
Vzácný případ DiGeorgeova syndromu s anomáliemi končetin: přínos vyšetření metodou SNP microarrayí?
Rare case of patient with DiGeorge syndrome and limbs anomalies: the benefit of SNP microarray analysis?
DiGeorge syndrome (velocardiofacial syndrome, VCFS, 22q11DS) is caused by a microdeletion of approximately 40 genes from one copy of chromosome 22. Expression of the syndrome is a variable combination of more than 190 phenotypic characteristics, the most common are congenital heart disease, cleft palate and velopharyngeal insufficiency. Limb anomalies of hands and feets are not a typical clinical symptoms of DiGeorge syndrome.
The aim of the study was to evaluate the extent of genomic changes in a 4.5 years old child with classical phenotypic features of 22q11DS in combination with severe ectrodactyly of all four limbs in addition; using microarray based single nucleotide polymorphism techniques in order to explain the origin of limb anomaly defects. FISH analysis, TP63 gene sequencing and whole genome SNP array analysis using HumanCytoSNP-12 DNA Analysis Beadchip (Illumina Inc.) were performed. FISH method revealed DiGeorge syndrome, TP63 gene sequencing did not detect any deleterious mutation, microarray SNP analysis established microdeletion of 22q11.2 region in standard defined range of 2,9 Mb. The original possible hypothesis, that in a patient it could exist a much more genomic changes than only these related to 22q11.2 region, involving genes associated with limb development and formation and thus explaining devastating limbs damage in patient, unfortunately was not confirmed. Although limb defects do not belong to the typical pathogenic signs of DiGeorge syndrome, this diagnosis should be considered in all neonate with congenital heart defect, cleft palate and limb anomalies. Even though variable phenotypical expressivity in DiGeorge syndrome is frequent, we can not exclude the possibility of coincidence of two independent genetic defects in our patient; DiGeorge syndrome due to 22q11.2 microdeletion and ectrodactyly which results of another gene/s mutation.
Key words:
DiGeorge syndrome, 22q11.2 deletion syndrome, VCFS, SNP microarray, ectrodactyly, syndactyly
Autoři:
A. Hladíková 1; P. Plevová 1; A. Balcar 1; D. Černá 1; P. Tvrdá 1; E. Šilhánová 1; D. Grečmalová 1; A. Křepelová 2
Působiště autorů:
Oddělení lékařské genetiky FN, Ostrava
primářka MUDr. E. Šilhánová
1; Ústav biologie a lékařské genetiky 2. LF UK a FN Motol, Praha
přednosta prof. MUDr. M. Macek Jr., DrSc.
2
Vyšlo v časopise:
Čes-slov Pediat 2015; 70 (5): 293-301.
Kategorie:
Kazuistika
Souhrn
DiGeorgeův syndrom (velokardiofaciální syndrom, VCFS, 22q11DS) je způsobený mikrodelecemi přibližně 40 genů na jedné kopii chromosomu 22. Projevem syndromu je variabilní kombinace více než 190 fenotypových příznaků, mezi nejčastější patří vrozené vady srdce, orofaciální rozštěp a velofaryngeální insuficience. Končetinové defekty v oblasti rukou a nohou nepatří do klasického klinického obrazu DiGeorgeova syndromu.
Cílem práce bylo u pacienta s typickým fenotypovým projevem 22q11DS a současně s raritním nálezem těžké ektrodaktylie na všech čtyřech končetinách zhodnotit rozsah genomické odchylky metodou celogenomových SNP arrayí za účelem vysvětlení končetinové vady. Bylo provedeno vyšetření metodou FISH, sekvenace genu TP63 a celogenomová SNP array pomocí kitu HumanCytoSNP-12 DNA Analysis Beadchip (Illumina Inc.). Metodou FISH byl potvrzen DiGeorgeův syndrom, sekvenace genu TP63 neodhalila patogenní mutaci, vyšetření metodou SNP arrayí potvrdilo deleci v oblasti 22q11.2 obvyklého rozsahu 2,9 Mb. Nebyla potvrzena původní hypotéza, že by se u pacienta mohlo jednat o deleci 22q11.2 většího rozsahu, než je obvyklé, která by zahrnovala gen asociovaný s vývojem končetin a vysvětlila těžký končetinový defekt. Ačkoli končetinové defekty nepředstavují typickou patologii v rámci DiGeorgeova syndromu, je nutno myslet na tuto diagnózu u každého novorozence s vrozenou srdeční vadou, rozštěpem obličeje a anomáliemi končetin. Přestože je u DiGeorgeova syndromu běžná značná variabilní expresivita, nelze u našeho pacienta vyloučit také náhodnou koincidenci dvou nezávislých genetických vad, DiGeorgeova syndromu v důsledku mikrodelece 22q11.2 a ektrodaktylie způsobené mutací jiného/jiných genů.
Klíčová slova:
DiGeorgeův syndrom, 22q11.2 deleční syndrom, VCFS, SNP microarray, ektrodaktylie, syndaktylie
Úvod
DiGeorgeův syndrom (velokardiofaciální syndrom, VCFS, 22q11 deleční syndrom – 22q11DS, MIM #192430) je nejčastěji se vyskytujícím mikrodelečním syndromem u člověka s prevalencí přibližně 1 : 2000 [1, 2], podle jiných autorů 1 : 4000 [3] až 1 : 8224 [4]. Syndrom je zapříčiněný delecí jedné kopie dlouhého ramene 22. chromosomu v oblasti q11.2 [5, 6]. Ztráta přibližně 3 milionů nukleotidových bází zahrnuje přibližně 40 genů [7]. Delece vzniká v meióze jako důsledek nealelické homologní rekombinace mezi segmentálními duplikacemi, které oblast ohraničují [8, 9]. Pacienti s genomickou delecí v oblasti 22q11 mají podobný fenotypový obraz, i když množství chybějících genů je odlišné [10]. DiGeorgeův syndrom vykazuje velmi variabilní klinické příznaky, od mírných forem až po velmi závažné postižení. Nejčastěji se vyskytují vrozené vývojové vady srdce, anomálie v orofaciální oblasti včetně rozštěpu rtu a/nebo patra, imunologické abnormality včetně aplazie nebo hypoplazie thymu a hypokalcémie. Klinický obraz je však mnohem variabilnější. V literatuře je uváděno více než 190 symptomů asociovaných s tímto syndromem [7, 11]. Podrobný přehled těchto symptomů je uveden v tabulce 1.
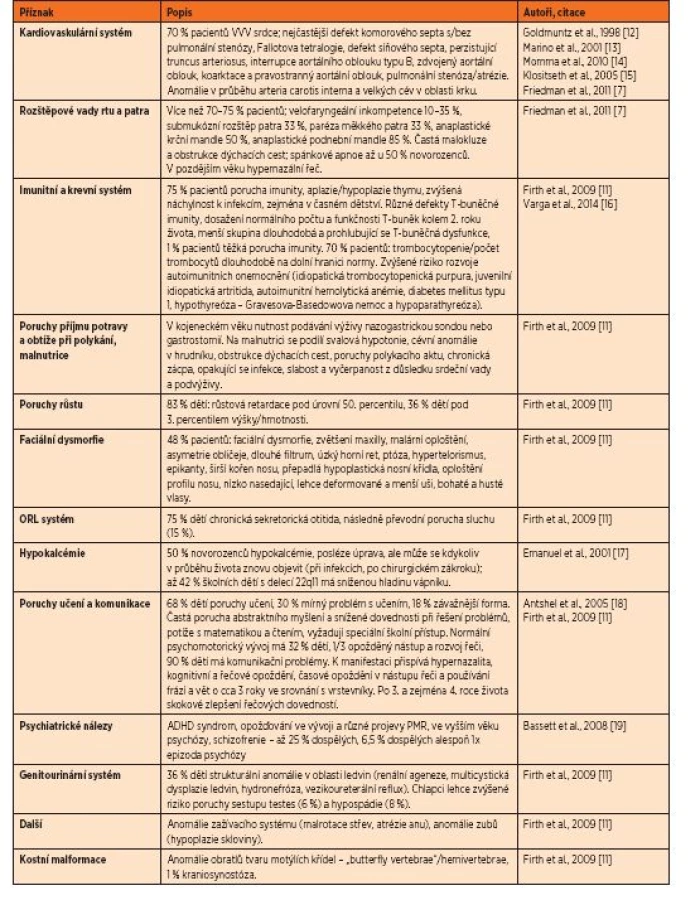
Dědičnost onemocnění je autozomálně dominantního typu s 50% rizikem pro potomstvo bez rozdílu pohlaví. U více než 93 % pacientů vzniká fenotypové postižení v důsledku de novo vzniklé delece oblasti 22q11, jen 7 % pacientů dědí stejnou deleci od některého z rodičů [20]. Riziko opakování u dalšího dítěte stejného rodičovského páru za předpokladu, že rodiče jsou zdraví a nenesou mikrodeleční syndrom 22q11.2, je relativně nízké, pod 1 %; v literatuře jsou popsány pouze dva případy, kdy došlo k opakování DiGeorgeova syndromu u dalšího dítěte, vysvětlením je gonadální mozaicismus [11].
Autoři prezentují kazuistiku pacienta s typickým fenotypovým projevem DiGeorgeova syndromu a současně s raritním nálezem těžké ektrodaktylie všech čtyřech končetin. Cílem sdělení je pokusit se objasnit genetickou příčinu závažné končetinové vady netypické pro DiGeorgeův syndrom.
Kazuistika
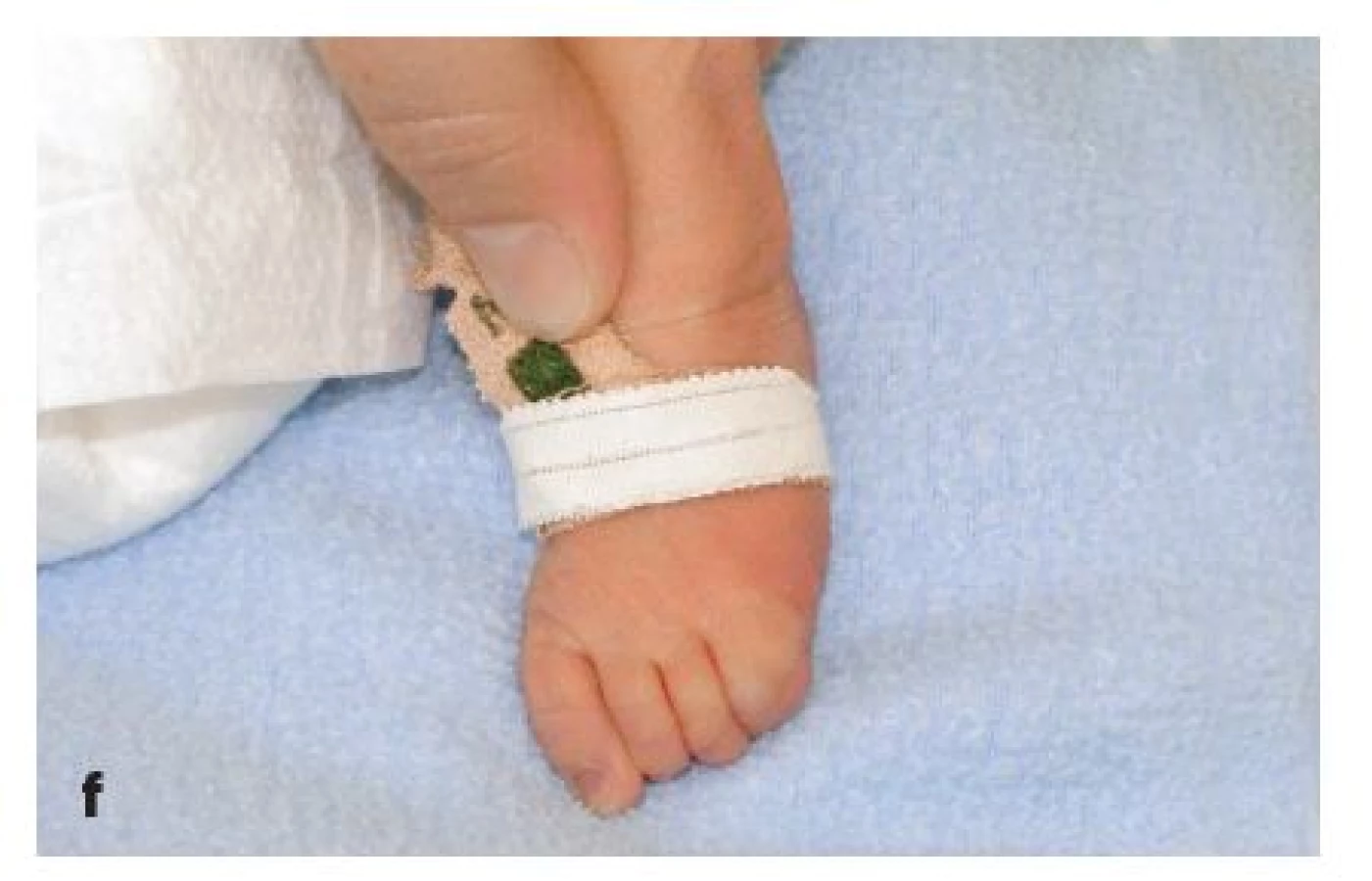
Jedná se o chlapce z 2. těhotenství nepříbuzných zdravých rodičů. Již prenatálně byla pomocí ultrazvukového vyšetření plodu diagnostikována rozštěpová vada obličeje a anomálie prstů končetin, z plodové vody odebrané amniocentézou byly vyloučeny nejčastější aneuploidie. Dítě se narodilo překotně záhlavím ve 37. gestačním týdnu, několik minut po odtoku zkalené plodové vody. Apgar skóre dosáhlo 6–8–9 bodů, porodní hmotnost byla 2220 g a porodní délka 46 cm. Dítě bylo umístěno na novorozeneckou stanici intenzivní péče, do 5. dne vyžadovalo oxygenoterapii. Při echokardiografickém vyšetření srdce byla zjištěna vrozená srdeční vada – subaortální defekt komorového septa; ultrazvukové vyšetření CNS a břicha ukázalo fyziologické nálezy. Při klinickém vyšetření genetikem bylo dítě hodnoceno jako hypotrofický lehce nezralý novorozenec, v obličeji s oboustranným kompletním rozštěpem rtu a patra (obr. 1a, 1d), faciálně širokým kořenem nosu, hypertelorismem, mírně dysplastickými, níže uloženými ušními boltci. Na končetinách byly patrné rozsáhlé defekty v oblasti obou rukou s výraznou stranovou odlišností. Pravá ruka měla charakter klepeta, 1. a 2. prst byly syndaktylické, 3. prst měl pouze jeden článek, který byl srostlý se 4. a 5. prstem (obr. 1b). Levá horní končetina měla normální palec a 1. prst s volným kloubem, 3. prst byl rudimentární, rozštěpený, částečně srostlý s 2. prstem a částečně se 4. prstem, 5. prst byl normální (obr. 1c). Na obou nohou byl rovněž asymetrický nález: pravá noha měla palec a 1. článek 2. prstu v syndaktylii, 3. prst byl hypoplastický a byla přítomna syndaktylie 1. a 2. článku 3. a 4. prstu, 5. prst byl normálně utvářený (obr. 1e). Na levé noze byla přítomna syndaktylie 4. a 5. prstu, 3. prst byl vyhnutý zevně, 1. a 2. prst byly normálně utvářené (obr. 1f).
Z klinického hlediska byl chlapec multioborově sledován, opakovaně byla provedena korekce a plastické řešení rozštěpové vady obličeje (obr. 1g) a rozrušení syndaktylií na obou rukou (obr. 1h).





V dětském kardiocentru FN Motol byla nutná operace defektu komorového septa, chlapec je i po plastice velké skrotální hernie. Během kojeneckého období byla výživa vzhledem k těžké malnutrici dítěte podávána cestou perkutánní endoskopické gastrostomie (PEG). S pomocí ga-stroenterologa a nutričního terapeuta bylo dosaženo adekvátních váhových přírůstků a dítě bylo ve věku 1,5 roku převedeno na běžnou stravu. Během prvních 4 let života byl komplikovaný vývoj řeči, stav vyžadoval otorinolaryngologickou a logopedickou péči. Zejména kvůli deformitám nohou stav vyžadoval intenzivní rehabilitaci a nácvik chůzového mechanismu. Z neurologického hlediska dominovala konstituční hypotonie s lehkou kyfózou, psychicky byl v útlém věku pacient hodnocen na úrovni lehké až středně těžké mentální retardace vzhledem k vývojové dysfázii, opožděnému vývoji řeči a omezené jemné motorice a dynamické praxe rukou; v následujícím období díky intenzivní rehabilitaci a individuální foniatricko-logopedické péči se vývoj upravil a prokázal se chlapcův normální intelekt. Vzhledem k opakovaným infekcím dýchacího traktu a recidivujícím otitidám bylo nutno opakované podávání antibiotik a imunologická léčba. Ve věku 4,5 let je chlapec plně soběstačný, s normálním stereotypem chůze, mluví srozumitelně s bohatou slovní zásobou, má adekvátní úchop rukou, kreslí bez problémů, sociálně je bez omezení oproti zdravým vrstevníkům, navštěvuje předškolní zařízení a připravuje se na běžnou povinnou školní docházku. Časná operace orofaciálního rozštěpu a pečlivá multioborová péče měla zásadní psychosociální vliv na pacienta i jeho rodinu [21].



Metodika a výsledky
Stanovení karyotypu bylo provedeno standardním postupem z kultivovaných lymfocytů periferní krve.
Bylo provedeno vyšetření na přítomnost mikrodelece oblasti 22q11.2 z kultivovaných lymfocytů periferní krve metodou fluorescenční in situ hybridizace (FISH) pomocí kitu VCFS TUPLE 1 (Cytocell Ltd., Cambridge, UK) standardním postupem podle návodu výrobce.
Izolace DNA z periferní krve byla provedena na přístroji MagNA Pure Compact Instrument (Roche, Basilej, Švýcarsko). Byla provedena sekvenace exonů 5, 6, 7, 8, 13, a 14 genu TP63 pomocí kitu BigDye Terminator v3.1 Cycle Sequencing Kit na genetickém analyzátoru ABI3130 podle návodu výrobce. Celogenomová SNP array byla provedena pomocí kitu HumanCytoSNP-12 v2.1 DNA Analysis Beadchip (Illumina Inc., San Diego, CA, USA) podle návodu výrobce. Získaná data byla zpracována pomocí softwaru BlueFuse Multi v4.1 (Illumina Inc., San Diego, CA, USA).
Studie byla schválena etickou komisí Fakultní nemocnice Ostrava, vyšetření dítěte proběhlo s informovaným souhlasem jeho rodičů.
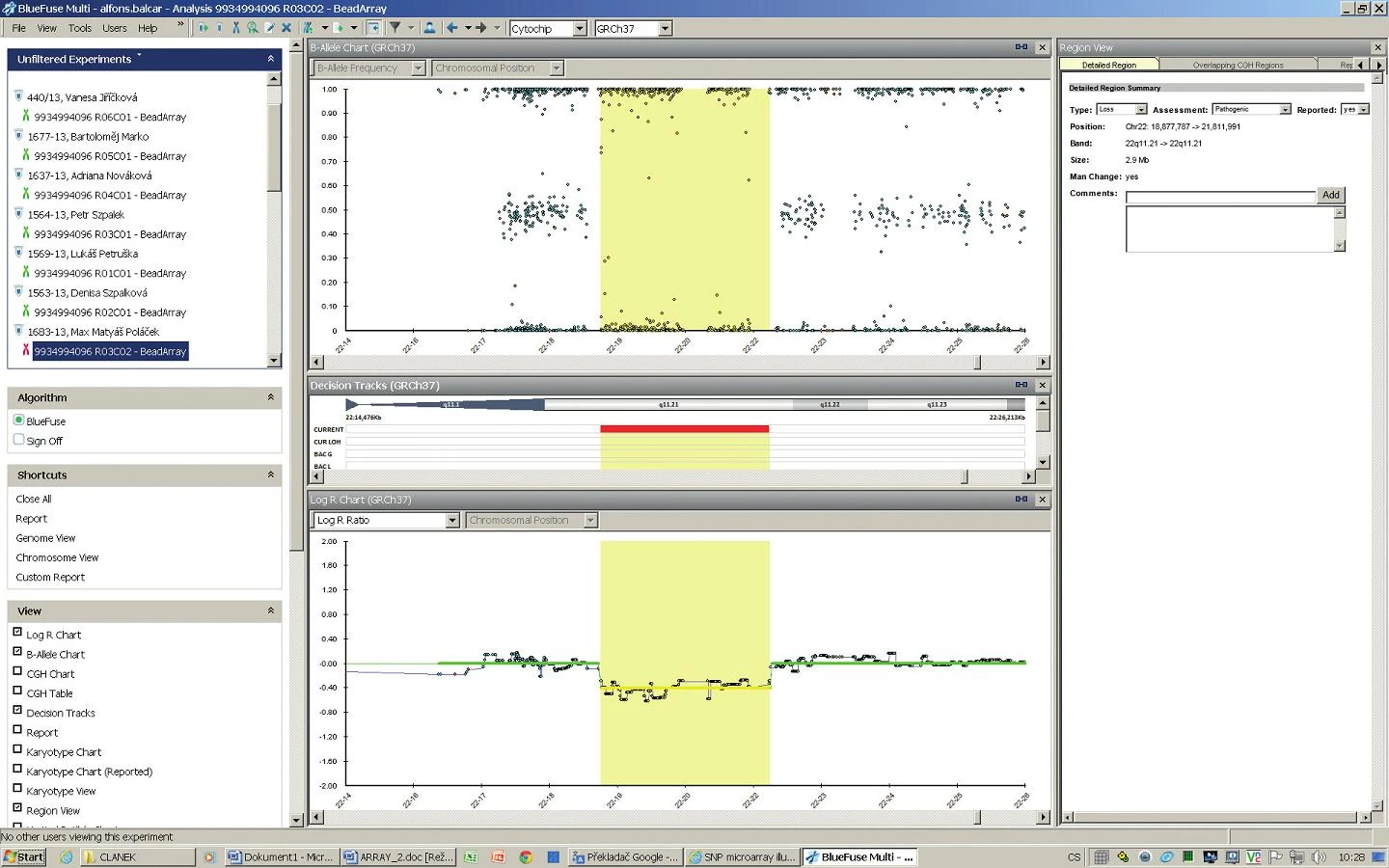
Klasické cytogenetické vyšetření prokázalo normální mužský karyotyp, 46,XY. Výsledek vyšetření metodou FISH: ish22q11.2(TUPLE1x1). Tento nález potvrzuje DiGeorgeův syndrom (obr. 2).

Sekvenace genu TP63 neodhalila patogenní mutaci ve vyšetřených oblastech.
Vyšetření metodou SNP arrayí potvrdilo deleci v oblasti 22q11.2 v rozsahu 2,9 Mb (obr. 3).
Diskuse
U pacienta s vrozeným rozštěpem rtu a patra, subaortálním defektem komorového septa a závažnými vrozenými amonáliemi končetin byl zjištěn normální mužský karyotyp. Diferenciálně diagnosticky byl zvažován syndrom EEC („ectrodactyly, ectodermal dysplasia, cleft lip/palate“) či jen ECP syndrom, u kterého není přítomna ektodermální dysplazie. Tyto syndromy mohou být spojeny s mutacemi v genu TP63, proto bylo provedeno molekulárně genetické vyšetření tohoto genu. Ve vyšetřených oblastech genu nebyla nalezena patogenní mutace, která by tento syndrom potvrdila. Pro vrozený orofaciální rozštěp a subaortální defekt komorového septa bylo provedeno vyšetření pomocí metody FISH a byla prokázána mikrodelece v oblasti 22q11.2, čímž byla potvrzena diagnóza DiGeorgeova syndromu.
Končetinové anomálie u DiGeorgeova syndromu jsou popisovány vzácně [22–29]. U pacienta s typickým fenotypovým projevem 22q11DS a současně s raritním nálezem těžké ektrodaktylie na všech čtyřech končetinách jsme předpokládali, že by rozsah delece oblasti 22q11 mohl být větší, než je obvyklé, a že by mohl zahrnovat gen, který je důležitý pro rozvoj distálních oblastí končetin. Z tohoto důvodu jsme u pacienta indikovali vyšetření metodou celogenomových SNP arrayí. Bylo však zjištěno, že delece je obvyklého rozsahu 2,9 Mb. Vyšetření tedy nepřineslo objasnění přítomnosti závažné končetinové vady u pacienta.
Přestože je u DiGeorgeova syndromu běžná značná variabilní expresivita a končetinové defekty byly v souvislosti s tímto syndromem popsány na úrovni kazuistických sdělení, nelze u našeho pacienta vyloučit také náhodnou koincidenci dvou nezávislých genetických vad, tj. DiGeorgeova syndromu v důsledku mikrodelece 22q11.2 a ektrodaktylie způsobené mutací v oblasti genu TP63, která nebyla vyšetřována, nebo mutací jiného/jiných genů. Ačkoli končetinové defekty nepředstavují typickou patologii v rámci DiGeorgeova syndromu, je nutno myslet na tuto diagnózu u každého novorozence s vrozenou srdeční vadou, rozštěpem obličeje a anomáliemi končetin.
Končetinové anomálie u DiGeorgeova syndromu jsou velmi vzácné, v literatuře jsou publikovány formou kazuistických sdělení. Velmi raritně byla popsána u pacientů s delecí 22q11 kamptodaktylie. Jedná se o flekční deformitu proximálních interfalangeálních kloubů, nejvíce bývá postižený 5. prst, méně časté je postižení všech prstů včetně palce. Kamptodaktylie bývá součástí mnoha syndromů, ale rovněž se může jednat o samostatnou nozologickou jednotku s autozomálně dominantním typem dědičnosti [28, 29]. Přehled dosud publikovaných prací věnovaných končetinovým anomáliím u pacientů s DiGeorgeovým syndromem je uveden v tabulce 2.

Široké spektrum malformací u DiGeorgeova syndromu souvisí s abnormálním vývojem neurální lišty a z ní vyplývajících orgánových patologií na úrovni III. a IV. fa-ryngeálního oblouku [30]. Tento abnormální vývoj vysvětluje aplazii (hypoplazii) thymu, příštítných tělísek a konotrunkálních defektů. Menší anomálie v oblasti obličeje jsou následkem poruchy vývoje v oblasti prvního branchiálního oblouku [31]. Přesný mechanismus vzniku malformací na embryonální úrovni však zatím není plně objasněn. Z dosud publikovaných prací týkajících se končetinových anomálií u pacientů s DiGeorgeovým syndromem vyplývá, že zatím nebyla zjištěna kauzální souvislost mezi delecí v oblasti 22q11 a klinickým nálezem končetinových anomálií u tohoto syndromu, nepodařilo se prokázat existenci genu ležícího v deletované oblasti, jehož ztráta by byla příčinou abnormálního vývoje končetin [25]. Hypoteticky je zvažována možná úloha několika pseudogenů ležících v oblasti 22q11, které pravděpodobně podléhají transkripci [32]. Také se uvažuje o roli pseudogenu SHFM3P1 v souvislosti s rozštěpovými anomáliemi ruky a nohy [25].
Vyšetření pomocí celogenomových arrayí se indikuje za účelem screeningu a upřesnění rozsahu submikroskopických chromosomálních přestaveb [33]. V literatuře byla použita metodika array CGH za účelem detekce defektů celého genomu u dvou pacientů s 22q11DS poprvé v r. 2007 [34] a 2008 [35], se závěrem, že nebyly zjištěny žádné další změny kromě delece inkriminované oblasti 22q11. U dalších 12 dětí s DiGeorgeovým syndromem [20] se rovněž neprokázala žádná další přidružená chromosomální dysbalance kromě změn v oblasti 22q11.
Pacienti s DiGeorgeovým syndromem představují 1 % pacientů se schizofrenií [19]. S cílem objasnit příčinu variabilní klinické exprese syndromu se zaměřením na riziko vzniku schizofrenie u pacientů s tímto syndromem byla zjišťována metodou celogenomových arrayí variabilita počtu kopií určitých sekvencí genomu (“copy number variations”, CNVs) a dále od kterého rodiče je chromosomový materiál v oblasti 22q11.2 deletován. Do studie bylo zařazeno 100 dospělých s DiGeorgeovým syndromem (z nich 44 mělo prokázanou schizofrenii) a kontrolní soubor. U pacientů se schizofrenií nebyly zjištěny oproti kontrolám nově vzniklé CNV ani abnormality ve výskytu CNV mimo oblast 22q11.2. Vliv neměl ani rodičovský původ delece, rozsah delece, věk rodičů nebo rodinná anamnéza výskytu schizofrenie. Za hlavní rizikový faktor schizofrenie je u pacientů s DiGeorgeovým syndromem považována hemizygozita pro CNV v oblasti 22q11.2 v důsledku delece. Zajímavým nálezem byl relativně malý počet mužů s nově vzniklou delecí 22q11.2 otcovského původu [19].
Jiní autoři se věnovali kazuistice chlapce s potvrzeným 22q11.2 delečním syndromem v kombinaci se vzácným nálezem těžké kraniosynostózy, artrogrypózy a kamptodaktylie. Indikovali metodiku SNP microarray (Affymetrix SNP 6,0), která potvrdila deleci o rozsahu 2,68 Mb v oblasti 22q11.21, ale překvapivě neodhalila žádnou jinou další abnormalitu [26], obdobně jako v našem případě.
Ektrodaktylie (ruka s rozštěpem, klepeto) představuje deficit nebo chybění jednoho nebo více centrálních prstů ruky nebo nohy a je známa též pod názvem „split hand/split foot malformation“ (SHFM) [36, 37]. Často se vyskytuje v kombinaci s jinými vrozenými anomáliemi [38]. Databáze the London Dysmorphology uvádí více než 50 syndromů s výskytem této abnormity. Příkladem lze uvést syndrom EEC (Ectrodactyly-Ectodermal Dysplasia--Clefting) zahrnující ektrodaktylii, ektodermální dysplazii a rozštěp rtu/patra, který se překrývá se syndromy ADULT s výskytem abnormit rukou a nohou a ektodermální dysplazií a LMS s abnormitami rukou a nohou a hypoplazií nebo aplazií mléčné žlázy a bradavky, ECP syndrom s ektrodaktylií a rozštěpem rtu/patra, syndrom ektrodaktylie – ektodermální dysplazie a dystrofie sítnice, syndrom EFA s ektrodaktylií a aplazií nebo hypoplazií fibuly a syndrom ektrodaktylie a polydaktylie [39].
Výsledky dosavadních vědeckých studií včetně naší ukazují, že metodika array (SNP, CGH) je příliš nákladná a nemá žádný praktický klinický přínos pro pacienty s již prokázanou diagnózou 22q11DS pomocí cytogenetických metod a FISH [20, 40].
Výskyt končetinových anomálií u našeho pacienta s DiGeorgeovým syndromem rozšiřuje klinické spektrum syndromu a ukazuje na nutnost pečlivé diferenciálně diagnostické rozvahy.
Závěr
I když končetinové defekty představují vzácně se vyskytující patologii v rámci DiGeorgeova syndromu, je nutno myslet na tuto diagnózu u každého novorozence s orofaciálním rozštěpem, konotrunkálním defektem a anomáliemi článků končetin. Metodika SNP microarrayí nepřinesla vysvětlení koincidence těžkých defektů v oblasti končetin u chlapce s DiGeorgeovým syndromem. V dalším období bude zapotřebí dalšího podrobného výzkumu za účelem objasnění konkrétního kauzálního mechanismu vzniku kombinovaných vzácných vad u obdobných pacientů, jaký je prezentován v uvedeném kazuistickém sdělení.
Poděkování:
Tato práce vznikla za veřejné finanční podpory dotačního programu „Podpora vědy a výzkumu v Moravskoslezském kraji 2013“, projekt č. 02486 2013 RRC.
Došlo: 3. 3. 2015
Přijato: 23. 6. 2015
MUDr. Andrea Hladíková, Ph.D.
Oddělení lékařské genetiky
FN Ostrava
17. listopadu 1790
708 52 Ostrava-Poruba
e-mail: andrea.hladikova@fno.cz
Zdroje
1. Shprintzen RJ, Higgins AM, Antshel K, et al. Velo-cardio-facial syndrome. Curr Opin Pediatr 2005; 17 (6): 725–730.
2. Robin NH, Shprintzen RJ. Defining the clinical spectrum of deletion 22q11.2. J Pediatr 2005; 147 (1): 90–96.
3. Schinke M, Izumo S. Deconstructing DiGeorge syndrome. Nat Genet 2001; 7 (3): 238–240.
4. Óskarsdóttir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11 deletion syndrome: a population based study in Western Sweden. Arch Dis Child 2004; 89: 148–151.
5. Morrow B, Goldberg R, Carlson C, et al. Molecular definition of the 22q11 deletions in velo-cardiofacial syndrome. Am J Hum Genet 1995; 56 (6): 1391–1403.
6. Carlson C, Sirotkin H, Pandita R, et al. Molecular definition of 22q11 deletions in 151 velo-cardiofacial syndrome patients. Am J Hum Genet 1997; 61 (3): 620–629.
7. Friedman MA, Miletta N, Roe CH, et al. Cleft palate, retrognathia and congenital heart disease in velo-cardio-facial syndrome: a phenotype correlation study. Int J Pediatr Otorhinolaryngol 2011; 75 (9): 1167–1172.
8. Edelmann L, Pandita RK, Morrow BE. Low-copy repeats mediate the common 3-mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet 1999; 64 (4): 1076–1086.
9. Shaikh TH, Kurahashi H, Saitta SC, et al. Chromosome 22 – specific low copy repeats and the 22q11.2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum Mol Genet 2000; 9 (4): 489–501.
10. Maynard TM, Haskell GT, Lieberman JA, et al. 22q11 DS: genomic mechanisms and gene function in DiGeorge/velocardiofacial syndrome. Int J Dev Neurosci 2002; 20: 407–419.
11. Firth HV, Hurst JA, Hall JG, et al. Velocardiofacial syndrome 22q11del syndrome. Oxford desk Reference: Clinical Genetics. Oxford, UK: Oxford University Press, 2009: 490–492.
12. Goldmuntz E, Clark BJ, Mitchell LE, et al. Frequency of 22q11 deletions in patients with conotruncal defects. J Am Coll Cardiol 1998; 32 (2): 492–498.
13. Marino B, Digilio MC, Toscano A, et al. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med 2001; 3 (1): 45–48.
14. Momma K. Cardiovascular anomalies associated with chromosome 22q11.2 deletion syndrome. Am J Cardiol 2010; 105 (11): 1617–1624.
15. Khositseth A, Tocharoentanaphol C, Khowsathit P, et al. Chromosome 22q11 deletions in patients with conotruncal heart defects. Pediatr Cardiol 2005; 26 (5): 570–573.
16. Varga I, Plank L, Mešťanová V, et al. Prehľad a návrh klasifikácie vrodených anomálií týmusu u detí. Čes-slov Pediat 2014; 69 (3): 178–190.
17. Emanuel BS, McDonald-McGinn D, Saitta SC, et al. The 22q11.2 deletion syndrome. Adv Pediatr 2001; 48: 39–73.
18. Antshel KM, Kates WR, Roizen N, et al. 22q11.2 deletion syndrome: genetics, neuroanatomy and cognitive/behavioral features keywords. Child Neuropsychol 2005; 11 (1): 5–19.
19. Bassett AS, Marshall CR, Lionel AC, et al. Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Hum Mol Genet 2008; 24 (17): 4045–4053.
20. Yang Ch, Huang CHH, Cheon ML, et al. Unambiguous molecular detections with multiple genetic approach for the complicated chromosome 22q11 deletion syndrome. BMC Medical Genetics 2009; 10: 16.
21. Vokurková J, Elstnerová L, Lukášová O, et al. Vývoj neonatální péče a zhodnocení zkušeností prvních pěti let operací rozštěpu rtu v neonatálním období. Čes-slov Pediat 2011; 66 (6): 356–362.
22. Wilson DI, Burn J, Goodship J. DiGeorge syndrome: part of CATCH 22. J Med Genet 1993; 30: 852–856.
23. Cormier-Daire V, Iserin L, Theophile D, et al. Upper limb malformations in DiGeorge syndrome. Am J Med Genet 1995; 56: 39–41.
24. Prasad C, Quackenbush EJ, Whiteman D, et al. Limb anomalies in DiGeorge and CHARGE syndromes. Am J Med Genet 1997; 68: 179–181.
25. Kokitsu-Nakata NM, Guion-Almeida ML, Richieri-Costa A. 22q11 deletion syndrome and limb anomalies: report on two brazilian patients. Cleft Palate Craniofac J 2008; 45 (5): 561–566.
26. Al-Hertani W, Hastings VA, McGowan-Jordan J, et al. Severe craniosynostosis in an infant with deletion 22q11.2 syndrome. Am J Med Genet 2012; 161A: 153–157.
27. Ming JE, McDonald-McGinn DM, Megerian TE, et al. Skeletal anomalies and deformities in patients with deletions of 22q11. Am J Med Genet 1997; 72: 210–215.
28. O´Driscoll MC, Clayton-Smith J. Bilateral camptodactyly and recurrent patellar dislocation: a new sign of 22q11 deletions or an independent dominant disorder? Clin Dysmorphol 2008; 17: 157–159.
29. Brites MM. Familial camptodactyly. Eur J Dermatol 1998; 8: 355–356.
30. Lammer EJ, Opitz JM. The DiGeorge anomaly as a developmental field defect. Am J Med Genet 1986; 2: 113–127.
31. Le Douarin N. Migration and differentiation of neural crest cells. Curr Top Dev Biol 1980; 16: 31–85.
32. Zheng D, Zhang Z, Harrison PM, et al. Integrated pseudogene annotation for human chromosome 22: evidence for transcription. J Mol Biol 2005; 349: 27–45.
33. Thuresson AC, Bondeson ML, Edeby C, et al. Whole-genome array-CGH for detection of submicroscopic chromosomal imbalances in children with mental retardation. Cytogenet Genome Res 2007; 118 (1): 1– 7.
34. Erickson RP, de Ståhl T, Bruder CE, et al. A patient with 22q11.2 deletion and Opitz syndrome-like phenotype has the same deletion as velocardiofacial patients. Am J Med Genet A 2007; 143A (24): 3302–3308.
35. Ben-Shachar S, Ou Z, Shaw CA, et al. 22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome. Am J Hum Genet 2008; 82 (1): 214–221.
36. Giele H, Cassell O. Plastic and Reconstructive Surgery. Oxford: Oxford University Press, 2008: 1–197.
37. Moerman P, Fryns JP. Ectodermal dysplasia, Rapp-Hodgkin type in a mother and severe ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC) in her child. Am J Med Genet 1998; 63 (3): 479–481.
38. Duijf PHG, van Bokhoven H, Brunner H. Pathogenesis of split-hand//split-foot malformation. Hum Mol Genet 2003;12 (1): 51–60.
39. Winter RM, Baraitser M. The London dysmorphology database. J Med Genet 1987; 24 (8): 509–510.
40. Weksberg R, Hughes S, Moldovan L, et al. A method for accurate detection of genomic microdeletions using real-time quantitative PCR. BMC Genomics 2005; 6: 180.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2015 Číslo 5
- Horní limit denní dávky vitaminu D: Jaké množství je ještě bezpečné?
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Syndrom Noonanové: etiologie, diagnostika a terapie
Nejčtenější v tomto čísle
- Metabolické kostní onemocnění při nezralosti
- Vzácný případ DiGeorgeova syndromu s anomáliemi končetin: přínos vyšetření metodou SNP microarrayí?
- Vybrané špecifiká ultrasonografie pľúc v detskom veku
- Akutní kašel