
Novinky ve WHO klasifikaci nádorů centrálního nervového systému 2016
2. část: Embryonální nádory CNS a ostatní skupiny nádorů (kromě difúzních gliomů)
Update on the 2016 WHO classification of tumors of the central nervous system.
Part 2: Embryonal tumors and other tumor groups (except for diffuse gliomas)
The 2016 revision of the WHO classification of tumors of the central nervous system is a conceptual advance over the 2007 classification system. Similarly to the group of diffuse gliomas, a significant shift in the understanding of the molecular background and tumor biology has recently occurred also in the category of embryonal CNS tumors, especially in medulloblastomas. The classification now incorporates new entities that are defined by both histology and molecular features. Updates in the group of gliomas (except for diffuse gliomas), in the meningeal tumors as well as in the tumors of peripheral nerve sheaths will also be discussed.
Keywords:
WHO classification – meduloblastoma – AT/RT – pleomorphic xanthoastrocytoma – pilocytic astrocytoma – peripheral nerve sheath tumors
Autoři:
Josef Zámečník 1; Boris Rychlý 2; Marián Švajdler 3,4
Působiště autorů:
Ústav patologie a molekulární medicíny 2. lékařské fakulty UK a FN v Motole, Praha, Česká republika
1; Cytopathos s. r. o., Bratislava, Slovenská republika
2; Šiklův ústav patologie, Univerzita Karlova v Praze, Lékařská fakulta v Plzni a Fakultní nemocnice Plzeň, Česká republika
3; Bioptická laboratoř, s. r. o., Plzeň, Česká republika
4
Vyšlo v časopise:
Čes.-slov. Patol., 53, 2017, No. 1, p. 22-28
Kategorie:
Přehledový článek
Souhrn
Revidovaná WHO klasifikace nádorů centrálního nervového systému z roku 2016 obsahuje výrazné koncepční změny v porovnání s klasifikací z roku 2007. Podobně jako ve skupině difúzních gliomů došlo v posledních letech k výraznému posunu v poznání molekulárního pozadí nádorové biologie také v kategorii embryonálních nádorů CNS, zejména u meduloblastomu. Klasifikace nyní uvádí nové jednotky, které definuje jak histologicky tak zároveň na podkladě jejich molekulárních změn. Tento článek dále diskutuje novinky i ve skupině gliomů (kromě difúzních gliomů), u nádorů mozkových plen a u nádorů z pochvy periferního nervu.
Klíčová slova:
WHO klasifikace – meduloblastom – AT/RT – pleomorfní xantoastrocytom – pilocytární astrocytom – nádory z pochvy periferního nervu
Kromě difúzních astrocytárních a oligodendrogliálních nádorů, kterým byl věnován první článek v tomto čísle časopisu, doznaly v revidované WHO klasifikaci z roku 2016 (1) nejvýznamnějších změn v diagnostické koncepci embryonální nádory CNS a mezi nimi zejména meduloblastom. Těmto jednotkám tedy bude věnována největší část tohoto sdělení. Změny v revidované klasifikaci u ostatních skupin nádorů budou shrnuty v druhé části článku.
MEDULOBLASTOM A OSTATNÍ EMBRYONÁLNÍ NÁDORY CNS
Podobně jako ve skupině difúzních gliomů došlo v posledních letech k výraznému posunu v poznání molekulárního pozadí nádorové biologie také v kategorii embryonálních nádorů CNS. Zároveň bylo prokázáno, že detekce řady z molekulárních změn má přímé klinické využití pro svou výraznou prognostickou (a potenciálně prediktivní) hodnotu, což by se mělo projevit v přesnější terapeutické stratifikaci pacientů a potenciálně i v lepších léčebných výsledcích.
Meduloblastomy jsou nově klasifikovány nejen podle svých histopatologických charakteristik, ale také podle svých molekulárních znaků. Vícečetné studie transkriptomu, microRNA a methylomu meduloblastomů (2,3) ve shodě prokázaly, že meduloblastomy lze rozdělit do několika molekulárních podskupin (4) a že tyto podskupiny představují nejen rozdílné jednotky co se týče jejich molekulárního pozadí, ale i jednotky s výrazně rozdílným biologickým chování a tedy i prognózou. Tzv. integrovaná diagnóza meduloblastomu by nově měla obsahovat zařazení nádoru do dvou klasifikací - jednak jako meduloblastom histologicky definovaný, jednak jako meduloblastom geneticky definovaný. Jednotlivé meduloblastomy mají podle kombinace svých histologických a genetických parametrů výrazně rozdílnou prognózu a správné zařazení nádoru bude mít velký vliv na terapeutickou strategii u konkrétního pacienta (viz tabulka č. 1).
Klasifikace připouští diagnózu meduloblastom, NOS, avšak pouze pro takové embryonální nádory IV. mozkové komory nebo mozečku, u kterých množství či kvalita bioptického vzorku neumožnila histologickou a molekulárně genetickou charakterizaci. Pro stanovení této diagnózy však musí být vyloučeny histopatologicky podobné jednotky jako high grade malobuněčné gliomy, embryonální nádor s mnohovrstevnatými rozetami (ETMR) či atypický teratoidní/rhabdoidní nádor (AT/RT). Nelze-li ani toto zajistit, doporučuje se obecnější diagnostické zařazení jako embryonální nádor CNS, NOS.
Meduloblastom, histologicky definovaný
Oproti minulé WHO klasifikaci došlo ke spojení velkobuněčného meduloblastomu a anaplastického+ meduloblastomu do jedné skupiny (neboť odlišení těchto dvou variant většinou stejně nebylo pro jejich průnik v praxi realizovatelné), a histologicky definované meduloblastomy tak obsahují čtyři kategorie (obr. 1):
- klasický meduloblastom
- desmoplastický/nodulární meduloblastom
- meduloblastom s extenzivní nodularitou
- velkobuněčný/anaplastický meduloblastom.

Meduloblastom, geneticky definovaný
Meduloblastomy lze z molekulárního pohledu rozčlenit do 4 základních skupin (4): WNT-aktivované meduloblastomy (asi 10 % případů), SHH-aktivované meduloblastomy (asi 30 % případů) a dvě skupiny meduloblastomů nonWNT-nonSHH, které jsou provizorně označeny jako „skupina 3“ (asi 30 % případů) a „skupina 4“ (asi 40 % případů). Skupina SHH-aktivovaných meduloblastomů se dále dělí na dvě prognosticky zcela rozdílné kategorie podle přítomnosti či absence mutace genu TP53. SHH-aktivované meduloblastomy s mutací TP53 (a většinou velkobuněčnou/anaplastickou morfologií) jsou vysoce agresivní nádory; naopak SHH-aktivované meduloblastomy bez mutace TP53 (a většinou nodulární/desmoplastickou morfologií) patří mezi low risk nádory.
Některé molekulární změny, které jsou v současné době vyšetřovány a jsou potřebné pro stratifikaci pacientů (zejména amplifikace MYC) nejsou součástí nové klasifikace. Autoři však doporučují tato vyšetření nadále provádět a jejich výsledky uvádět v závěru pro zvýšení diagnostické a prognostické přesnosti.
Revidovaná klasifikace bohužel neuvádí konkrétní praktická doporučení, jakými metodami by měla být molekulární klasifikace meduloblastomů dosažena a nechává tedy prostor pro zvyklosti jednotlivých laboratoří. Obecně je doporučen i panel imunohistochemických protilátek, které fungují na parafinovém materiálu, a kombinace výsledků jejich vyšetření v celém panelu by mělo umožnit jejich molekulární zařazení. Jedná se o protilátky proti proteinům beta-catenin, GAB1, YAP1, FILA, GLI1, SFRP1, NPR3 a KCNA1. Zkušenost autora tohoto textu (J.Z.) s uvedeným imunohistochemickým panelem je však pro zdaleka nikoli stoprocentní senzitivitu ani specificitu jednotlivých protilátek spíše ambivalentní.
Meduloblastom, WNT-aktivovaný
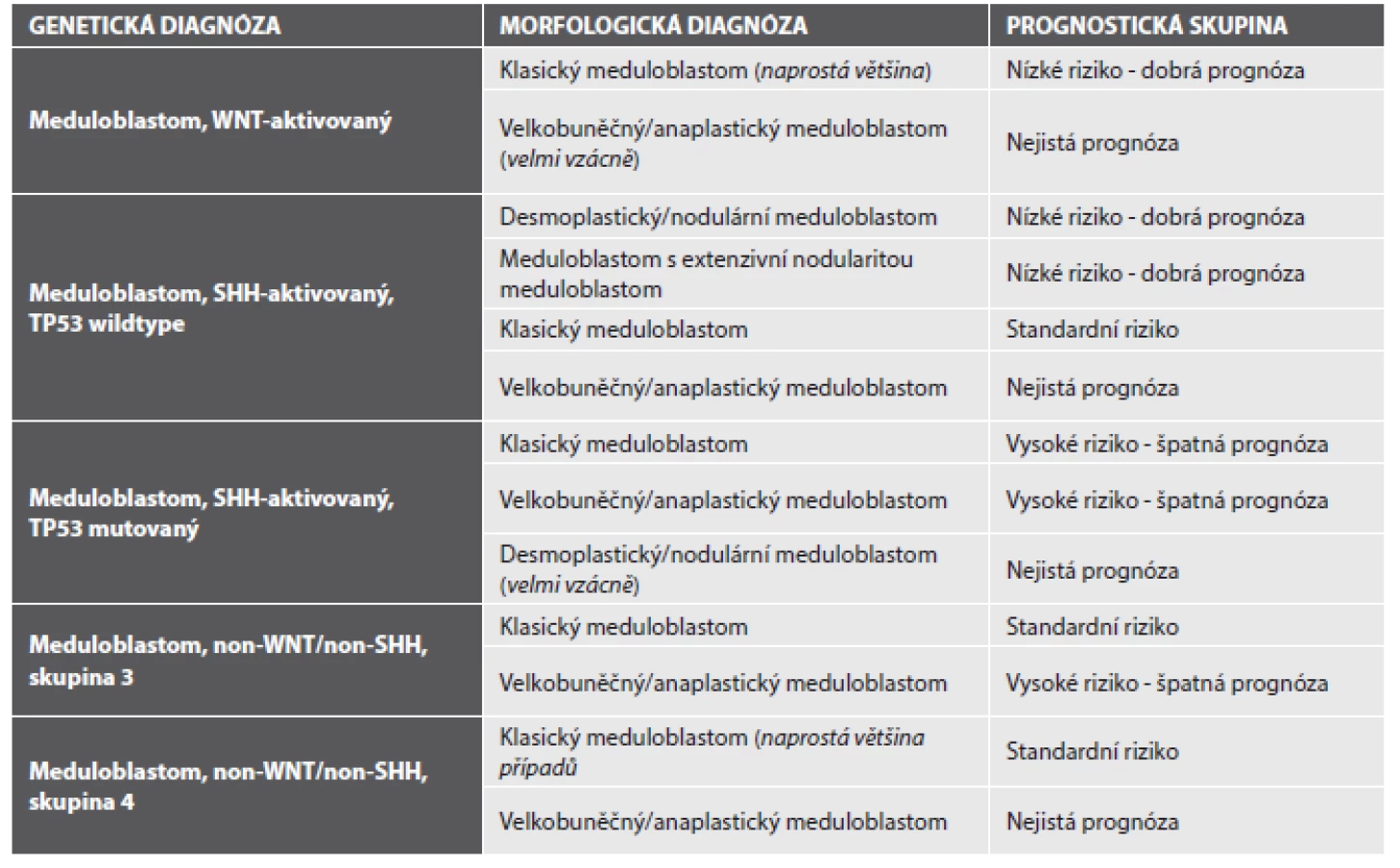
Aktivace WNT dráhy, která tuto skupinu nádorů charakterizuje, může být prokázána imunohistochemicky, a to jadernou akumulací beta-cateninu. Nejen podle našich zkušeností však nemusí být tento postup zcela bez problémů. Jaderná pozitivita beta-cateninu ve WNT-aktivovaných meduloblastomech totiž často nebývá difúzní a navíc velmi kolísá při použití různých klonů protilátek (obr. 2). Klasifikace proto doporučuje kombinovat imunohistochemii s vyšetřením hot spot mutací v exonu 3 genu CTNNB1 (3) a/nebo s detekcí monosomie 6. chromozomu (5,6).
Dalšími mutovanými geny u WNT-aktivovaných meduloblastomů jsou DDX3X (v 50 % případů), SMARCA4 (27 %), KMT2D (13 %) a TP53 (13 %). Na rozdíl od SHH-aktivovaných meduloblastomů není mutace TP53 známkou horší prognózy.
WNT-aktivované meduloblastomy jsou typicky meduloblastomy s klasickou morfologií a excelentní prognózou - při současném léčebném režimu zahrnujícím chirurgické odstranění s adjuvantní terapií je celkové přežití téměř 100 % (7,8). Velmi vzácně se v této skupině mohou vyskytnout meduloblastomy s velkobuněčnou/anaplastickou morfologií a prognóza je pak nejistá.
Meduloblastom, SHH-aktivovaný
Ačkoli je pro stanovení aktivace dráhy SHH (sonic hedgehoc) u meduloblastomů jako zlatý standard doporučena analýza genové exprese a metylační profilování (9), bylo nalezeno několik proteinů, jejich exprese je signaturou této dráhy a může být analyzována imunohistochemicky (10). Jedná se zejména o proteiny GAB1, který je markerem SHH-aktivovaných meduloblastomů a YAP1, který je pozitivní jak v SHH- aktivovaných tak v WNT-aktivovaných nádorech, je však negativní ve skupině non-WNT/non-SHH meduloblastomů. Jinak je ale pro zařazení doporučována molekulární analýza genů zapojených s SHH dráze (11,12).
Meduloblastom, SHH-aktivovaný, TP53 wildtype
Aktivace SHH dráhy je u meduloblastomů bez mutace TP53 spojena s germinální nebo somatickou mutací v genech PTCH1, SUFU a SMO. Do této skupiny nejčastěji spadají meduloblastomy s desmoplastickou/nodulární morfologií a meduloblastomy s extenzivní nodularitou (8,13) a řadí se k low-risk nádorům s velmi dobrou prognózou. Méně často ale moho mít SHH-aktivované meduloblastomy také morfologii klasickou či velkobuněčnou/anaplastickou a prognóza je pak nejistá.
Meduloblastom, SHH-aktivovaný, TP53 mutovaný
Aktivace SHH dráhy je u TP53-mutovaných nádorů spojena s amplifikací genů GLI2, MYCN nebo SHH. Mutace v genech PTCH1, SUFU a SMO většinou chybí.
U těchto nádorů bývá nejčastěji velkobuněčná/anaplastická morfologie a prognóza je velmi nepříznivá.
Meduloblastom, non-WNT/non-SHH
Do této skupiny spadá asi 60 % meduloblastomů a jsou to meduloblastomy molekulární „skupiny 3“ nebo „skupiny 4“. Označení „skupina 3“ a „skupina 4“ je provizorní, neboť na rozdíl od WNT- a SHH-aktivovaných meduloblastomů není zatím jejich molekulární charakterizace jednoznačná (14,15).
Nejčastěji mají non-WNT/non-SHH meduloblastomy klasickou morfologii a jsou to nádory standardního rizika. Vzácněji se může jednat o velkobuněčný/anaplastický meduloblastom (většinou spadá do molekulární „skupiny 3“) a pak nádor patří do kategorie high-risk tumorů.
WHO klasifikace doporučuje pro klasifikaci těchto tumorů panel imunohistochemických protilátek, viz výše (10).
ATYPICKÝ TERATOIDNÍ/RHABDOIDNÍ NÁDOR (AT/RT)
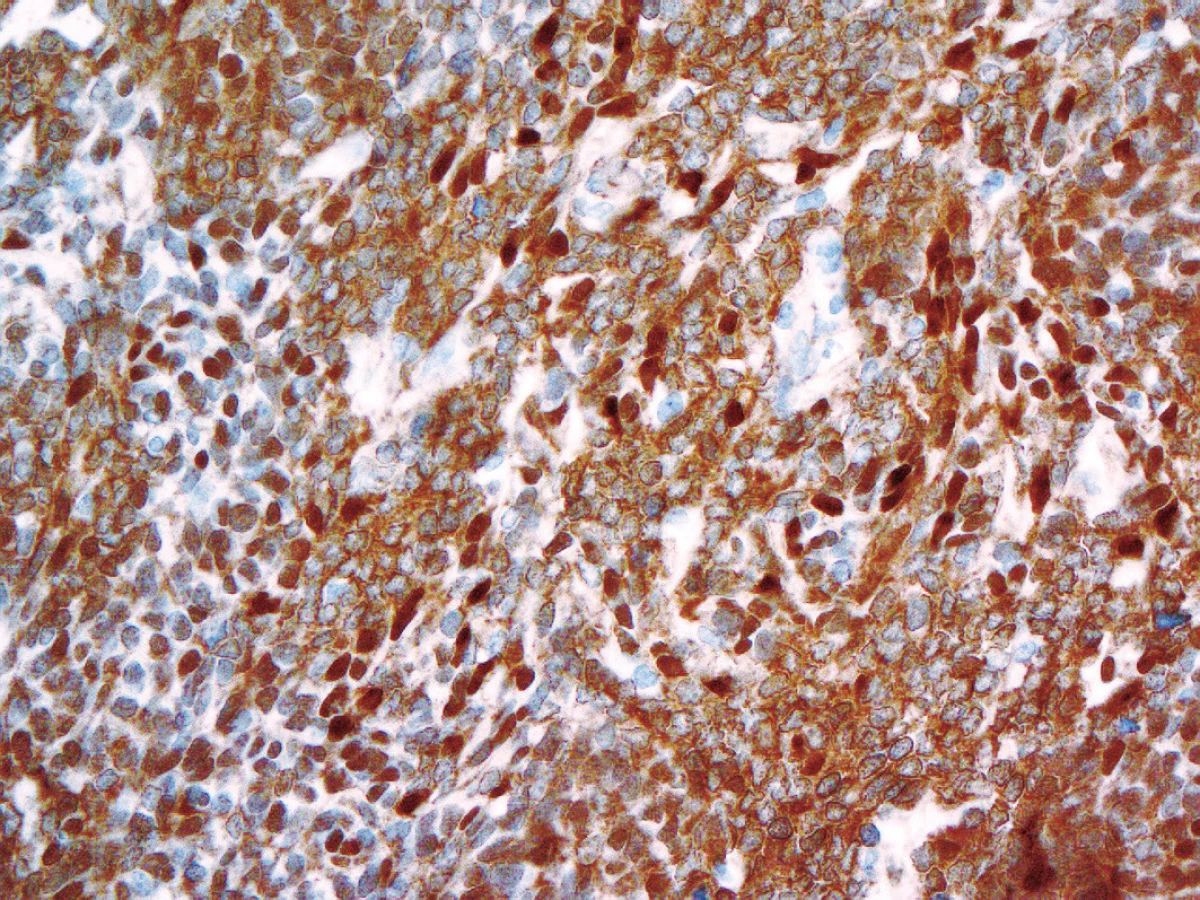
Novinkou v revidované klasifikaci je rozšíření molekulární definice nádoru - kromě inaktivace genu SMARCB1 (INI1) může být v pozadí rozvoje AT/RT i mutace genu SMARCA4 (BRG1) (16-19). Proti oběma proteinům (INI1, BRG1) jsou dostupné protilátky pro imunohistochemické vyšetření, které by měly při inaktivaci genu dávat v jádrech nádorových buněk negativní výsledek (při pozitivní vnitřní kontrole v nenádorových buňkách) (obr. 3). Imunohistochemický nález by měl být vždy verifikován na úrovni molekulárně genetické mutační analýzou příslušných genů.
Pokud není prokázána mutace ani jedno z těchto genů a nádor má přesto morfologii AT/RT, je doporučeno nádor klasifikovat jako tzv. embryonální nádor CNS s rhabdoidními rysy.
OSTATNÍ EMBRYONÁLNÍ NÁDORY CNS
Nejvýraznější změnou mezi ostatními embryonálními nádory CNS je zrušení jednotky primitivní neuroektodermální nádor (PNET) CNS a zavedení nové jednotky - embryonální nádor s mnohovrstevnatými rozetami (embryonal tumor with multilayered rosettes - ETMR), s alterací C19MC.
Embryonální nádor s mnohovrstevnatými rozetami (ETMR), C19MC alterovaný
Mezi tyto velmi agresivní nádory, které jsou charakterizované alterací (zahrnující amplifikace a fúze) C19MC lokusu na 19. chromozomu (19q13.42), patří některé nádory, které byly dříve označovány jako ETANTR (embryonální nádor s abundantním neuropilem a pravými rozetami), ependymoblastom či meduloepiteliom (20,21) (obr. 4). Mohou vzniknout prakticky v jakékoli lokalizaci CNS.

Pokud má embryonální nádor morfologii některé z těchto jednotek, ale alterace C19MC lokusu na 19. chromozomu není prokázána, doporučuje WHO klasifikace tyto nádory zařadit do jedné z těchto skupin:
- Embryonální nádor s mnohovrstevnatými rozetami, NOS
- Meduloepiteliom
- CNS neuroblastom
- CNS ganglioneuroblastom.
Pokud morfologie embryonálního nádoru CNS neumožňuje jeho zařazení, lze použít poslední možnost („wastebasket“): embryonální nádor CNS, NOS.
ASTROCYTOMY (KROMĚ DIFÚZNÍCH ASTROCYTOMŮ)
Základní rozdělení těchto gliových nádorů zůstává v revidované klasifikace stejné.
Pilocytární astrocytom (grade I)
Nově je v definici nádoru charakterizováno molekulární pozadí nádoru, a to přítomnost mutací genů kódující proteiny dráhy MAPK (22,23). Nejčastěji (ve více než 70 %) je to tandemová duplikace vedoucí ke vzniku transformujícího fúzního genu KIAA1549-BRAF. Méně často to mohou být i jiné RAF fúze či mutace BRAF (včetně BRAF V600E), mutace genu KRAS a dalších genů zahrnutých v dráze MAPK (24). Molekulární vyšetření není v revidované WHO klasifikaci pro diagnózu pilocytárního astrocytomu vyžadováno, je však doporučeno jako nástroj podporující diagnózu.
Pilomyxoidní astrocytom
Tato převážně supraselární varianta pilocytárního astrocytomu, které byl v minulé WHO klasifikaci pro údajně agresivnější biologické chování s častějšími recidivami a možností rozsevu v likvorových cestách (25) přirazen grade II, v klasifikaci zůstává. Vznikly však pochybnosti o jeho skutečně horším biologickém chování než je tomu u klasického pilocytárního astrocytomu, a tak klasifikace doporučuje do vyřešení této otázky pilomyxoidnímu astrocytomu žádný zvláštní grade nestanovovat.
Pleomorfní xantoastrocytom (PXA, grade II).
V definici PXA je nyní uvedena jeho nejčastější genetická změna - mutace BRAF V600E, která při absenci mutace genů IDH podporuje diagnózu.
Pro PXA s výraznějšími známkami anaplazie bylo v minulé klasifikaci doporučeno označení „pleomorfní xantoastrocytom s anaplastickými rysy“. Nově byla vyčleněna nová jednotka - anaplastický PXA, grade III - a je definována jako PXA s mitotickým indexem ≥ 5/10 HPF. Může být přítomna nekróza, ale pokud není přítomna zvýšená mitotická aktivita má přítomnost nekrózy nejednoznačný význam (26). Frekvence mutace BRAF V600E u anaplastického PXA je podstatně nižší než u konvenčního PXA (26,27).
Subependymální obrovskobuněčný astrocytom (SEGA, grade I)
Jednotka zůstává beze změn.
EPENDYMÁLNÍ NÁDORY
Oproti očekávání zatím kapitola ependymálních nádorů nedoznala výraznějších změn. Problematický grading ependymomů v revidované WHO klasifikaci zůstává. Vágní doporučení pro odlišení ependymomu, grade II, od jeho anaplastického protějšku (grade III) zůstalo bohužel beze změn, a tak grading ependymomů pro svou velmi nízkou reproducibilitu (28,29) nadále nejspíše nebude mít význam na klinický management těchto nádorových onemocnění.
V poslední době se vyskytla řada studií, které detekovaly u ependymomů mnoho molekulárních změn, které by mohly mít v budoucnu relevantní prognostický a/nebo prediktivní význam (30-32). Výsledky však doposud nebyly reprodukovány a tak autoři WHO klasifikace volí vyčkávací strategii. Potenciální rozdělení ependymomů do 9 podskupin podle jejich molekulárních charakteristik a lokalizace je sice v revidovaném vydání klasifikace uvedeno, není však pro klasifikaci aplikováno.
Přesto dvě změny v klasifikaci ependymálních nádorů nalezneme. Jednak byl zrušen tzv. „buněčný ependymom“, a tak ze zvláštních morfologických variant ependymomu zůstávají pouze ependymomy papilární, světlobuněčné a tanycytické.
Nově byla definována jednotka - ependymom, RELA-fúze pozitivní - jako supratentoriální ependymom dětského věku s mimořádně špatnou prognózou (31). Přítomnost definující fúze C11orf95-RELA vede k imunohistochemické expresi proteinu L1CAM (33); tento protein však může být detekován i v jiných mozkových nádorech. Pro detekci RELA-fúze je ve WHO klasifikaci doporučeno vyšetření metodou FISH. Morfologicky se tyto nádory od konvenčních ependymomů neliší (33).
NEURONÁLNÍ A SMÍŠENÉ GLIONEURONÁLNÍ NÁDORY
Kategorie glioneuronálních nádorů, snad kromě zavedení morfologické varianty gangliocytomu - tzv. “multinodulárního a vakuolizujícího neuronální nádoru“ (34) - nedoznala výraznějších změn. Nově byla do této skupiny zařazena klinicko-patologická jednotka difúzní leptomeningeální glioneuronální tumor (35), což je vzácný nádor dětí a adolescentů s převážně leptomeningeálním šířením, který je tvořen buňkami připomínající oligodendroglii.
NÁDORY MOZKOVÝCH PLEN
Jedinou výraznější změnou je změna definice atypického meningiomu, grade II. Kromě mitotického indexu ≥ 4/10HPF nebo přítomnosti tří z pěti následujících rysů: zvýšená buněčnost, přítomnost malých buněk s vysokým nukleocytoplazmatickým indexem, prominentní jadérka, přítomnost spontánních nekróz a tzv. sheeting (tj. růst bez organoidního uspořádání do pruhů či vírovitých struktur), je dalším kriteriem pro označení meningiomu jako nádoru atypického (grade II) přítomnost invaze do mozkové tkáně.
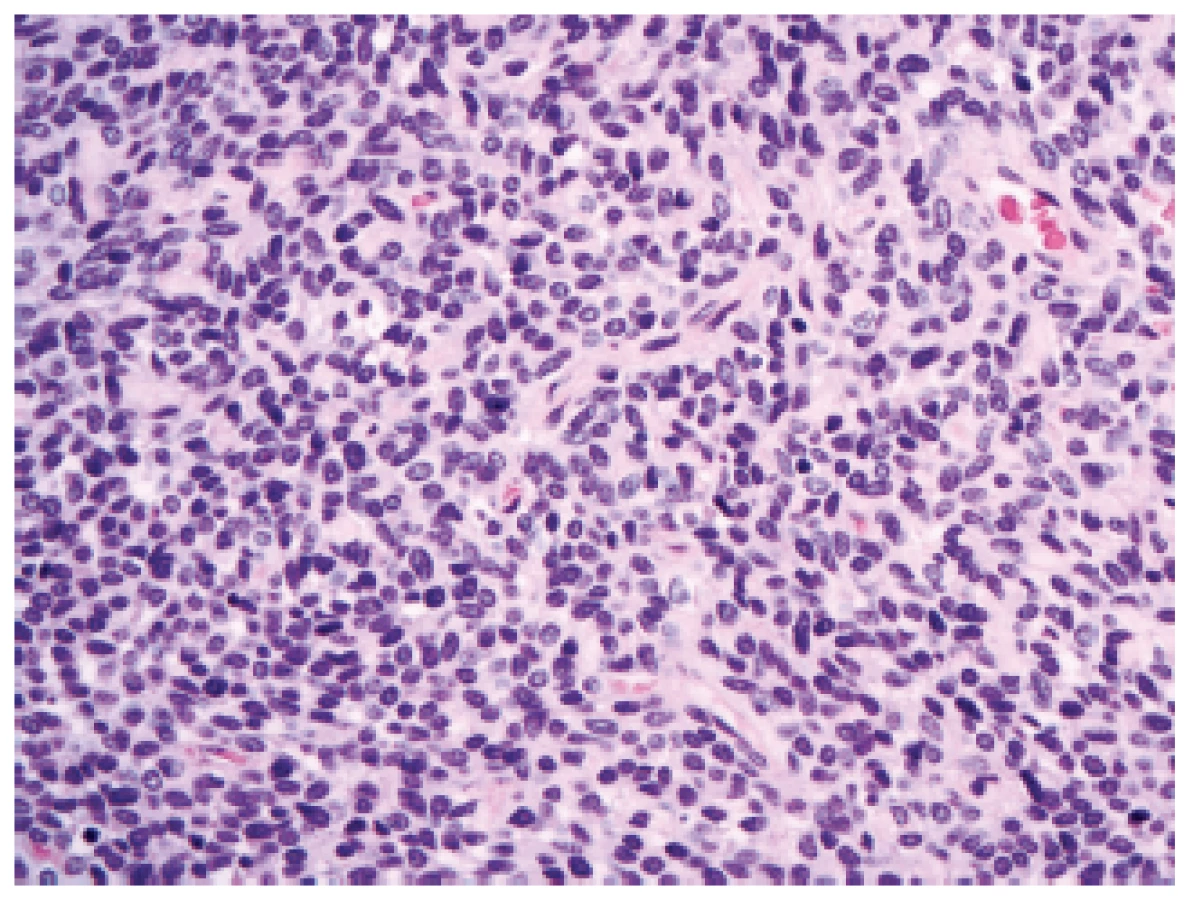
V revidované klasifikaci se (polo)ustoupilo od diagnózy leptomeningeálního hemangiopericytomu a byla zavedena nová jednotka se smíšeným označením - „solitární fibrózní tumor / hemangiopericytom“. Novinkou v oblasti nádorů CNS je možnost přidělit tomuto nádoru grade podobně jako to je u většiny maligních nádorů mimo CNS, a to ze spektra grade I - III, podle buněčnosti a mitotické aktivity. Jako grade I by měly být označeny nádory hypocelulární s bohatou kolagenizací intersticia (dříve označované spíše jako solitární fibrózní nádory). Jako grade II by se měly označit nádory méně kolagenizované, buněčnější s charakteristickou sítí větvených kapilár (dříve označované spíše jako hemangiopericytomy)(obr. 5). A pro nádory s vysokou buněčností a mitotickým indexem ≥ 5/10 HPF (dříve anaplastický hemangiopericytom) je rezervován grade III.
NÁDORY Z POCHVY PERIFERNÍHO NERVU
Mezi těmito nádory došlo v revidované klasifikaci k několika změnám. Melanotický schwannom už není jen morfologickou variantou schwannomu, ale je vedle celulárního a plexiformního schwannomu další samostatnou klinicko-patologickou jednotkou. Důvodem k tomu je jednak jeho častá asociace s Carneyho komplexem a mutací genu PRKAR1A, jednak skutečnost, že více než 10 % případů melanotického schwannomu malignizuje (36).
Dále byla zavedena nová jednotka „hybridní nádor z pochvy periferního nervu“, kam spadají benigní nádory s kombinací morfologie více než jednoho nádoru z pochvy periferního nervu (schwannom, neurofibrom, perineuriom). Nejčastější kombinací je hybridní nádor obsahující schwannom/perineuriom, ten bývá nejčastěji sporadický, a dále neurofibrom/schwannom a neurofibrom/perineuriom (obr. 6), které bývají typicky asociované s neurokutánními syndromy (schwannomatóza, neurofibromatózy) (37,38).
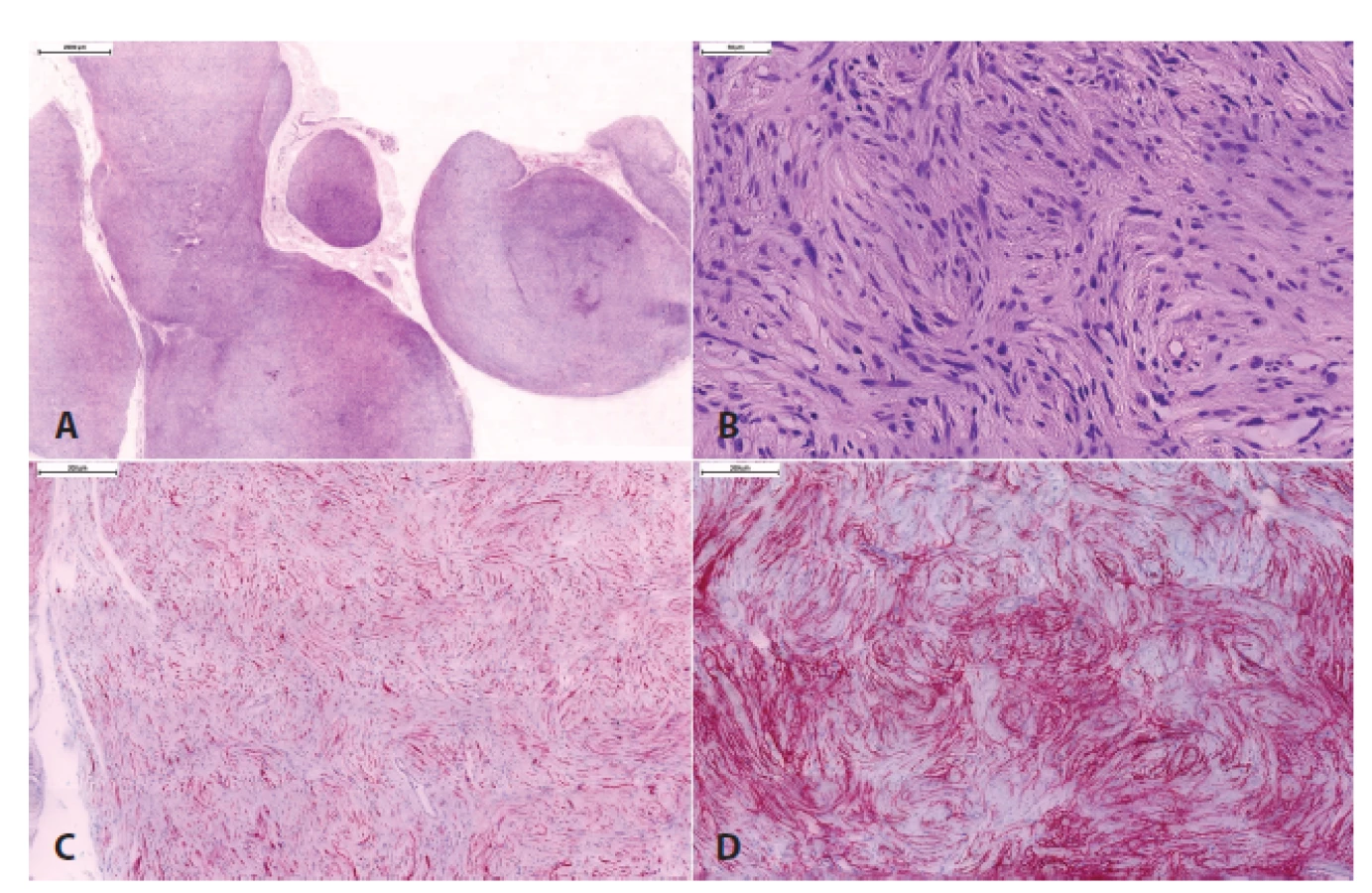
V rámci maligních nádorů z pochvy periferního nervu (MPNST) se nyní pro svoji zřetelně lepší prognózu než v případě konvenčního MPNST (39-41) vydělují dva subtypy - epiteloidní MPNST a perineurální MPNST. Naproti tomu je MPNST s divergentní diferenciací (tj. maligní Tritonský nádor) považován jen za morfologickou variantu konvenčního MPNST.
PROHLÁŠENÍ
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Adresa pro korespondenci:
Prof. MUDr. Josef Zámečník, Ph.D.
Ústav patologie a molekulární medicíny
2. LF UK a FN v Motole
V Úvalu 84,
150 06 Praha 5
tel.: 224 435 635
e-mail: josef.zamecnik@lfmotol.cuni.cz
Zdroje
1. Louis DN et al. World Health Organization classification of tumours of the central nervous system (revised 4th ed). Lyon, IARC, 2016.
2. Batora NV, Sturm D, Jones DT, Kool M, Pfister SM, Northcott PA. Transitioning from genotypes to epigenotypes: why the time has come for medulloblastoma epigenomics. Neuroscience 2014; 264: 171-185.
3. Northcott PA, Jones DT, Kool M, et al. Medulloblastomics: the end of the beginning. Nat Rev Cancer 2012; 12(12): 818-834.
4. Taylor MD, Northcott PA, Korshunov A, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 2012; 123(4): 465-472.
5. Clifford SC, Lusher ME, Lindsey JC, et al. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006; 5(22): 2666-2670.
6. Kool M, Korshunov A, Remke M, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 2012; 123(4): 473-484.
7. Ellison DW, Onilude OE, Lindsey JC, et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children‘s Cancer Study Group Brain Tumour Committee. J Clin Oncol 2005; 23(31): 7951-7957.
8. Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 2006; 24(12): 1924-1931.
9. Kool M, Jones DT, Jager N, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014; 25(3): 393-405.
10. Ellison DW, Dalton J, Kocak M, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 2011; 121(3): 381-396.
11. Northcott PA, Shih DJ, Remke M, et al. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol 2012; 123(4): 615-626.
12. Shou Y, Robinson DM, Amakye DD, et al. A five-gene hedgehog signature developed as a patient preselection tool for hedgehog inhibitor therapy in medulloblastoma. Clin Cancer Res 2015; 21(3): 585-593.
13. Pomeroy SL, Tamayo P, Gaasenbeek M, et al. Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature 2002; 415(6870): 436-442.
14. Cho YJ, Tsherniak A, Tamayo P, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 2011; 29(11): 1424-1430.
15. Kool M, Koster J, Bunt J, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 2008; 3(8): e3088.
16. Biegel JA. Molecular genetics of atypical teratoid/rhabdoid tumor. Neurosurg Focus 2006; 20(1): E11.
17. Hasselblatt M, Gesk S, Oyen F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 2011; 35(6): 933-935.
18. Judkins AR. Immunohistochemistry of INI1 expression: a new tool for old challenges in CNS and soft tissue pathology. Adv Anat Pathol 2007; 14(5): 335-339.
19. Woehrer A, Slavc I, Waldhoer T, et al. Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 2010; 116(24): 5725-5732.
20. Korshunov A, Sturm D, Ryzhova M, et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 2014; 128(2): 279-289.
21. Spence T, Sin-Chan P, Picard D, et al. CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol 2014; 128(2): 291-303.
22. Jones DT, Hutter B, Jager N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 2013; 45(8): 927-932.
23. Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013; 45(6): 602-612.
24. Collins VP, Jones DT, Giannini C. Pilocytic astrocytoma: pathology, molecular mechanisms and markers. Acta Neuropathol 2015; 129(6): 775-788.
25. Tihan T, Fisher PG, Kepner JL, et al. Pediatric astrocytomas with monomorphous pilomyxoid features and a less favorable outcome. J Neuropathol Exp Neurol 1999; 58(10): 1061-1068.
26. Ida CM, Rodriguez FJ, Burger PC, et al. Pleomorphic Xanthoastrocytoma: Natural History and Long-Term Follow-Up. Brain Pathol 2015; 25(5): 575-586.
27. Schmidt Y, Kleinschmidt-DeMasters BK, Aisner DL, Lillehei KO, Damek D. Anaplastic PXA in adults: case series with clinicopathologic and molecular features. J Neurooncol 2013; 111(1): 59-69.
28. Ellison DW, Kocak M, Figarella-Branger D, et al. Histopathological grading of pediatric ependymoma: reproducibility and clinical relevance in European trial cohorts. J Negat Results Biomed 2011; 10: 7.
29. Godfraind C. Classification and controversies in pathology of ependymomas. Childs Nerv Syst 2009; 25(10): 1185-1193.
30. Mack SC, Witt H, Piro RM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014; 506(7489): 445-450.
31. Pajtler KW, Witt H, Sill M, et al. Molecular Classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 2015; 27(5): 728-743.
32. Witt H, Mack SC, Ryzhova M, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 2011; 20(2): 143-157.
33. Parker M, Mohankumar KM, Punchihewa C, et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014; 506(7489): 451-455.
34. Huse JT, Edgar M, Halliday J, Mikolaenko I, Lavi E, Rosenblum MK. Multinodular and vacuolating neuronal tumors of the cerebrum: 10 cases of a distinctive seizure-associated lesion. Brain Pathol 2013; 23(5): 515-524.
35. Rodriguez FJ, Perry A, Rosenblum MK, et al. Disseminated oligodendroglial-like leptomeningeal tumor of childhood: a distinctive clinicopathologic entity. Acta Neuropathol 2012; 124(5): 627-641.
36. Carney JA. Psammomatous melanotic schwannoma. A distinctive, heritable tumor with special associations, including cardiac myxoma and the Cushing syndrome. Am J Surg Pathol 1990; 14(3): 206-222.
37. Harder A, Wesemann M, Hagel C, et al. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. Am J Surg Pathol 2012; 36(5): 702-709.
38. Kacerovská D, Michal M, Kuroda N, et al. Hybrid peripheral nerve sheath tumors, including a malignant variant in type 1 neurofibromatosis. Am J Dermatopathol 2013; 35(6): 641-649.
39. Jo VY, Fletcher CD. Epithelioid malignant peripheral nerve sheath tumor: clinicopathologic analysis of 63 cases. Am J Surg Pathol 2015; 39(5): 673-682.
40. Hirose T, Scheithauer BW, Sano T. Perineurial malignant peripheral nerve sheath tumor (MPNST): a clinicopathologic, immunohistochemical, and ultrastructural study of seven cases. Am J Surg Pathol 1998; 22(11): 1368-1378.
41. Rosenberg AS, Langee CL, Stevens GL, Morgan MB. Malignant peripheral nerve sheath tumor with perineurial differentiation: „malignant perineurioma“. J Cutan Pathol 2002; 29(6): 362-367.
Štítky
Patologie Soudní lékařství ToxikologieČlánek vyšel v časopise
Česko-slovenská patologie

2017 Číslo 1
Nejčtenější v tomto čísle
-
Novinky vo WHO klasifikácii nádorov centrálneho nervového systému 2016
– 1. časť: Difúzne infiltrujúce gliómy -
Novinky ve WHO klasifikaci nádorů centrálního nervového systému 2016
2. část: Embryonální nádory CNS a ostatní skupiny nádorů (kromě difúzních gliomů) - Neobvyklý histopatologický obraz akútneho poškodenia pľúc v rôznej fáze rezorpcie s prevahou organizujúcej sa pneumónie u jedinca s chrípkou A(H1N1)
- Sebaceozní adenom vzniklý ve zralém cystickém teratomu ovária. Kazuistika