
Hereditární feochromocytom a paragangliom
Hereditary Pheochromocytoma and Paraganglioma
Pheochromocytomas and paragangliomas are tumors arising from chromaffin cells. These tumors produce catecholamines and are typically found with symptoms and signs that may include hypertension (persistent or episodic), palpitations, headache and sweating. So far, 10 different genes have been associated with both tumors and other genes are expected to be detected. Pheochromocytoma and paraganglioma can occur as a part of genetic syndromes – familial paragangliomas (SDH genes, SDHAF2 gene), von Hippel-Lindau syndrome (VHL gene), multiple endocrine neoplasia type 2 (RET gene), and neurofibromatosis type 1 (NF1 gene). These tumors may be the first and only manifestation of these genetic syndromes. Patients with SDHB mutations are at high risk to develop malignant disease and unfortunately current therapeutic options for malignant form of disease are poor. Genetic testing plays a key role in the management of these tumors and therefore not only index patients with pheochromocytoma but also relatives should be tested. Management of this disease requires multidisciplinary cooperation and should be performed in the specialized medical centres.
Key words:
pheochromocytoma – paraganglioma – genetic testing – follow up
This study was supported by PRVOUK-P27/LF1/1, sVV–2012–264514 grant, by a project of the research organisation 00064203 and by IGaMZ ČR NT12336-4/2011 grant.
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE “uniform requirements” for biomedical papers.
Submitted:
30. 5. 2012
Accepted:
27. 7. 2012
Autoři:
Z. Musil 1,2; A. Vícha 2; T. Zelinka 3; H. Turková 3; T. Labudová 2; M. Kohoutová 1; K. Pacák 4
Působiště autorů:
Ústav biologie a lékařské genetiky, 1. LF UK a VFN v Praze
1; Klinika dětské hematologie a onkologie, UK 2. LF a FN Motol (KDHO), Praha
2; III. interní klinika – klinika endokrinologie a metabolismu, 1. LF UK a VFN v Praze
3; Program in Reproductive and Adult Endocrinology, Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health, Bethesda, Maryland, USA
4
Vyšlo v časopise:
Klin Onkol 2012; 25(Supplementum): 21-26
Souhrn
Feochromocytomy a paragangliomy jsou nádory vznikající z chromafinních buněk, mohou metabolizovat, skladovat, ale ne vždy vylučovat katecholaminy. Typickými projevy feochromocytomu nebo paragangliomu jsou hypertenze (trvalá i záchvatovitá), palpitace, bolesti hlavy a pocení. Se vznikem těchto nádorů je dnes spojeno deset genů a předpokládá se, že další budou objeveny. Oba typy nádorů se vyskytují také v rámci genetických syndromů: syndromu familiární paragangliomatózy (geny SDH, SDHAF2), syndromu von Hippel-Lindau (gen VHL), syndromu mnohočetné endokrinní neoplazie typu 2 (gen RET) a neurofibromatózy typu 1 (gen NF1). U některých syndromů jsou tyto nádory prvním a jediným manifestovaným onemocněním. Některé typy mutací, především v genu SDHB, jsou spojeny s vysokým počtem maligních onemocnění, která jsou v současné době standardními postupy nevyléčitelná. Z těchto důvodů je nezbytné provádět genetické vyšetření nejen u pacienta, ale v celé rodině, a nabídnout nositelům mutací dlouhodobé nebo celoživotní sledování a případně včasnou léčbu. Péče o pacienty s těmito onemocněními proto vyžaduje multidisciplinární spolupráci a měla by být prováděna pouze ve specializovaných centrech, která mají s tímto druhem onemocnění dostatečné zkušenosti.
Klíčová slova:
feochromocytom – paragangliom – genetické vyšetření – sledování
Zodpovědné geny: SDHA, SDHB, SDHC, SDHD, SDHAF2 (SDH5), VHL, NF1, RET, MAX, TMEM127
Typ dědičnosti: autozomálně dominantní
Pracoviště poskytující analýzu genů v ČR: Ústav biologie a lékařské genetiky, 1. LF UK a VFN, Albertov 4, 128 00 Praha 2
Klinika dětské hematologie a onkologie, UK 2. LF a FN Motol (KDHO), V Úvalu 84, 150 06 Praha 5
Mgr. Zdeněk Musil, musil.z@seznam.cz, mutační analýza genů SDHB, SDHD, RET, zavádíme analýzu genů SDHC a MAX
Klinika dětské hematologie a onkologie, UK 2. LF a FN Motol (KDHO), V Úvalu 84, 150 06 Praha 5
MUDr. Aleš Vícha, ales.vicha@lfmotol.cuni.cz, SNP array, CGH, FISH
Ústav biologie a lékařské genetiky, FN Motol Praha, V Úvalu 84, 150 06 Praha 5
MUDr. Anna Křepelová, CSc., anna.krepelova@fnmotol.cz, mutační analýza genu VHL
Pracoviště sloužící pro konzultaci klinických otázek: III. interní klinika, 1. LF UK a VFN, U nemocnice 1, 128 08 Praha 2
doc. MUDr. Tomáš Zelinka, CSc., Tomas.Zelinka@lf1.cuni.cz
Charakteristika onemocnění
Feochromocytomy jsou relativně vzácné tumory vycházející z chromafinních buněk a produkující katecholaminy. Incidence feochromocytomu se odhaduje v rozmezí 0,5–1 diagnostikovaný případ na 100 000 obyvatel za rok. Klasické feochromocytomy se nachází v dřeni nadledvin, extraadrenální feochromocytomy, které nazýváme paragangliomy, se mohou nacházet v oblasti břicha, pánve, hrudníku a krku. Ačkoli tyto nádory vycházejí ze stejné tkáně, mají odlišné klinické projevy, genetický základ a prognózu. Feochromocytomy se objevují přibližně u 0,5 % pacientů s hypertenzí a u 4 % pacientů s adrenálním incidentalomem. V dnešní době je asi jedna třetina feochromocytomů diagnostikována jako náhodně zjištěný tumor nadledvin [1–4].
V odborné veřejnosti byly feochromocytomy známy jako nádory 10 %. Do roku 2000 bylo 10 % těchto nádorů považováno za dědičné v rámci genetických syndromů. Výsledky současných studií ukazují přítomnost kauzálních mutací u přibližně 30–40 % feochromocytomů nebo paragangliomů. Familiární formy jsou často multifokální nebo bilaterální a objevují se v mladším věku než sporadické případy [1,3,5,6].
Klinický obraz
Nejčastějšími příznaky feochromocytomu bývá hypertenze (80–90 % případů), která může být jak setrvalá, tak i záchvatovitá, palpitace (60 %), bolesti hlavy (50 %), bledost (40 %), pocení a psychické obtíže zahrnující úzkost a paniku (35 %). Typický je také váhový úbytek [7–10].
Malignita je u feochromocytomu a paragangliomu definována jen dle výskytu vzdálených metastáz (nejčastěji lymfatické uzliny, kosti, játra, plíce). Doba přežití u pacientů s metastatickým feochromocytomem a paragangliomem je velmi variabilní – někdy je průběh velmi fulminantní a v některých případech mohou pacienti přežívat i více než 20 let [8,9].
V dnešní době však není vzácností, že feochromocytom probíhá zcela asymptomaticky, na druhé straně jeho první manifestací může být i život ohrožující komplikace, jako je arytmie, infarkt myokardu, hypertenzní krize nebo cévní mozková příhoda. Feochromocytomy jsou geneticky velmi odlišné nádory. Jejich vznik je spojen s řadou dnes známých genů a předpokládá se objevení dalších, jak tomu bylo například v loňském roce u genu MAX [11]. Feochromocytomy mohou být také součástí genetických syndromů: syndromu mnohočetné endokrinní neoplazie 2. typu (zárodečné mutace v RET proto-onkogenu), von Hippel-Lindauova syndromu (mutace v tumor supresorovém genu VHL) a také Recklinghausenovy neurofibromatózy (mutace genu NF1). V rámci těchto syndromů se jen vzácně vyskytují paragangliomy břicha nebo krku [1,6,12].
Další geny, které jsou příčinou především paragangliomů, kódují podjednotky mitochondriálního enzymu sukcinátdehydrogenázy (geny SDHA, SDHB, SDHC , SDHD a SDHAF2) a způsobují syndrom PGL1-5 [1,12]. Jednotlivé geny jsou vyšetřovány postupně podle klinického obrazu onemocnění (obr. 1). Jedná se o tumor supresorové geny a příčinou je ztráta heterozygozity. Ztráta wild type alely v tumoru společně se zárodečnou mutací znamená destabilizaci sukcinátdehydrogenázového komplexu a nefunkčnost enzymu [13,14]. Charakteristika jednotlivých genů podílejících se na vzniku feochromocytomu nebo paragangliomu je uvedena v tab. 1.

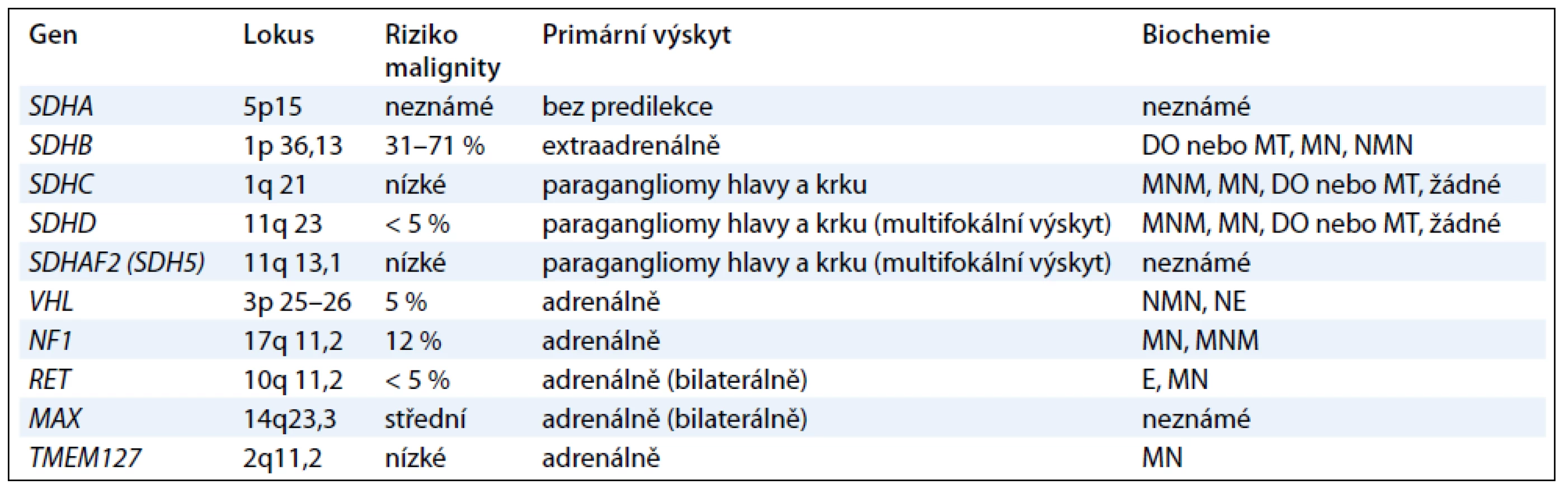
Sukcinátdehydrogenáza (též komplex II) je enzymatický komplex, který katalyzuje oxidaci sukcinátu na fumarát. Sukcinátdehydrogenáza se skládá ze čtyř různých podjednotek, SDHA je flavoprotein obsahující flavinadenindinukleotid, SDHB je FeS protein (obsahuje komplex železa a síry), SDHC a SDHD jsou hydrofobní proteiny kotvící komplex k membráně. Ačkoli jednotlivé podjednotky jsou součástí stejného proteinového komplexu, mutace v jednotlivých genech vedou k rozdílnému klinickému fenotypu [13].
Gen SDHD , syndrom paragangliomu typ 1 (PGL1); (OMIM 602690): mutace genu SDHD vedou téměř výhradně k rozvoji onemocnění, jsou-li zděděny od otce (parent-of-origin efekt) [15], vyskytují se především u krčních paragangliomů, méně často u paragangliomů hrudníku a břicha a feochromocytomů, metastatická forma onemocnění se vyskytuje zřídka [16]. Penetrance u nosičů SDHD mutace je vcelku vysoká a pohybuje se mezi 87 a 100 %. Průměrný věk pacientů s PGL1 syndromem se pohybuje mezi 20. a 40. rokem [13].
Gen SDHAF2 ( SDH5), syndrom paragangliomu typ 2 (PGL2); (OMIM 601650): gen SDH5 kóduje protein, který zajišťuje inkorporaci FAD kofaktoru SDHA podjednotky sukcinátdehydrogenázy, která je nezbytná pro správnou funkci SDH komplexu [16]. Mutace genu SDHAF2 vykazují stejně jako změny v genu SDHDparent-of-origin efekt a vedou nejčastěji k multifokálním krčním paragangliomům v mladém věku [17]. Výskyt mutací není častý a genetické vyšetření se provádí, nejsou-li nalezeny změny v genech SDHD, SDHCnebo SDHB [13].
Gen SDHC, syndrom paragangliomu typ 3 (PGL3); (OMIM 602413): mutace genu SDHC vedou nejčastěji k rozvoji solitárních krčních paragangliomů, ale vzácně též byly zaznamenány případy sympatických paragangliomů a feochromocytomů. Paragangliomy vzniklé vlivem mutací genu SDHC jsou však daleko méně časté než SDHB nebo SDHD paragangliomy a vyskytují se v méně než 1 % případů. Většinou se jedná o benigní formu onemocnění. Popsán byl pouze jeden případ malignity. Změny v tomto genu jsou vzácné a genetické testování se provádí u SDHD a SDHB negativních pacientů [12,13].
Gen SDHB, syndrom paragangliomu typ 4 (PGL4); (OMIM 185470): tento syndrom se nejčastěji projevuje přítomností sympatických paragangliomů, méně často se vyskytují parasympatické krční paragangliomy. Na rozdíl od převážně benigních SDHC a SDHD tumorů, SDHB paragangliomy mohou velmi často metastazovat a vyskytují se v mladém věku. Nedávno publikovaná data prokázala, že u dětí s metastatickým paragangliomem se mutace SDHB genu vyskytovala až v 83 % případů [18]. Mutace genu SDHB mohou predisponovat nosiče k rozvoji dalších nádorových onemocnění jako např. Carney-Stratakisova syndromu, gastrointestinálních stromálních tumorů (GIST) [19,20], renálního karcinomu různých typů, neuroblastomu a papilárnímu karcinomu štítnice [21,22].
Carney-Stratakis syndrom (OMIM 606864) je nedávno popsaný syndrom, který zahrnuje výskyt multicentrického paragangliomu a multifokálního GISTu. U pacientů s tímto syndromem se nachází alelické ztráty chromozomálních lokusů pro geny SDHB nebo SDHC. Germinální mutace v genech SDH se nachází u 15 % pacientů s GIST bez rodinného výskytu paragangliomu [23]. Obdobně ve 14 % případů pacientů s renálním karcinomem byla prokázána přítomnost germinální mutace genu SDHB.
Gen SDHA, syndrom paragangliomu typ 5 (PGL5); (OMIM 600857): zárodečné mutace tohoto genu způsobují především neurodegenerativní onemocnění, tzv. Leighnův syndrom. Výskyt mutací u sporadických feochromocytomů a paragangliomů je vzácný a objevuje se méně než ve 3 % případů [16].
Gen TMEM127 ; (OMIM 613403): tento gen kóduje vysoce konzervovaný a široce exprimovaný transmembránový protein a nachází se na cytoplazmatické membráně i v cytoplazmě. Protein má tři transmembránové domény, ale nemá zatím žádnou známou funkční doménu a hraje roli v přenosu proteinů mezi cytoplazmatickou membránou, Golgiho komplexem a lyzozomy [24]. Mutace genu TMEM127 vedou téměř výhradně k rozvoji feochromocytomů (často bilaterálních) [16], nicméně byly i zaznamenány případy extraadrenálních a krčních paragangliomů, zřídka jsou však maligní [25].
Gen MAX (MYC – associated factor X); (OMIM 154950): tento gen kóduje transkripční faktor, jako homodimer nemá transkripční doménu a působí inhibičně, naopak s c-myc onkogenem vytváří heterodimer, který se váže na DNA a je aktivační pro řadu buněčných pochodů. Zárodečné mutace v genu MAX byly nalezeny u 1 % pacientů s feochromocytomem, u nichž nebyla nalezena mutace v jiných genech [11].
Gen VHL, syndrom von Hippel-Lindau; (OMIM 608537): příčinou vzniku von Hippel-Lindauova syndromu jsou zárodečné mutace VHL tumor supresorového genu. Klinickými příznaky VHL syndromu je přítomnost hemangioblastomů retiny a CNS, světlobuněčný karcinom ledvin, feochromocytom, cysty pankreatu a ledvin, tumory saccus endolymphaticus a papilární cystadenomy. Zárodečné mutace VHL vykazují autozomálně dominantní charakter dědičnosti. U 90 % nosičů se vyvine onemocnění do 60 let věku, 3–5 % pacientů se sporadickým feochromocytomem může mít zárodečnou mutaci ve VHL genu. Feochromocytomy asociované s VHL syndromem jsou nejčastěji adrenální a bilaterální, mohou se objevit i extraadrenálně. V rámci VHLsyndromu bylo nalezeno přibližně 5 % maligních feochromocytomů [2,4,26].
Gen RET, syndrom mnohočetné endokrinní neoplazie 2. typu (MEN-2); (OMIM 171400): syndrom mnohočetné endokrinní neoplazie je podmíněn mutacemi v RET protoonkogenu. Klinickými projevy tohoto syndromu jsou medulární karcinom štítné žlázy, primární hyperparatyreóza (MEN 2A) a další znaky jako např. ganglioneuromy a marfanoidní habitus (MEN 2B). Medulární karcinom štítné žlázy mají téměř všichni pacienti MEN-2 syndromu a většinou předchází diagnóze feochromocytomu vyskytující se s penetrancí více než 90 % do 50 let věku. Feochromocytom se objevuje přibližně u 50 % pacientů s MEN-2. Protoonkogen RET kóduje transmembránový tyrosinkinázový receptor [4]. Mutace v tomto genu se nejčastěji nachází v exonu 10 a 11 u MEN 2A a v exonu 16 u MEN 2B. Paragangliom se v rámci syndromu MEN 2 vyskytuje zcela vzácně. Feochromocytomy se nachází uni- i bilaterálně [2].
Gen NF1, neurofibromatóza – typ 1 (NF-1); (OMIM 162200): neurofibromatóza – typ 1 (nazývaná též morbus von Recklinghausen či periferní typ neurofibromatózy). Jde o tumor-supresorový gen, jehož produkt (neurofibromin) je součástí intracelulární signální kaskády spojené s RAS-kinázou. Jedná se o relativně časté AD dědičné onemocnění (1 : 2 500–4 000 novorozenců). Projevuje se abnormálním růstem podpůrných buněk centrální a periferní nervové soustavy (gliomy, Schwannovy buňky aj.) s výraznou predispozicí ke vzniku benigních i maligních nádorů. Klinický průkaz neurofibromatózy 1. typu je v současné době považován u feochromocytomu za dostatečně průkazný pro zařazení pacienta do skupiny feochromocytomů asociovaných s neurofibromatózou [4].
Mezi klinické projevy neurofibromatózy patří tzv. „café-au-lait spots“ (skvrny barvy „bílé kávy“, v 90 % se objeví do pěti let věku), neurofibromy (mnohočetné tumorózní uzlíky; kutánní, subkutánní a plexiformní; hlavně v axilách a tříslech), Lischovy uzlíky (hamartomy duhovky), NF 1 je asociován s větším počtem různých neuroendokrinních tumorů včetně feochromocytomu, který ovšem není příliš častý a vyskytuje se přibližně v 0,1–5,5 % případů [2].
Biochemické vyšetření
Jak již bylo zmíněno, feochromocytomy nebo paragangliomy produkují, ale ne vždy vylučují, katecholaminy. Naopak jejich metabolity metanefriny bývají vylučovány prakticky vždy, a vykazují tedy lepší senzitivitu než mateřské katecholaminy. Dle posledních prací vykazují nejlepších výsledků plazmatické metanefriny s ohledem na lepší specificitu ve srovnání s močovými metanefriny [27]. Z tohoto důvodu je pravděpodobně nejvýhodnější používat stanovení plazmatických metanefrinů. Odpadá tak horší spolupráce pacientů při sběrech moči. Pro volbu způsobu stanovení metanefrinů je však nejdůležitější zkušenost pracoviště s danou metodou. Některé tumory (především ty méně diferencované, jako jsou paragangliomy na podkladě genu SDHB nebo paragangliomy hlavy a krku) mohou produkovat pouze dopamin nebo methoxytyramin. I přes výrazné zlepšení biochemické diagnostiky však u některých tumorů (typicky paragangliomy hlavy a krku a pak i nádorů na podkladě mutace genu SDHB) nemusíme prokázat jakékoli zvýšení metanefrinů a tyto nádory považujeme za sekrečně němé. Zde nám může pomoci i stanovení jiného markeru neuroendokrinních nádorů – chromograninu, který může být jediným zvýšeným nálezem u pacientů s jinak sekrečně němým paragangliomem na podkladě mutace genu SDHB [7–10,28].
Zobrazovací metody
K lokalizaci tumorů by se mělo přistoupit až po předchozím potvrzení biochemickými metodami, pokud nebyl nádor zjištěn dříve. Základním diagnostickým nástrojem pro nádory v oblasti břicha je v našich podmínkách CT. MR použijeme jedině při alergii na kontrastní látku nebo u dětských pacientů (pro snížení radiační zátěže). Naopak pro oblast hlavy a krku je vhodné použití MR. Pro potvrzení diagnózy (CT i MR nejsou specifické) a také k vyloučení mnohočetného (a metastatického) postižení využíváme metod nukleární medicíny – pro feochromocytomy scintigrafii s [123I]- metajodobenzylguanidinem a pro paragangliomy hlavy a krku s [111In]-octreotidem. V dnešní době se ale dostávají do popředí metody založené na positronové emisní tomografii – jednak [18F]-fluorodopa (ta je nejvýhodnější pro paragangliomy hlavy a krku) a pro pacienty s mutací genu SDHB nebo metastatickým postižením [18F]-fluorodeoxyglukóza [8–10,28].
Pooperační sledování
Všichni pacienti by měli být po operaci sledováni. Zvláštní pozornost zaslouží pacienti s největším rizikem možné recidivy nebo vzniku metastáz. Na prvním místě to jsou pacienti s mutací genu SDHB [14,29,30]. U nich je vhodné pravidelné pooperační sledování s maximálním intervalem šesti měsíců s biochemickým (stanovení metanefrinů) a případně i morfologickým (funkčním) vyšetřením. Pokud by se však jednalo o nádor sekrečně němý, tak v tomto případě je nutné se spolehnout jen na morfologické (funkční) vyšetření. Mezi rizikové z hlediska vzniku metastáz řadíme dále pacienty s funkčním paragangliomem a s objemným feochromocytomem, naopak příznivý průběh můžeme očekávat u starších nemocných s malými feochromocytomy, které produkují adrenalin [30]. Vyšší pravděpodobnost recidivy můžeme očekávat i u pacientů s feochromocytomy vzniklými na podkladě mutace genů VHL, RET či NF1, u nichž je vysoká pravděpodobnost bilaterálního postižení. I zde je vhodné pravidelné, nejlépe šestiměsíční sledování. Toto vyšetřovací schéma volíme zpočátku i u ostatních pacientů s feochromocytomy těsně po operaci s možností prodloužení na roční interval v dalším průběhu sledování. Pacienty s paragangliomem hlavy a krku bychom měli sledovat i po operaci, především ty s mutací SDHD genu, neboť je zde velké riziko vzniku dalšího nádoru. Pro všechny pacienty s těmito nádory doporučujeme sledování ve specializovaných centrech s dostatečnou zkušeností v léčbě těchto nádorů.
Preventivní sledování u zdravých nosičů patogenní mutace s vysokým rizikem feochromocytomu a paragangliomů
- Klinické vyšetření specialistou včetně kontroly krevního tlaku a případně další rutinní vyšetření, jako je stanovení krevního obrazu a základní biochemie jednou ročně.
- Zobrazovací metody: sonografie krku, břicha po dvou letech; magnetická rezonance hlavy a krku, hrudníku, břicha a malé pánve (v závislosti na druhu mutace, u SDHB nosičů zvážit celotělové vyšetření) po dvou letech; u pozitivního nálezu potvrzení PET/CT vyšetřením. Metody lze v ročním intervalu střídat.
- Plazmatické, případně močové metanefriny včetně metoxytyraminu (metoxytyramin vždy u SDHC nosičů) jednou ročně. Pokud byl jediným zvýšeným laboratorním ukazatelem u probanda chromogranin, tak i stanovení chromograninu (chromogranin je nutný u SDHB nosičů).
Návrhy preventivní péče pro osoby s von Hippel-Lindau syndromem, NF1 syndromem a MEN2 syndromem byly publikovány v Klinické onkologii 2009, Suppl 22.
Péče o pacienty s feochromocytomem a paragangliomem nebo o rizikové osoby vyžaduje multidisciplinární přístup (endokrinolog, pediatr, chirurg včetně urologa, otorinolaryngolog, cévní chirurg nebo neurochirurg, anesteziolog, klinický genetik, klinický onkolog a další). Při nízké incidenci, obtížné předoperační přípravě a možnosti maligního onemocnění je nepochybně s výhodou pro pacienta centralizace těchto pacientů včetně genetického vyšetření.
Preventivní péče o děti ve vysokém riziku by měla být prováděna ve FN Motol a FN Brno. Preventivní péče o dospělé by měla probíhat v centrech, která mají dostatečnou zkušenost s diagnostikou a léčbou těchto nádorů.
Práce byla podpořena PRVOUK-P27/LF1/1, grantu SVV–2012–264514, projektu koncepčního rozvoje výzkumné organizace 00064203 a grantu IGA MZ ČR NT12336-4/2011.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
Mgr. Zdeněk Musil
Ústav biologie a lékařské genetiky 1. LF UK a VFN v Praze
Albertov 4
128 00 Praha 2
e-mail: musil.z@seznam.cz
Obdrženo: 30. 5. 2012
Přijato: 27. 7. 2012
Zdroje
1. Boedeker CC. Paragangliomas and paraganglioma syndromes. Laryngorhinootologie 2011; 90 (Suppl 1): S56–S82.
2. Fishbein L, Nathanson KL. Pheochromocytoma and paraganglioma: understanding the complexities of the genetic background. Cancer Genet 2012; 205(1–2): 1–11.
3. Karasek D, Frysak Z, Pacak K. Genetic testing for pheochromocytoma. Curr Hypertens Rep 2010; 12(6): 456–464.
4. Opocher G, Schiavi F. Genetics of pheochromocytomas and paragangliomas. Best Pract Res Clin Endocrinol Metab 2010; 24(6): 943–956.
5. Bausch B, Malinoc A, Maruschke L et al. Genetics of pheochromocytoma. Chirurg 2012; 83(6): 511–518.
6. Neumann HP, Bausch B, McWhinney SR et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346(19): 1459–1466.
7. Jafri M, Maher ER. The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol 2012; 166(2): 151–158.
8. Widimsky J Jr, Zelinka T, Petrak O et al. Pheochromocytoma: diagnosis and treatment. Cas Lek Cesk 2009; 148(8): 365–369.
9. Widimsky J Jr, Zelinka T, Petrak O et al. Diagnostic and therapeutic procedures in pheochromocytoma: current trends. Vnitr Lek 2007; 53(4): 428–433.
10. Zelinka T, Eisenhofer G, Pacak K. Pheochromocytoma as a catecholamine producing tumor: implications for clinical practice. Stress 2007; 10(2): 195–203.
11. Burnichon N, Cascon A, Schiavi F et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res 2012; 18(10): 2828–2837.
12. Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta 2011; 1807(11): 1432–1443.
13. Hensen EF, Bayley JP. Recent advances in the genetics of SDH-related paraganglioma and pheochromocytoma. Fam Cancer 2011; 10(2): 355–363.
14. Musil Z, Puchmajerova A, Krepelova A et al. Paraganglioma in a 13-year-old girl: a novel SDHB gene mutation in the family? Cancer Genet Cytogenet 2010; 197(2): 189–192.
15. Baysal BE, McKay SE, Kim YJ et al. Genomic imprinting at a boundary element flanking the SDHD locus. Hum Mol Genet 2011; 20(22): 4452–4461.
16. Opocher G, Schiavi F. Functional consequences of succinate dehydrogenase mutations. Endocr Pract 2011; 17 (Suppl 3): 64–71.
17. Kunst HP, Rutten MH, de Monnink JP et al. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res 2011; 17(2): 247–254.
18. King KS, Prodanov T, Kantorovich V et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol 2011; 29(31): 4137–4142.
19. Celestino R, Lima J, Faustino A et al. A novel germline SDHB mutation in a gastrointestinal stromal tumor patient without bona fide features of the Carney-Stratakis dyad. Fam Cancer 2011; 11(2): 189–194.
20. Miettinen M, Wang ZF, Sarlomo-Rikala M et al. Succinate dehydrogenase-deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol 2011; 35(11): 1712–1721.
21. Raygada M, Pasini B and Stratakis CA. Hereditary paragangliomas. Adv Otorhinolaryngol 2011; 70: 99–106.
22. Ricketts CJ, Forman JR, Rattenberry E et al. Tumor risks and genotype-phenotype-proteotype analysis in 358 patients with germline mutations in SDHB and SDHD. Hum Mutat 2010; 31(1): 41–51.
23. Pantaleo MA, Astolfi A, Indio V et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 2011; 103(12): 983–987.
24. Jiang S, Dahia PL. Minireview: the busy road to pheochromocytomas and paragangliomas has a new member, TMEM127. Endocrinology 2011; 152(6): 2133–2140.
25. Neumann HP, Sullivan M, Winter A et al. Germline mutations of the TMEM127 gene in patients with paraganglioma of head and neck and extraadrenal abdominal sites. J Clin Endocrinol Metab 2011; 96(8): E1279–E1282.
26. Eisenhofer G, Vocke CD, Elkahloun A et al. Genetic Screening for von Hippel-Lindau Gene Mutations in Non-syndromic Pheochromocytoma: Low Prevalence and False-positives or Misdiagnosis Indicate a Need for Caution. Horm Metab Res 2012; 44(5): 343–348.
27. Lenders JW, Pacak K, Walther MM et al. Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002; 287(11): 1427–1434.
28. Timmers HJ, Gimenez-Roqueplo AP, Mannelli M et al. Clinical aspects of SDHx-related pheochromocytoma and paraganglioma. Endocr Relat Cancer 2009; 16(2): 391–400.
29. Vicha A, Holzerova M, Krepelova A et al. Molecular cytogenetic characterization in four pediatric pheochromocytomas and paragangliomas. Pathol Oncol Res 2011; 17(4): 801–208.
30. Zelinka T, Musil Z, Duskova J et al. Metastatic pheochromocytoma: does the size and age matter? Eur J Clin Invest 2011; 41(10): 1121–1128.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2012 Číslo Supplementum
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- Specifika v komunikaci s pacienty s ránou – laická doporučení
- MUDr. Lenka Klimešová: Multioborová vizita může být klíčem k efektivnější perioperační léčbě chronické bolesti
Nejčtenější v tomto čísle
- Syndrom Birt-Hogg-Dubé
- Klinický význam analýz genů středního rizika pro hodnocení rizika vzniku karcinomu prsu a dalších nádorů v České republice
- Hereditární difuzní karcinom žaludku
- Klinické dysmorfické syndrómy s tumorigenézou