointestinální dysfunkce. V léčbě se uplatňují stejné postupy jako při terapii gastrointestinálního postižení u nemocných bez sklerodermie. Tzn. dietní a režimová opatření, inhibitory protonové pumpy, prokinetika, cyklické podávání antibiotik u pacientů se SIBO, případně protiprůjmová léčba (např. loperamid) nebo léčba laxativy u pacientů se zácpou [31].
Komplexní léčba systémové sklerodermie
Přes medicínský pokrok dosud neexistuje univerzální účinná léčba SSc. Farmakologická terapie tohoto onemocnění je pouze orgánově specifická a symptomatická, proto je popsána u jednotlivých projevů nemoci. V léčbě SSc se užívají také nefarmakologické postupy. Patří k nim běžně užívané metody jako parafínové zábaly, manuální lymfodrenáž, protahovací cvičení rukou a obličeje, masáž, cvičení k udržení kloubního rozsahu, mobilizace kloubů, silový tréning a aerobní cvičení [43] (Matucci-Cerinic).
Závěr
Tíha včasné primární diagnostiky dopadá na lékaře mnoha odborností (praktické lékaře, ambulantní revmatology, angiology, pneumology atd). Důležité je rozpoznání prvních příznaků SSc, jako je Raynaudův fenomén, první typické změny kůže, postižení plic. Na začátku diagnostiky je velmi užitečné kapilaroskopické vyšetření. Praktickým doporučením je indikace kapilaroskopického vyšetření nehtových valů v případě Raynaudova fenoménu ve spojení s pozitivitou ANA. Dále bychom měli mít na paměti, že při izolovaném nálezu intersticiálního plicního postižení je nutno také pátrat po primární mimoplicní chorobě. Role revmatologa spočívá v časném zjištění diagnózy a odhalení orgánových manifestací. Nutnost specializovaných vyšetření (každoroční echokardiografie a funkčního plicního vyšetření s vyšetřením plicní difuze) tuto nemoc posouvá do péče specializovaných center s cílem včas diagnostikovat a léčit závažné, bez léčby prognosticky nepříznivé orgánové projevy, jako je plicní arteriální hypertenze a intersticiální postižení plic.
Práce vznikla za podpory projektu PROGRES Q40–15 (Karlovy Univerzity).
Doručeno do redakce 26. 9. 2017
Přijato po recenzi 19. 11. 2017
doc. MUDr. Tomáš Soukup, Ph.D.
Subkatedra revmatologie II. interní gastroenterologické kliniky LF UK a FN Hradec Králové
Zdroje
1. Curzio C. Discussioni anatomico-pratiche di un raro, estravagante morbo cutaneo in una giovane donna felice-mente curato in questo grande ospedale degl‘incurabili. Pressa Giovanni di Simone, Napoli 1753. [Curzio C. An account of an extraordinary disease of the skin and its cure. Philosoph Trans 1754; 48: 579. Translated by R. Watson].
2. Raynaud M. Thése de Médicine. De l´asphyxie locale et de la gangréne symétrique des extremitiés. Laclerc: Paris 1862.
3. Štork J. Sklerodemie. Galen: Praha 1996. ISBN 80–85824–41–8.
4. D´Angelo W, Fries JF, Masi AR et al. Pathologic observations in systemic sclerosis. Am J Med 1969; 46(3): 428–440.
5. Ferri C, Valentini G, Cozzi F et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(2): 139–153.
6. LeRoy EC, Medsger TA Jr.Criteria for the classification of early systemic sclerosis. J Rheumatol 2001; 28(7): 1573–1576.
7. Bečvář R, Štork J, Jansa P. New trends in diagnosis and treatment of systemic sclerosis. Vnitř Lék 2006; 52(7–8): 712–717.
8. Poormoghim H, Lucas M, Fertig N et al. Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum 2000; 43(2): 444–451. Dostupné z DOI: <http://dx.doi.org/10.1002/1529–0131(200002)43:2<444::AID-ANR27>3.0.CO;2-G>.
9. Van den Hoogen F, Khanna D, Fransen J et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2013; 72(11): 1747–1755. Dostupné z DOI: <http://dx.doi.org/10.1136/annrheumdis-2013–204424>.
10. Joven BE, Almodovar R, Carmona L et al. Survival, causes of death, and risk factors associated with mortality in Spanish systemic sclerosis patients: results from a single university hospital. Semin Arthritis Rheum 2010; 39(4): 285–293. Dostupné z DOI: <http://dx.doi.org/10.1016/j.semarthrit.2009.06.002>.
11. Ferri C, Valentini G, Cozzi F et al. Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 2002; 81(2): 139–153.
12. Steen VD, Medsger TA Jr. Long-term outcomes of scleroderma renal crisis. Ann Intern Med 2000; 133(8): 600–603.
13. Agarwal SK, Tan FK, Arnett FC Genetics and genomic studies in scleroderma (systemic sclerosis). Rheum Dis Clin North Am 2008; 34(1): 17–40. Dostupné z DOI: <http://dx.doi.org/10.1016/j.rdc.2007.10.001>.
14. Abraham DJ, Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol 2005; 26(11): 587–595. Dostupné z DOI: <http://dx.doi.org/10.1016/j.it.2005.09.004>.
15. Bečvář R. Systémová sklerodermie. In Pavelka K, Vencovský J, Horák P et al. Revmatologie. Maxdorf: Praha 2012: 374–382. ISBN 978–80–7345–295–7.
16. Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest 2007; 117(3): 557–567. Dostupné z DOI: <http://dx.doi.org/10.1172/JCI31139>.
17. Kowal-Bielecka O, Fransen J, Avouac J et al. [EUSTAR co-authors]. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017; 76(8): 1327–1339. Dostupné z DOI: <http://dx.doi.org/10.1136/annrheumdis-2016–209909>.
18. Denton CP, Black CM. Scleroderma and related disorders: therapeutic aspects. Baillieres Best Pract Res Clin Rheumatol 2000; 14(1): 17–35. Dostupné z DOI: <http://dx.doi.org/10.1053/berh.1999.0075>.
19. Boin F, Wigley FM. Clinical features and treatment of scleroderma. In: Firestein GS, Budd RC, McInnes IB et al. Kelley´s textbook of rheumatology. 9th ed. Elsevier: Philadelphia 2013: 1366–1303. ISBN: 978–1-4377–1738–9.
20. Korn JH, Mayes M, Matucci CM et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum 2004; 50(12): 3985–3993. Dostupné z DOI: <http://dx.doi.org/10.1002/art.20676>.
21. Mukerjee D, St George D, Coleiro B et al. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: application of a registry approach. Ann Rheum Dis 2003; 62(11): 1088–1093.
22. Fisher MR, Mathai SC, Champion HC et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum 2006; 54(9): 3043–3050. Dostupné z DOI: <http://dx.doi.org/10.1002/art.22069>.
23. McGoon M, Gutterman D, Steen V et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension. Chest 2004; 126(1 Suppl): 14S-34S. Dostupné z DOI: <http://dx.doi.org/10.1378/chest.126.1_suppl.14S>.
24. Galie N, Humbert M, Vachiery JL et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37(1): 67–119. Dostupné z DOI: <http://dx.doi.org/10.1093/eurheartj/ehv317>.
25. Soukup T, Pudil R, Kubinova K et al. Application of the DETECT algorithm for detection of risk of pulmonary arterial hypertension in systemic sclerosis: data from a Czech tertiary centre. Rheumatology (Oxford) 2016; 55(1): 109–114. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/kev327>.
26. Aschermann M, Jansa P. Drug therapy of pulmonary arterial hypertension in 2014. Vnitř Lék 2014; 60(4): 282–288.
27. Matucci-Cerinic M, Denton CP, Furst DE et al. Bosentan treatment of digital ulcers related to systemic sclerosis: results from the RAPIDS-2 randomised, double-blind, placebo-controlled trial. Ann Rheum Dis 2011; 70(1): 32–38. Dostupné z DOI: <http://dx.doi.org/10.1136/ard.2010.130658>.
28. Khanna D, Denton CP, Merkel PA et al. Effect of macitentan on the development of new ischemic digital ulcers in patients with systemic sclerosis: DUAL-1 and DUAL-2 Randomized Clinical Trials. JAMA 2016; 315(18): 1975–1988. Dostupné z DOI: <http://dx.doi.org/10.1001/jama.2016.5258>.
29. Humbert M, Barst RJ, Robbins IM et al. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur Respir J 2004; 24(3): 353–359. Dostupné z DOI: <http://dx.doi.org/10.1183/09031936.04.00028404>.
30. Jansa P Development of interest of pulmonary hypertension in the Czech Republic. Vnitř Lék 2014; 60(4): 271–274.
31. Denton CP, Hughes M, Gak N et al. BSR and BHPR guideline for the treatment of systemic sclerosis. Rheumatology (Oxforrd) 2016; 55(10): 1906–1910. Dostupné z DOI: <http://dx.doi.org/10.1093/rheumatology/kew224>.
32. Bečvář R, Šterclová M. Systémová sklerodemie. In: Plicní postižení u systémových nemocí pojiva, vaskulitid a idiopatických zánětů v gastroenterologii. Mladá fronta: Praha 2016: 63–75. ISBN 978–80–204–4044–0.
33. Fan MH, Feghali-Bostwick CA, Silver RM. Update on scleroderma-associated interstitial lung disease. Curr Opin Rheumatol 2014; 26(6): 630–636. Dostupné z DOI: <http://dx.doi.org/10.1097/BOR.0000000000000111>.
34. Burt RK, Shah SJ, Dill K et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): an open-label, randomised phase 2 trial. Lancet 2011; 378(9790): 498–506. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(11)60982–3>.
35. Van Laar JM, Farge D, Sont JK et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: a randomized clinical trial. JAMA 2014; 311(24): 2490–2498. Dostupné z DOI: <http://dx.doi.org/10.1001/jama.2014.6368>.
36. Denton CP, Hudson M Renal cisis and other renal manifestations of scleroderma. In: Varga J, Denton CP, Wigley FM et al (eds) Scleroderma. From pathogenesis to compherensive management. 2nd ed. Springer: New York 2017: 317–331. ISBN 978–3319314051.
37. Penn H, Howie AJ, Kingdon EJ et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. QJM 2007; 100(8): 485–494. Dostupné z DOI: <http://dx.doi.org/10.1093/qjmed/hcm052>.
38. Walker KM, Pope J. [Scleroderma Clinical Trials Consortium. Canadian Scleroderma Research Group]. Treatment of systemic sclerosis complications: what to use when first-line treatment fails – consensus of systemic sclerosis experts. Semin Arthritis Rheum 2012; 42(1): 42–55. Dostupné z DOI: <http://dx.doi.org/10.1016/j.semarthrit.2012.01.003>.
39. Mann JF, Schmieder RE, McQueen M et al. Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet 2008; 372(9638): 547–553. Dostupné z DOI: <http://dx.doi.org/10.1016/S0140–6736(08)61236–2>.
40. Lopez-Ovejero JA, Saal SD, D’Angelo WA et al. Reversal of vascular and renal crises of scleroderma by oral angiotensin converting enzyme blockade. N Engl J Med 1979; 300(25): 1417–1419. Dostupné z DOI: <http://dx.doi.org/10.1056/NEJM197906213002505>.
41. Marie I. Gastrointestinal involvement in systemic sclerosis. Presse Med 2006; 35(12): 1952–1965.
42. Korn JH. Scleroderma: a treatable disease. Cleve Clin J Med 2003; 70(11): 954–958.
43. Bečvář R, Soukup T, Tomčík M et al. Doporučení České revmatologické společnosti pro léčbu systémové skleorodermie. Čes Revmatol 2017; 25(2): 68–84.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2018 Číslo 2
- MINISERIÁL: Když ženám stoupá tlak...
- Tirzepatid – nová éra v léčbě nadváhy a obezity
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- Specifika v komunikaci s pacienty s ránou – laická doporučení
Nejčtenější v tomto čísle
- Axiální spondylartritida
- Využitie MRI vyšetrenia pri diagnostike axiálnej spondylartritídy
- Idiopatické zánětlivé myopatie
- Difuzní alveolární hemoragie – akutní, život ohrožující stav v revmatologii