
Prenatální diagnostika chromozomálních aberací Česká republika: 1994 – 2007
Prenatal Diagnostics of Chromosomal Aberrations Czech Republic: 1994 – 2007
Aim of study:
An analysis of prenatal diagnostics efficiency of selected types of chromosomal aberrations in the Czech Republic in 2007. Update of 1994 – 2007 data according to particular selected diagnoses.
Typ of study:
Retrospective epidemiological analysis of pre- and postnatal chromosomal aberrations diagnostics and its efficiency.
Material and methods:
Data on pre- and postnatally diagnosed birth defects in the Czech Republic during 1994 – 2007 were used. Data on prenatally diagnosed birth defects (and for terminated pregnancies) were collected from particular departments of prenatal diagnostics, medical genetics and ultrasound diagnostics in the Czech Republic, data on birth defects in births from the National Birth Defects Register (Institute for Health Information and Statistics). Total numbers over the period under the study, mean incidences of selected types of chromosomal aberrations and mean prenatal diagnostics efficiencies were analyzed. Following chromosomal aberrations were studied: Down, Edwards, Patau, Turner and Klinefelter syndromes and syndromes 47,XXX and 47,XYY.
Results:
A relative proportion of Down, Edwards and Patau syndromes as well as other autosomal and gonosomal aberration is presented in figures. Recently, trisomies 13, 18 and 21 present around 70 % of all chromosomal aberrations in selectively aborted fetuses, in other pregnancies, “other chromosomal aberrations” category (mostly balanced reciprocal translocations and inversions) present more than 2/3 of all diagnoses.
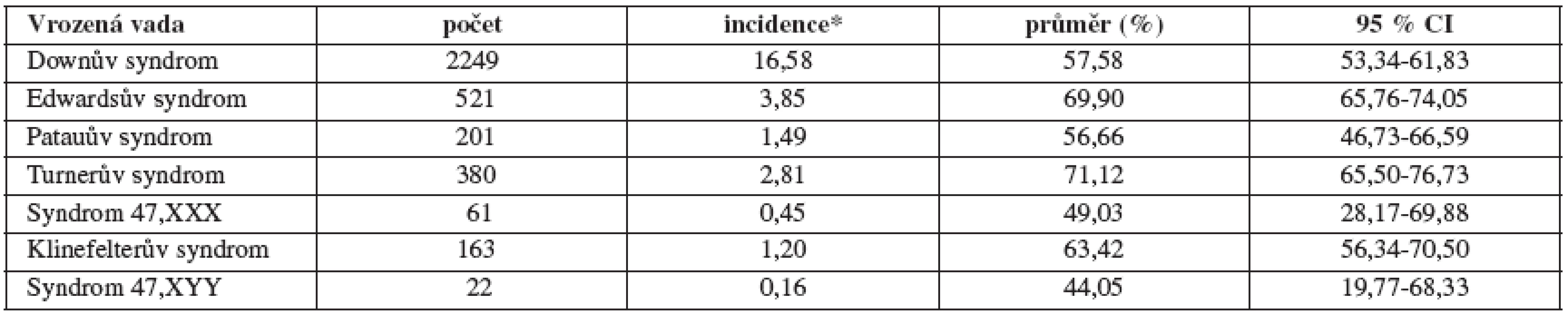
During the period under the study, following total numbers, mean relative incidences (per 10 000 live births, in brackets) and mean prenatal diagnostics efficiency (in %) were found in following chromosomal syndromes: Down syndrome 2 244 (16.58) and 63.37 %, Edwards syndrome 521 (3.85) and 79.93 %, Patau syndrome 201 (1.49) and 68.87 %, Turner syndrome 380 (2.81) and 79.89 %, 47,XXX syndrome 61 (0.45) and 59.74 %, Klinefelter syndrome 163 (1.20) and 73.65 % and 47,XYY syndrome 22 (0.16) and 54.76 %.
Conclusions:
The study gives updated results of incidences analysis of both pre- and postnatally diagnosed chromosomal birth defects in the Czech Republic during the 1994 - 2007 period. Incidences found in our study correspond (in case of trisomies 13, 18 and 21) with those published widely in literature as well as with those found in large-scale international studies (ICBDSR, EUROCAT). In case of gonosomal aberrations, incidences found in this study are lower that those published, most probably due to a later registration (over 15 years of age of the child) of these diagnoses.
Key words:
birth defect, incidence, prenatal diagnostics, chromosomal aberration, Down syndrome, Edwards syndrome, Patau syndrome, Turner syndrome, Klinefelter syndrome, Czech Republic.
Autoři:
V. Gregor 1,2; A. Šípek 1,3; A. Šípek jr. 4; J. Horáček 1,5; P. Langhammer 6; L. Petržílková 6; P. Calda 4,7
![]()
Působiště autorů:
Oddělení lékařské genetiky, Fakultní Thomayerova nemocnice, Praha, ředitel MUDr. K. Filip, CSc., MBA
1; Katedra lékařské genetiky, Institut postgraduálního vzdělávání ve zdravotnictví, Praha, ředitel MUDr. Z. Hadra
2; 3. lékařská fakulta Univerzity Karlovy, Praha, děkan doc. MUDr. B. Svoboda, CSc.
3; 1. lékařská fakulta Univerzity Karlovy, Praha, děkan prof. MUDr. T. Zima, DrSc., MBA
4; Gennet, Praha, vedoucí MUDr. D. Stejskal
5; Ústav zdravotnických informací a statistiky České republiky, Praha, ředitelka Mgr. V. Mazánková
6; Gynekologicko-porodnická klinika 1. LF UK a VFN, Praha, přednosta prof. MUDr. A. Martan, DrSc.
7
Vyšlo v časopise:
Ceska Gynekol 2009; 74(1): 44-54
Souhrn
Cíl studie:
Analýza úspěšnosti prenatální diagnostiky a sekundární prevence vybraných typů chromozomálních aberací v České republice v roce 2007. Aktualizace dat za období 1994 - 2006 podle jednotlivých vybraných diagnóz.
Typ studie:
Retrospektivní epidemiologická analýza prenatálního a postnatálního záchytu vybraných typů chromozomálních aberací a efektivity jejich prenatální diagnostiky.
Materiál a metodika:
V práci byla použita data o prenatálně a postnatálně diagnostikovaných vrozených vadách v České republice v období let 1994 – 2007. Data o prenatálně diagnostikovaných a pro tuto vrozenou vadu předčasně ukončených těhotenství byla získána z jednotlivých pracovišť prenatální diagnostiky a lékařské genetiky v ČR. Data o diagnózách u narozených dětí byla získána z Národního registru vrozených vad vedeného v Ústavu zdravotnických informací a statistiky České republiky. Byly analyzovány absolutní počty a stanoveny incidence za celé sledované období. Dále byla vyhodnocena úspěšnost sekundární prevence těchto vad. V naší analýze jsme sledovali incidence těchto vybraných typů chromozomálních aberací: Downův syndrom, Edwardsův syndrom, Patauův syndrom, Turnerův syndrom, syndrom 47,XXX, Klinefelterův syndrom a syndrom 47,XYY.
Výsledky:
Grafy ukazují vzájemné zastoupení těchto diagnóz: Downův syndrom, Edwardsův syndrom, Patauův syndrom, jiné autozomální odchylky a jiné gonozomální odchylky. V České republice v současné době u případů ukončených těhotenství významně dominují trizomie chromozomů 13, 18 a 21 – představují zhruba 70 % všech chromozomálních aberací; u případů neukončených je nejvíce zastoupená skupina „jiné chromozomální odchylky“, která představuje více než dvě třetiny diagnóz.
Výsledky jednotlivých analyzovaných diagnóz jsou uvedeny v grafech. Každé diagnóze jsou věnovány dva grafy – první (a) ukazuje absolutní počty a druhý (b) relativní incidence na 10 000 živě narozených. Údaje jsou uvedeny jak pro případy u narozených, tak pro případy prenatálně diagnostikované a pro tuto vadu ukončené. Doplňující údaje ke všem sledovaným diagnózám jsou pak uvedeny v tabulce 1. První část je věnována numerickým aberacím autozomů (Downův, Edwardsův a Patauův syndrom), druhá pak numerickým aberacím gonozomů (Turnerův a Klinefelterův syndrom, syndrom 47,XXX a 47,XYY).
V případě Downova syndromu bylo zachyceno celkem 2 244 diagnóz (16,58 na 10 000 živě narozených) a sekundární prevence byla 63,37 % (95% CI 56,65 – 70,10). Edwardsův syndrom - celkem registrováno 521 diagnóz (3,85 na 10 000 živě narozených) při sekundární prevenci 79,93 % (95% CI 75,34 – 84,53). Třetí sledovanou aberací je Patauův syndrom, celkem bylo zachyceno 201 diagnóz (1,49 na 10 000 živě narozených) a sekundární prevence této vady byla v průměru 68,87 % (95% CI 58,83 – 78,91).
Druhou sledovanou a prezentovanou skupinou jsou vybrané gonozomální aberace. První z této skupiny – Turnerův syndrom byl zachycen v počtu 380 diagnóz (2,81 na 10 000 živě narozených) a sekundární prevence vady byla v průměru 79,89 % (95% CI 73,00 – 86,79). Významně méně zastoupená diagnóza syndromu 47,XXX byla nalezena pouze v 61 případech (0,45 na 10 000 živě narozených), sekundární prevence je 59,74 % (95% CI 40,21 – 79,27). Třetí sledovanou vadou v této skupině je Klinefelterův syndrom. Celkem bylo v tomto období diagnostikováno 163 případů a byla zjištěna průměrná incidence 1,20 na 10 000 živě narozených. Průměrná sekundární prevence byla 73,65 % (95% CI 66,63 – 80,68). Poslední sledovanou vadou je další gonozomální aberace – Syndrom 47,XYY. Z registrovaných případů vyplývá, že je z této skupiny nejméně častá – 22 případů a relativní incidence je 0,16 na 10 000 živě narozených. Sekundární prevence je v průběhu díky nízké četnosti rozkolísaná, s průměrem 54,76% (95% CI 31,24 – 78,28).
Závěr:
Práce předkládá aktualizované výsledky analýz absolutního počtu, frekvence, incidence a procenta prenatálního záchytu 7 vybraných závažných vrozených chromozomálních aberací v České republice v období 1994 – 2007. Stanovili jsme incidence těchto sedmi závažných aberací v České republice pro celkové počty těchto diagnóz (prenatálně + postnatálně diagnostikované případy): Downův syndrom 16,58 na 10 000 živě narozených; Edwardsův syndrom 3,85 na 10 000 živě narozených; Patauův syndrom 1,49 na 10 000 živě narozených; Turnerův syndrom 2,81 na 10 000 živě narozených; syndrom 47,XXX 0,45 na 10 000 živě narozených; Klinefelterův syndrom 1,20 na 10 000 živě narozených a syndrom 47,XYY 0,16 na 10 000 živě narozených. Tyto nalezené incidence odpovídají v případě trizomií 13, 18 a 21 jak literárně uváděným incidencím, tak i v současnosti publikovaným incidencím z populačních přehledů mezinárodních organizací sledujících incidence vrozených vad v mezinárodním měřítku (International Cleringhouse for Birth Defects Surveillance and Research /ICBDSR/ a European Surveillance of Congenital Anomalies /EUROCAT/). V případě syndromu Turnerova, Klinefelterova a syndromů 47,XXX a 47,XYY jsou námi zjištěné incidence odpovídající údajům z populačních epidemiologických studií (EUROCAT a ICBDSR), jsou však nižší než některé literární údaje, což odpovídá možnému pozdějšímu záchytu těchto diagnóz (nad 15 let věku dítěte).
Klíčová slova:
vrozená vada, incidence, prenatální diagnostika, chromozomální aberace, Downův syndrom, Edwardsův syndrom, Patauův syndrom, Turnerův syndrom, Klinefelterův syndrom, Česká republika.
ÚVOD
Prenatální diagnostika je založená na komplexní spolupráci několika medicínských oborů – klinické genetiky, gynekologie a porodnictví, ultrazvukové diagnostiky a klinické biochemie. Vzhledem k náročnosti takovéto integrace je výhodné provádět prenatální diagnostiku v multidisciplinárních centrech s nepostradatelným zastoupením klinické genetiky. Může se zdát, že hlavním úkolem prenatální diagnostiky je včasné zachycení abnormalit ve vývoji plodu a nabídka umělého ukončení patologicky probíhajícího těhotenství.
V návaznosti na klinicko-genetické poradenství je zde však úkolů více:
- Poskytnout párům s rizikem narození dítěte s vadou možnost informovaného výběru dalšího postupu.
- Psychologicky podpořit rodiče, zvláště u případů závažných diagnóz či vysokých rizik.
- Umožnit párům s konkrétním rizikem vrozené vady započít těhotenství s vědomím, že případné postižení plodu je diagnostikovatelné ještě před narozením.
- Poskytnout párům v situaci před narozením postiženého dítěte optimální postup z hlediska těhotenské péče, vedení porodu a postnatální péče.
- Umožnit případnou prenatální léčbu postiženého plodu. Ta je zatím k dispozici pouze pro velice malý počet vrozených vad a onemocnění. Například ultrazvukově diagnostikovaná významná infravezikální obstrukce močových cest, kde zavedení shuntu do močových cest intra utero může zabránit nezvratnému postižení vyvíjejících se plic a ledvin.
Mezi genetikou a prenatálním vývojem lidského jedince existuje velmi složitý vztah, který má zásadní a praktické důsledky pro lidské zdraví a lidské nemoci. Pochopení normálních vývojových mechanismů a jejich genetické kontroly je nezbytné pro porozumění vzniku vrozených vývojových vad a geneticky podmíněných onemocnění. Pro úspěšné zavedení preventivních a terapeutických postupů je potom nezbytné co nejlepší porozumění patogenezi onemocnění na molekulárně-genetické úrovni. Jedinci s vrozenými chromozomálními aberacemi představují v současné době nadále závažný zdravotnický sociální, etický, ale i ekonomický problém. Vrozené vady stále patří k významným příčinám perinatální, novorozenecké a kojenecké úmrtnosti.
Chromozomální aberace představují odchylky karyotypu ve smyslu nestandardního počtu chromozomů (numerické aberace) či ve smyslu narušené struktury jednotlivých chromozomů (strukturální aberace). Tyto odchylky se mohou týkat jak nepohlavních (autozomální aberace), tak i pohlavních chromozomů (gonozomální aberace). Mezi klinicky definovanými syndromy způsobenými autozomálními aberacemi převažují trizomie – Downův syndrom (trizomie 21. chromozomu), Edwardsův syndrom (trizomie 18. chromozomu) a Patauův syndrom (trizomie 13. chromozomu). V případě syndromů způsobených gonozomálními aberacemi jde převážně o znásobení počtu gonozomů – syndrom Klinefelterův (47,XXY), syndrom 47,XXX či 47,XYY, případně o monozomii – Turnerův syndrom (45,X). Tento výčet odpovídá klasickému pohledu na chromozomální etiologii těchto syndromů. Klinická praxe je složitější, kdy se pod příslušnými klinickými diagnózami mohou skrývat mozaikové karyotypy, parciální duplikace či delece nebo jiné strukturální abnormality zodpovědných chromozomů.
Cílem primárního screeningu je identifikace těhotenství s vyšším rizikem vrozené vady a poskytnutí cílené prenatální diagnostiky. Screeningová vyšetření sice nedokážou určit, zdali je plod skutečně postižen, ale pomohou zúžit okruh těhotných se zvýšeným rizikem. V současné době neexistuje žádný univerzální screeningový test, který by byl schopen odhalit všechny druhy možného postižení plodu. Z tohoto důvodu v praxi využíváme v prenatální péči různé screeningové strategie, které se navzájem doplňují. Indikací ke stanovení karyotypu mohou být:
- a) věk matky, anamnestické údaje z rodinné anamnézy, nález balancované chromozomální aberace aj.;
- b) pozitivita kombinovaného testu v I. trimestru (stanovení rizika chromozomální aberace na základě kombinace věku matky, hladin volné β podjednotky hCG a PAPP-A a hodnoty tzv. šíjového ztluštění u plodu v 11.–13.+6 týdnu těhotenství);
- c) pozitivita biochemického screeningu ve II. trimestru (alfafetoprotein, choriový gonadotropin, estriol);
- d) přítomnost ultrazvukových markerů chromozomálních aberací při ultrazvukovém vyšetření ve II. trimestru.
Pro diagnózu chromozomálních aberací je stále zlatým standardem cytogenetické vyšetření, tj. vyšetření karyotypu. Pro získání materiálu k vyšetření se v rámci prenatální diagnostiky stále uplatňují klasické invazivní metody – amniocentéza, odběr choriových klků a kordocentéza. V postnatální diagnostice se v drtivé většině případů jedná o odběr periferní krve. Mimo standardní cytogenetické analýzy se využívají i metody molekulárně cytogenetické (PCR, FISH). Stále větší uplatnění v prenatální diagnostice chromozomálních aberací nachází tzv. rychlá diagnostika pomocí metody kvantitativní fluorescenční PCR (QF-PCR), byť v určitých oblastech (zejména v diagnostice strukturálních abnormalit) zatím nemůže klasické vyšetření karyotypu nahradit.
MATERIÁL A METODIKA
V práci byla použita data o prenatálně a postnatálně diagnostikovaných chromozomálních aberacích v České republice v období 1994 – 2007. Data o prenatálně diagnostikovaných a pro tuto aberaci předčasně ukončených těhotenství byla získána z jednotlivých pracovišť prenatální diagnostiky a lékařské genetiky v ČR. Data o diagnózách u narozených dětí byla získána z Národního registru vrozených vad vedeného v Ústavu zdravotnických informací a statistiky České republiky. Byly analyzovány absolutní počty a stanoveny incidence za celé sledované období. Dále byla vyhodnocena úspěšnost sekundární prevence chromozomálních aberací. V naší analýze jsme sledovali incidence těchto vybraných typů chromozomálních aberací: Downův syndrom, Edwardsův syndrom, Patauův syndrom, Turnerův syndrom, syndrom 47,XXX, Klinefelterův syndrom a syndrom 47,XYY. V práci předkládáme celkové absolutní počty příslušných klinických diagnóz, bez rozlišení jednotlivých karyotypových variant.
VÝSLEDKY
Na grafu 1 je ukázán vývoj prenatální diagnostiky v České republice v období 1994 – 2007 v relativních incidencích (na 10 000 živě narozených), a to zvláště pro těhotenství předčasně ukončená po pozitivní prenatální diagnostice; zvláště pro těhotenství pokračující po pozitivní prenatální diagnostice. V grafu je současně ukázán vývoj absolutního počtu narozených dětí v České republice. Graf 2 je věnován invazivní prenatální diagnostice v České republice za období 1990 – 2007. Z grafu je patrný nárůst relativních počtů provedených výkonů invazivní prenatální diagnostiky vztažený k počtům živě narozených dětí. V roce 2007 je relativní počet výkonů oproti roku 1990 téměř 6krát vyšší. Na dalším grafu, číslo 3, je ukázáno rozdělení invazivní prenatální diagnostiky podle jednotlivých použitých metod. Převažující metodou je v průběhu celého období let 1998 – 2007 amniocentéza. Počet punkcí pupečníku pro stanovení karyotypu (cordocentesis=CC) má ve sledovaném období víceméně sestupnou tendenci, protože toto vyšetření bylo většinou nahrazeno rychlým PCR stanovením z vody plodové. Počet biopsií choria (chorion villus sampling=CVS) ze začátku sledovaného období klesá, v průběhu dalších let se však trend obrací a v posledních letech počet provedených odběrů CVS významně narůstá. Je to především díky významnějšímu zastoupení prvotrimestrálního screeningu v systému screeningových testů těhotných žen a tím i stoupající potřeby časnější prenatální diagnostiky.

Následující grafy jsou věnovány aktuálnímu výsledku prenatální diagnostiky nejčastějších chromozomálních aberací. Grafy ukazují vzájemné zastoupení těchto diagnóz: Downův syndrom, Edwardsův syndrom, Patauův syndrom, jiné autozomální odchylky a jiné gonozomální odchylky. Jsou prezentovány aktuální výsledky v roce 2007 za celou Českou republiku. Graf 4 ukazuje případy ukončených těhotenství po pozitivní prenatální diagnostice, graf 5 pak případy pokračujících gravidit po pozitivní prenatální diagnostice a graf 6 pak případy celkem. Z grafů je patrné, že zatímco v případech ukončených těhotenství významně dominují trizomie chromozomů 13, 18 a 21 – představují zhruba 70 % všech chromozomálních aberací, v případech neukončených je nejvíce zastoupená skupina „jiné chromozomální odchylky“, která představuje v těchto případech více než dvě třetiny diagnóz. V přehledu celkem (ukončené + neukončené případy těhotenství po pozitivní prenatální diagnostice) představují nejzávažnější trizomie chromozomů 13, 18 a 21 zhruba 55 % všech diagnóz, skupina jiných autozomálních aberací pak asi jednu třetinu případů a jiné gonozomální aberace pak asi 16 %.





Výsledky sledování u jednotlivých analyzovaných diagnóz jsou uvedeny na následujících grafech. Každé vadě jsou věnovány dva grafy – první (a) ukazuje absolutní počty a druhý (b) relativní incidence na 10 000 živě narozených. Údaje jsou uvedeny jak pro případy u narozených, tak pro případy prenatálně diagnostikované a pro tuto diagnózu ukončené. Doplňující údaje ke všem sledovaným chromozomálním aberacím jsou pak uvedeny v tabulce 1. První část je věnována autozomálním aberacím (Downův, Edwardsův a Patauův syndrom), druhá pak gonozomálním aberacím (Turnerův a Klinefelterův syndrom, syndrom 47,XXX a 47,XYY).
Graf 7 ukazuje absolutní počty (7a) a relativní incidence (7b) Downova syndromu – případy prenatálně a postnatálně diagnostikované. Celkem bylo zachyceno 2 244 diagnóz Downova syndromu (16,58 na 10 000 živě narozených). Průměr sekundární prevence byl v období 1994 – 2007 63,37 % (95% CI 56,65 – 70,10). Další grafy, 8a a 8b, ukazují hodnoty pro Edwardsův syndrom. V absolutních počtech bylo celkem registrováno 521 diagnóz (3,85 na 10 000 živě narozených). Průměr sekundární prevence této aberace byl 79,93 % (95% CI 75,34 – 84,53). Třetí sledovanou chromozomální aberací je Patauův syndrom. Grafy 9a ukazují absolutní počty a graf 9b relativní incidence této chromozomální aberace. Tato aberace je oproti předchozím dvěma nejméně četná, celkem bylo zachyceno 201 diagnóz (1,49 na 10 000 živě narozených). Sekundární prevence této vady byla v průměru 68,87 % (95% CI 58,83 – 78,91).



Druhou sledovanou a prezentovanou skupinou jsou vybrané gonozomální aberace. První z této skupiny – Turnerův syndrom je na grafech 10a a 10b. V absolutních počtech to bylo 380 diagnóz a v relativních pak 2,81 na 10 000 živě narozených. Sekundární prevence byla v průměru 79,89 % (95% CI 73,00 – 86,79) a v průběhu sledovaného čtrnáctiletého období se procento prenatálně diagnostikovaných a pro tuto diagnózu předčasně ukončených těhotenství nemění. Významně méně zastoupená diagnóza syndromu 47,XXX (Triple X syndrom) je pak ukázána na grafech 11a a 11b. Celkově bylo zachyceno 61 těchto diagnóz, což představuje relativní incidenci 0,45 na 10 000 živě narozených. Průměrná sekundární prevence této vady je 59,74 % (95% CI 40,21 – 79,27). Třetí sledovanou vadou v této skupině je Klinefelterův syndrom. Celkem bylo diagnostikováno v tomto období 163 případů a byla zjištěna průměrná incidence 1,20 na 10 000 živě narozených. Průměrná sekundární prevence byla v tomto případě 73,65 % (95% CI 66,63 – 80,68). Poslední sledovanou vadou je další gonozomální aberace – Syndrom 47,XYY. Z registrovaných případů vyplývá, že je z této skupiny nejméně častá – 22 případů a relativní incidence je 0,16 na 10 000 živě narozených. Sekundární prevence je v průběhu díky nízké četnosti rozkolísaná, s průměrem 54,76 % (95% CI 31,24 – 78,28).


DISKUSE
V naší práci jsme sledovali sedm vybraných diagnóz vrozených chromozomálních aberací a analyzovali jsme prenatálně a postnatálně diagnostikované případy. V případě prenatální diagnostiky jsou uvedeny pouze ty plody, kdy došlo k ukončení gravidity na základě diagnostikované vrozené vady. U postnatálně diagnostikovaných případů jsou pak uvedeny všechny narozené děti s příslušnou chromozomální aberací, ať již došlo k diagnostice této odchylky prenatálně, či postnatálně. Výsledná efektivita prenatálního záchytu odpovídá tedy sekundární prevenci a také použitým screeningovým a prenatálně diagnostickým metodám chromozomálních aberací v České republice.
V polovině 80. let minulého století byla nalezena souvislost mezi výskytem trizomie 21. chromozomu a nízkými hladinami mateřského sérového alfafetoproteinu (MS – AFP, α-fetoproteinu), zvýšenými hladinami lidského choriového gonadotropinu (hCG) a nízkými hladinami sérového nekonjugovaného estriolu (μE3). Následně bylo vypočteno, že s navrženým postupem kombinujícím mateřský věk s biochemickými hodnotami MS-AFP, hCG a μE3 se podaří místo 30 % (detection rate věkového screeningu) zachytit až 66 % všech plodů s trizomií 21.chromozomu. Při tomto postupu je u 5 – 7 % vyšetřených žen screening pozitivních (riziko vyšší než 1 z 270) a je jim doporučena invazivní prenatální diagnostika. Aplikací tohoto kombinovaného biochemického screeningu se podařilo zvýšit procento detekovaných chromozomálních aberací ve všech věkových kategoriích. Tomu odpovídají nálezy prenatálního záchytu závažných vrozených chromozomálních aberací v České republice v období let 1994 – 2004.
V devadesátých letech se začíná provádět screening kombinující věk matky a tloušťky prosáknutí záhlaví (NT – nuchal translucence) plodu v 11.–13.+6 týdnu těhotenství při ultrazvukovém vyšetření. Zjistilo se, že s využitím této metody lze identifikovat zhruba 75 % postižených plodů při falešné pozitivitě kolem 5 %. V dalších letech se kombinací věku matky, ultrazvukového stanovení NT plodu a biochemických hodnot séra matky – volné β-podjednotky hCG a těhotenského plazmatického proteinu (PAPP-A) v prvním trimestru podařilo zvýšit původní záchyt ze 75 % až na 90 – 95 % postižených plodů. Z toho pak vyplývala snaha zavést a rozšířit od roku 2003 v České republice kombinovaný screening v I. trimestru tak, aby se zvýšil záchyt závažných chromozomálních abnormalit (trizomií 13, 18, 21). V současnosti je v ČR kolem sta certifikovaných ultrasonografistů a postupně se certifikují i jednotlivé laboratoře. Stále je však v populaci nejrozšířenější tzv. biochemický screening ve II. trimestru, čemuž odpovídá také v České republice v současné době průměrný týden při diagnostice těchto výše uvedených vrozených vad, který je za posledních deset let zhruba 18. týden těhotenství a například pro Downův syndrom se v průběhu let 1994 – 2007 snížil z 19,49 (1994) na 17,59 týdne těhotenství (2007). Lze předpokládat, že v blízké budoucnosti se výrazně zvýší počet záchytů již na konci prvého trimestru.
Případy závažných trizomií u narozených dětí – Downův, Edwardsův a Patauův syndrom – jsou zřejmě diagnostikovány a následně hlášeny do centrálního registru bez většího úniku. Naproti tomu, incidence syndromů podmíněných gonozomálními aberacemi bude s nejvyšší pravděpodobností ve skutečnosti vyšší, než ukazuje naše analýza. Tato situace má několik příčin:
- Variabilita fenotypu těchto syndromů je veliká a existuje určité procento případů, kdy pro nedostatek symptomů není syndrom diagnostikován nikdy (především syndromy 47,XXX a 47,XYY).
- V České republice jsou v současné době (od roku 1994) hlášeny případy vrozených vad zjištěných do dokončeného 15. roku věku. U všech 4 sledovaných diagnóz (Turnerův syndrom, Klinefelterův syndrom, syndrom 47,XXX a syndrom 47,XYY) může být prvním zjištěným symptomem porucha fertility, která je určitě vyšetřována až po 15. roce věku. Z tohoto důvodu určitá část zmiňovaných syndromů uniká současné registraci.
- Nástup některých dalších typických symptomů uvedených syndromů je rovněž opožděn, například do doby akcelerovaného tělesného růstu, či do puberty (Turnerův syndrom, Klinefelterův syndrom). Přestože v tomto případě může být definitivní diagnóza potvrzena na základě cytogenetického vyšetření ještě do 15. roku věku, k nahlášení vady do Registru nakonec nedojde, neboť pacient často přicházejí s (nepotvrzenou) pracovní diagnózou, která může vzbudit dojem již provedeného hlášení.
ZÁVĚR
Práce předkládá aktualizované výsledky analýz absolutního počtu, incidence a procenta prenatálního záchytu sedmi vybraných chromozomálních aberací v České republice v období let 1994 – 2007.
Stanovili jsme incidence příslušných sedmi diagnóz v České republice pro celkové počty případů (prenatálně + postnatálně diagnostikované případy): Downův syndrom 16,58 na 10 000 živě narozených; Edwardsův syndrom 3,85 na 10 000 živě narozených; Patauův syndrom 1,49 na 10 000 živě narozených; Turnerův syndrom 2,81 na 10 000 živě narozených; syndrom 47,XXX 0,45 na 10 000 živě narozených; Klinefelterův syndrom 1,20 na 10 000 živě narozených a syndrom 47,XYY 0,16 na 10 000 živě narozených. Tyto nalezené incidence odpovídají v případě trizomií 13, 18 a 21, jak literárně uváděným incidencím, tak i v současnosti publikovaným incidencím z populačních přehledů mezinárodních organizací sledujících incidence vrozených vad v mezinárodním měřítku (European Surveillance of Congenital Anomalies /EUROCAT/). V případě syndromu Turnerova, syndromu Klinefelterova a syndromů 47,XXX a 47,XYY jsou námi zjištěné incidence odpovídající údajům z populačních epidemiologických studií (EUROCAT), jsou však nižší než některé literární údaje, což odpovídá pozdějšímu záchytu těchto diagnóz (nad 15 let věku dítěte).
Práce byla podpořena grantem IGA MZ ČR – NR/9005 – 3.
Na závěr by autoři rádi poděkovali všem lékařům a spolupracovníkům, kteří se účastní sběru dat nezbytných pro celorepublikovou evidenci údajů o vrozených vadách a jejich prenatální diagnostice, bez jejichž trpělivé a spolehlivé práce by nemohlo vzniknout toto zpracování.
MUDr. Vladimír Gregor
Oddělení lékařské genetiky
Fakultní Thomayerova nemocnice s poliklinikou
Vídeňská 800
140 59 Praha 4
e-mail: vladimir.gregor@ftn.cz
http://www.vrozene-vady.cz
Zdroje
1. Calda, P., et al. Ultrazvuková diagnostika v těhotenství (pro praxi). Praha: Aprofema, 2007.
2. Dolk, H., et al. EUROCAT Annual Report to WHO 2004-2005, online [http://www.eurocat.ulster.ac.uk/pdf/], staženo 26. 10. 2008.
3. Gregor, V., Šípek, A., Mašátová, D. Podíl prenatální diagnostiky na snižování výskytu vrozených vad v ČR. Čes Gynek, 2003, 68, 6, s. 395-400.
4. Gregor, V., Šípek, A., Horáček, J. Vrozené vady v České republice – prenatální diagnostika. Čes Gynek, 2007, 72, 4, s. 262-268.
5. Gregor, V., Šípek, A., Horáček, J., et al. Prenatální diagnostika vybraných typů vrozených vad v České republice v období 1994 – 2006. Čes Gynek, 2008, 73, 3, s. 169-178.
6. Gregor, V., Šípek, A., Horáček, J., et al. Přežívání dětí s vybranými vrozenými vadami v průběhu prvního roku života. Čes Gynek, 2008, 73, 3, s. 163-169.
7. Gregor, V., Šípek, A., Horáček, J., et al. Analýza incidencí vrozených vad v České republice podle četnosti těhotenství. Čes Gynek, 2008, 73, 4, s. 199-208.
8. Stockholm, K., Juul, S., Juel, K., et al. Prevalence, incidence, diagnostic delay and mortality in turner syndrome. J Clin Endocrinol Metab, 2006, 91, 10, p. 3897-3902.
9. Šípek, A. Výskyt vrozených vad u narozených ve vybraných oblastech a státech světa v období 1988 –1998. Čes Gynek, 2002, 67, 4, s. 202-209.
10. Šípek, A, Gregor, V., Horáček, J., Mašátová, D. Analýza vrozených vad podílejících se na perinatální úmrtnosti v ČR. Čes Gynek, 2003, 68, 6, s. 389-394.
11. Šípek, A., Gregor, V., Rychtaříková, J., et al. Stav dětí po asistované reprodukci v ČR za období 1995 – 1999. Čes Gynek, 2004, 69, s. 358-365.
12. Šípek, A., Gregor, V., Horáček, J., et al. Výskyt a přežívání dětí s vybranými typy vrozených vad v České republice, 1.část. Čes Gynek, 2004, 69, s. 59-65.
13. Šípek, A., Gregor, V., Horáček, J., et al. Výskyt a přežívání dětí s vybranými typy vrozených vad v České republice. 2. část. Čes Gynek, 2004, 69, s. 149-155.
14. Šípek, A, Gregor, V., Horáček, J., Mašátová, D. Podíl prenatální diagnostiky na výskytu vrozených vad v České republice v roce 2004. Čes Gynek, 2006, 71, 5, s. 373-380.
15. Šípek, A., Gregor, V., Horáček, J. Vrozené vady v České republice v období 1961 až 2005 – průměrné incidence. Čes Gynek, 2007, 72, 3, s. 185-191.
16. Šípek, A., Gregor, V., Horáček, J. Čtvrtletní incidence vybraných typů vrozených vad v České republice v období 1994-2005: dvanáctileté období. Čes Gynek, 2007, 72, 4, s. 254-261.
17. Visootsak, J., Graham, JM. Jr. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J Rare Dis, 2006, 1:42, doi: 10.1186/1750-1172-1-42.
Štítky
Dětská gynekologie Gynekologie a porodnictví Reprodukční medicínaČlánek vyšel v časopise
Česká gynekologie

2009 Číslo 1
- Tirzepatid – nová éra v léčbě nadváhy a obezity
- Obezita je nemoc, kterou je třeba diagnostikovat a léčit
- Horní limit denní dávky vitaminu D: Jaké množství je ještě bezpečné?
- Postupné vysazování inhibitorů protonové pumpy
- Moje zkušenosti s Magnosolvem podávaným pacientům jako profylaxe migrény a u pacientů s diagnostikovanou spazmofilní tetanií i při normomagnezémii - MUDr. Dana Pecharová, neurolog
Nejčtenější v tomto čísle
- Štítna žľaza v gravidite
- Spontánní ruptura symfýzy následovaná profúzním krvácením do dutiny břišní během vaginálního porodu
- Význam hysterosalpingografie v průkazu tubárního faktoru neplodnosti
- Novinky v patofyziologii a managementu předčasného porodu