
Vybrané kapitoly z dětské onkologie a transplantace kostní dřeně
Autoři:
J. Starý
Působiště autorů:
Klinika dětské hematologie a onkologie UK 2. LF a FN Motol, Praha
Vyšlo v časopise:
Čes-slov Pediat 2011; 66 (3): 177-187.
Kategorie:
Vybrané kapitoly z nové učebnice Klinická pediatrie
Nakladatelství Galén připravuje k vydání novou učebnici „Klinická pediatrie“, která bude určena jak pro pregraduální studium na lékařských fakultách, tak i v rámci postgraduálního vzdělávání pro přípravu na atestaci z dětského lékařství a z praktického dětského lékařství. Autoři věří, že učebnici ocení i dětští lékaři v praxi.
Ve spolupráci nakladatelství Galén a redakce Česko-slovenské pediatrie na stránkách našeho časopisu v letošním roce postupně uveřejňujeme jednotlivé stati z nové učebnice – a jako bonus také některé kapitoly, které se do nové učebnice už „nevešly“, protože učebnice má přesně stanovený rozsah.
Čes-slov Pediat 2011; 66(3): 177–191.
1.0 Úvod
Ročně je v České republice diagnostikován zhoubný nádor přibližně u 350 dětí a dospívajících do 19 let. U dětí mladších 15 let je nejčastějším zhoubným nádorovým onemocněním akutní lymfoblastická leukemie (cca 25 % všech nádorů), následují nádory mozku (22 %) a neuroblastom (8 %), který je nejčastějším nádorem u kojenců. U dospívajících mezi 15–19 lety dominují maligní lymfomy (25 %), následované germinálními nádory (13 %) a nádory mozku (10 %). V tomto věku přibývá karcinomů – malignit typických pro věk dospělých.
Nejčastějšími příznaky nádorových onemocnění jsou horečka, bolest hlavy, zvracení, bledost, únava, bolest kostí, kulhání, hubnutí, krvácení a/nebo přítomnost nádorové rezistence. Většina těchto projevů se vyskytuje u daleko častějších, méně hrozivých onemocnění. K diagnóze akutní leukemie či nefroblastomu uplynou od prvních příznaků nejčastěji 2–3 týdny. Diagnóza kostních nádorů či nádorů mozku může být stanovena za řadu měsíců od počátku příznaků. Věk hraje důležitou roli. Malé děti navštěvují pediatra častěji než dospívající, kde role rodičů v záchytu příznaků nemoci ustupuje do pozadí. Příkladem jsou dorostenci s germinálními testikulárními nádory, kteří často měsíce nevěnují pozornost zvětšujícímu se, nebolestivému varleti a přicházejí k lékaři s pokročilým onemocněním.
Stanovení správné diagnózy vyžaduje odběr nádorové tkáně k histologickému vyšetření. U pacientů se solidními nádory má zásadní význam volba správné techniky biopsie, která nesmí kontaminovat nepostižené tělní dutiny, lymfatický systém a musí splnit zásady chirurgického stagingu, charakterizujícího rozsah nemoci a postižení spádových lymfatických uzlin. Moderní diagnostika dětských zhoubných nádorových onemocnění vyžaduje kombinaci metod imunohistochemických, in situ hybridizace a metod molekulárních. Odebraná tkáň nádoru nesmí být na operačním sále dána do formaldehydu, ale do sterilní nádobky s fyziologickým roztokem. Patolog zhodnotí rozsah nekróz a krvácení, extrahuje z čerstvé tkáně nádoru DNA a RNA pro molekulárně biologická vyšetření, odešle vzorky na vyšetření cytogenetické, průtokovou cytometrií, část vzorku zamrazí a část fixuje ve formaldehydu pro vyšetření mikroskopické. Neadekvátní chirurgický výkon (např. transskrotální biopsie nádoru varlete či rozsáhlá operace nehodgkinského lymfomu) poškozuje pacienta. Chybná fixace získaného vzorku znemožňuje diagnostický postup lege artis.
Moderní komplexní léčbou zhoubných nádorů se daří vyléčit více než 75 % dětí. Správná klasifikace rozsahu nemoci, volba a aplikace optimální léčby jsou základním předpokladem pro dosažení těchto léčebných výsledků. Děti jsou léčeny v centrech, splňujících požadavky pro moderní diagnostiku, komplexní léčbu a výzkum. Zkušenost multidisciplinárního týmu je v péči o pacienty s těmito vzácnými onemocněními nenahraditelná. Přežití není u dětí jediným cílem léčby nádorů. Stejný význam má kvalita budoucího života, výskyt a tíže pozdních následků. Většina dětí a dospívajících s nádory je léčena v klinických, léčbu optimalizujících studiích, kontrolovaných a díky vzácnosti dětských nádorů mezinárodních. Formou randomizované studie jsou standardní postupy léčby se známou pravděpodobností vyléčení a výskytu vedlejších účinků srovnávány s léčbou novou, experimentální, mající za cíl zlepšit výsledky léčby a/nebo snížit výskyt jejích časných a pozdních komplikací při zachování jejího efektu. Perspektivou je pro dětské pacienty aplikace cílené léčby se sníženým výskytem komplikací.
2.0 Nádory centrálního nervového systému
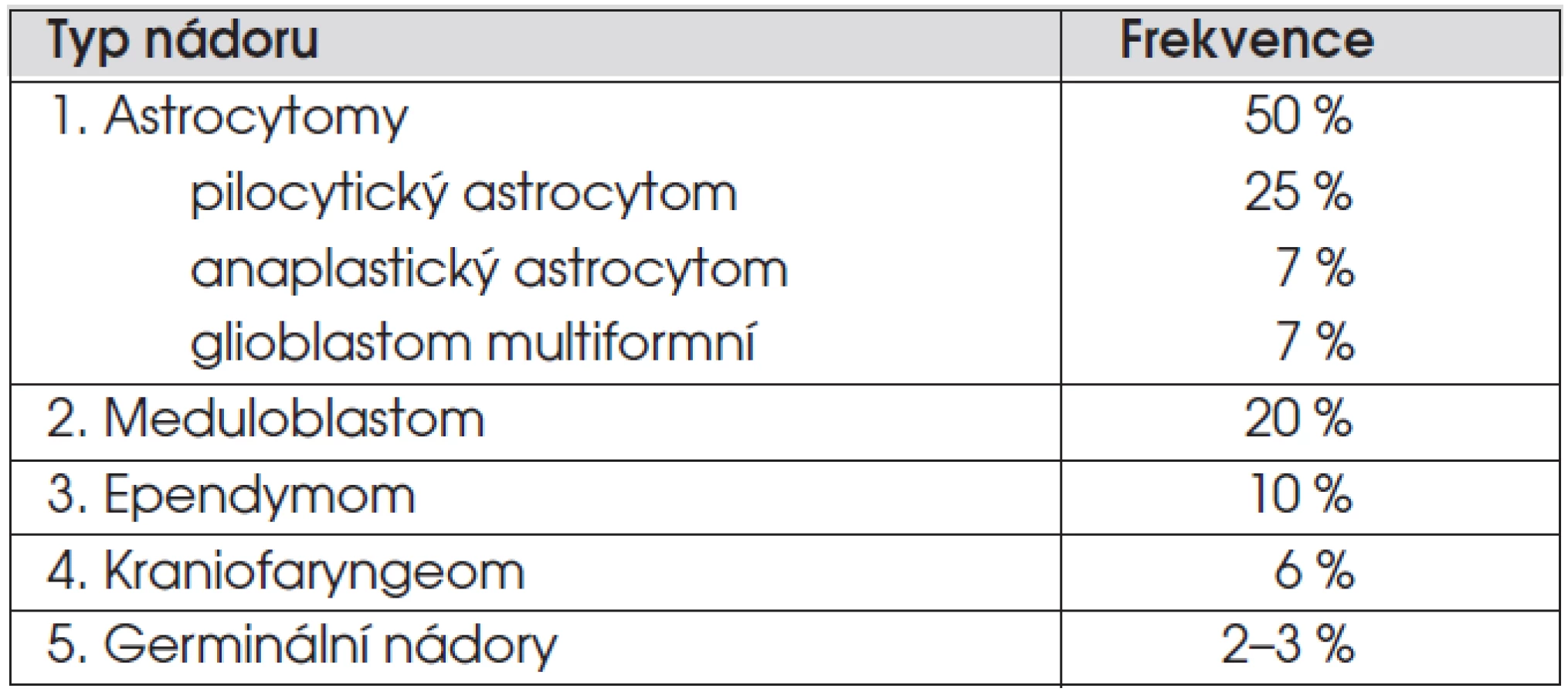
Nádory centrálního nervového systému tvoří 20–25 % dětských nádorů a jsou tak po leukemiích nádorem nejčastějším. Incidence je 3 případy/100 000 dětí. Zastoupení nejčastějších nádorů je uvedeno v tabulce 1. Nejčastějším nádorem je astrocytom, který tvoří 50 % všech nádorů mozku, nejčastějším maligním nádorem je meduloblastom (20 % nádorů). V míše se nejčastěji vyskytují astrocytomy a ependymomy. Diagnostika a léčba nádorů CNS je velmi komplexní záležitost a vyžaduje erudovaný tým složený z neurochirurgů, neurologů, radiologů, patologů, radioterapeutů, onkologů, neuropsychologů a rehabilitačních a sociálních pracovníků.
Etiopatogeneze
Méně než 5 % dětských nádorů se vyskytuje u genetických onemocnění, nejčastěji u neurofibromatózy 1. typu (NF-1) a tuberózní sklerózy. Asi 20–50 % pacientů s NF-1 postihne gliom mozku nízkého stupně malignity, často v oblasti zrakového nervu. Nejvýznamnějším rizikovým faktorem pro vznik sekundárních mozkových nádorů je radioterapie (zvýšený výskyt nádorů mozku po preventivním ozáření krania u ALL).
Klinický obraz
Projevy nádorů mozku jsou často zpočátku necharakteristické a od prvních příznaků k diagnóze zpravidla uplyne několik měsíců. Klasická trias příznaků – bolesti hlavy, ranní zvracení a edém papily zrakového nervu – se vyskytuje u třetiny pacientů. Jedná se o projev nitrolební hypertenze. Bolesti hlavy se v čase stupňují a mohou budit dítě ze spaní. V této situaci musí následovat radiodiagnostické vyšetření. Děti mladší čtyř let mají často makrocefalii. Poruchy chůze a rovnováhy postihují třetinu dětí, změny osobnosti až polovinu. Děti mladší dvou let bývají spavé, dráždivé, batolata a předškolní děti mění stravovací a spánkové návyky, mají problémy s řečí, starší děti zhorší školní prospěch, špatně se koncentrují, rychle mění nálady. Děti s nádory mozkového kmene častěji vykazují obrny hlavových nervů a pyramidové příznaky. Nádory míchy se manifestují bolestí v zádech, poruchou chůze a koordinace, sfinkterovými problémy.
Obecně platí, že děti mladší 10 let mají častěji maligní nádory zadní jámy lební (meduloblastom), které vedou časně k nitrolební hypertenzi a diagnóze. Nejčastějšími nádory dětí mladších tří let jsou meduloblastom, ependymom a astrocytom. Dospívající mají častěji supratentoriální nádory mozkových hemisfér, optiku, oblasti selly. Nejčastěji se jedná o astrocytomy s nízkým stupněm malignity, jejichž klinické příznaky se manifestují později a k diagnóze tak uplyne delší čas.
Diagnostika
Při podezření na mozkový nádor je indikováno oční a neurologické vyšetření, zhodnocení růstu (včetně obvodu hlavy u dětí mladších 4 let) a puberty. Zobrazovací metodou první volby je magnetická rezonance. Histologická diagnóza je zlatým standardem diagnostiky s výjimkou sekrečních germinálních nádorů, difuzních gliomů mozkového kmene a gliomů optiku u pacientů s NF-1, kde histologické ověření není nutné. Podle WHO doporučení je stupeň malignity (grading) hodnocen škálou I (nejvíce benigní) – IV (nejvíce maligní). Polovinu astrocytomů u dětí tvoří pilocytický astrocytom, což je nádor nízkého (I.) stupně malignity. Čtvrtinu astrocytomů tvoří anaplastický astrocytom (grade III) a glioblastom (grade IV), který je nejčastějším maligním nádorem dospělých. Vyšetření mozkomíšního moku je indikováno u maligních nádorů s tendencí metastazovat do kanálu páteřního (meduloblastom, germinální nádory). Při podezření na germinální nádor je nutné vyšetřit v krvi a moku alfa-1-fetoprotein a HCG.
Diferenciální diagnóza
Přibližně 50 % mozkových nádorů se vyskytuje supratentoriálně a 50 % infratentoriálně (v zadní jámě). Ze supratentoriálních nádorů jsou nejčastější astrocytomy v hemisférách, gliomy optiku a chiasmatu, kraniofaryngeom, germinom a sekreční germinální nádory v oblasti glandula pinealis a supraselárně. V zadní jámě se nachází meduloblastom a astrocytom mozečku, dvě třetiny ependymomů, gliomy mozkového kmene.
Terapie a prognóza
Základní léčebnou modalitou je operace. Cílem je kompletní resekce nádoru, nikoliv však za cenu trvalého neurologického postižení. MR provedená do 48 hodin po operaci vyhodnotí radikalitu výkonu. Náročné technické vybavení neurochirurgických operačních sálů zvyšuje radikalitu i bezpečnost operace. Operace je jedinou kurativní léčbou u astrocytomů nízkého stupně malignity, zatímco u maligních nádorů, jako je meduloblastom, musí navazovat pooperační radioterapie, event. chemoterapie. Zevní drenáž a ventrikuloperitoneální shunt jsou řešením obstrukčního hydrocefalu.
Radioterapie je aplikována na lůžko nádoru, v případě nádorů s vysokým rizikem metastazování do kanálu páteřního, jako je meduloblastom, se ozařuje kraniospinální osa. Chemoterapie doplňuje záření u pacientů s meduloblastomem, kde u pacientů standardního rizika (s kompletní resekcí nádoru a bez metastáz) umožňuje snížit dávku kraniospinálního záření a u pacientů vysokého rizika (pooperační reziduum a/nebo metastázy) v podobě vysokodávkované léčby zlepšuje prognózu. U recidivujících či parciálně resekovaných astrocytomů nízkého stupně malignity může chemoterapie oddálit radioterapii. U pacientů s gliomy a NF-1 je na místě konzervativní přístup. U germinomu není nutná radikální operace, pouze biopsie, radioterapie v kombinaci s chemoterapií dává 90% šanci na přežití. Naopak sekreční germinální nádory vyžadují komplexní léčbu, přesto je prognóza nejistá. Klíčovou léčbou ependymomu je operace. Kraniofaryngeom má sklon k recidivám, není--li operace radikální. Radikální operace ale může vést k poškození hypotalamu s trvalými následky pro pacienta, jako je porucha paměti, změna chování, spánkového rytmu, excesivní obezita. I zde je radioterapie metodou volby. Z cytostatik se uplatňují vinkristin, deriváty platiny, cyklofosfamid, metotrexát, CCNU.
Vzhledem k vysokému riziku pozdních následků záření je léčebným přístupem u dětí mladších 3–5 let s maligními nádory operace a chemoterapie s cílem oddálit ozáření za tuto věkovou hranici. Vzhledem k biologii nádorů malých dětí není tento přístup příliš úspěšný a nádory předčasně recidivují.
Pravděpodobnost přežití dětí s mozkovými nádory je 60–70%, více než 70% u astrocytomů, 90% u kraniofaryngeomu, 70% u meduloblastomů standardního rizika, horší u meduloblastomů vysokého rizika. Infaustní prognózu mají děti s difuzními gliomy kmene, kde není operace možná. Malé děti mají vzhledem k biologii svých nádorů horší výsledky léčby než dospívající. Špatnou prognózu mají děti mladší 3 let s maligními embryonálními nádory.
Pozdní následky postihují 60 % dětí léčených pro nádor centrálního nervového systému. Největším zdrojem pozdních následků je pro vyvíjející se dětský mozek radioterapie, což platí zejména pro děti mladší 8 let. Největším rizikem je ozáření kraniospinální osy. Podíl na pozdních následcích má i nádor sám a jeho operace. Důsledkem je ztráta bílé hmoty mozkové a snížení vývojového kvocientu, které je tím vyšší, čím je pacient při ozáření mladší. Postiženy jsou zejména kognitivní funkce, jako je pozornost, schopnost soustředit se, paměť. Děti po léčbě nádorů mozku mají více problémů ve školní výuce, hůře hledají odpovídající pracovní zařazení, častěji mají invalidní důchod. Velmi důležitou stránkou péče o děti s nádory mozku je komplexní rehabilitace, systematické monitorování hladiny růstového hormonu, funkce štítné žlázy a gonád, hormonální substituce, léčba epilepsie.
3.0 Maligní nádory kostí
Nejčastějšími zhoubnými nádory kostí jsou osteosarkom (OS) a Ewingův sarkom (ES). Osteosarkom se vyskytuje dvakrát častěji než ES. Metastázy jsou při diagnóze častěji přítomny u ES (25 %) než u OS (12 %). Lokalizace nádoru v axiálním skeletu (pánev, obratle) je u OS méně častá (10 %) než u ES (35 %). Vrchol výskytu je u obou nádorů u dospívajících, u dětí mladších 10 let se daleko častěji vyskytuje ES. Délka od prvních příznaků k diagnóze je u obou nádorů několik měsíců. Nejčastější projev kostních nádorů – bolest a zduření – často navazuje na sportovní úraz, faktor, který ne vzácně oddaluje stanovení správné diagnózy. OS je diagnostikován dříve, díky své predilekční lokalizaci v dlouhých kostech. Necharakteristické projevy axiálních nádorů (bolest v zádech bez nádorové masy) oddalují diagnózu. Dospívající s nádory vyhledají lékaře většinou později než menší děti, kde hrají větší roli rodiče.
3.1 Osteosarkom
Osteosarkom (OS) je nejčastější zhoubný kostní nádor u dětí. Pochází z primitivního kost vytvářejícího mezenchymu a charakterizuje ho produkce osteoidu nebo nezralé kosti maligními buňkami stromatu nádoru. Incidence je 5,6 na milion dětí. Vrchol výskytu je v dospívání. Chlapci jsou postiženi častěji. Výskyt nádoru je spojen s pubertálním růstovým zrychlením v druhé dekádě života, u děvčat se vyskytuje dříve než u chlapců, protože pubertální růstový spurt u nich začíná dříve. Děti s osteosarkomem jsou vyšší než jejich vrstevníci. Predilekční lokalizací je pro nádor metafýza nejrychleji rostoucích dlouhých kostí – distálního femuru a proximální tibie.
Etiopatogeneze
Etiologie OS je neznámá. Zvýšené riziko vzniku OS mají některá benigní kostní onemocnění – osteochondrom, enchondromatóza, fibrózní dysplazie. Rizikovým faktorem pro vznik OS je ozáření. Pacienti s vrozeným retinoblastomem mají významně zvýšené riziko vzniku osteosarkomu.
Klinický obraz
Osteosarkom se u většiny dětí projevuje bolestí postižené kosti. Zduření okolních měkkých tkání může a nemusí být přítomné. Postiženy jsou nejčastěji dlouhé kosti v okolí kolenního kloubu – distální femur a proximální tibie. Následují proximální humerus a femur. Kosti axiálního skeletu, nejčastěji pánev, jsou postiženy pouze v 10 % případů. Patrné metastázy má při diagnóze 10 až 15 % pacientů – nejčastěji do plic, méně často do kostí, výjimečně do mízních uzlin. Patologická fraktura, i přes značnou destrukci architektoniky postižené kosti, je iniciálním příznakem pouze v 1 % případů.
Diagnostika
Na nativní rentgenový snímek, prokazující destrukci kosti, periostální reakci a zduření měkkých tkání, navazuje MRI znázorňující rozsah nádorového postižení. Kostní scintigrafie vyloučí metastázy do kostí, rtg a CT plic metastázy plicní. Potvrzením diagnózy je histologické vyšetření biopsie nádoru. Nález novotvořeného osteoidu a kosti v maligním sarkomovém stromatu odlišuje OS od příbuzných nádorů, chondrosarkomu a fibrosarkomu.
Terapie a prognóza
Šance na vyléčení pacientů s lokalizovanými OS dlouhých kostí je 60–70%. Primárně metastatické onemocnění má prognózu významně horší (přežití 10–40%). Špatnou prognózu mají rovněž OS axiálního skeletu, kde většinou není možná radikální operace. Histologický grading a velikost nádoru jsou dalšími prognostickými faktory. Obecně platí, že šance na vyléčení je významně vyšší u těch nemocných, kde se podaří radikálně chirurgicky odstranit veškerou nádorovou masu, a to jak primárního, tak i metastatického procesu.
Základním léčebným přístupem k osteosarkomu je kombinace neoadjuvantní předoperační chemoterapie, radikální operace a následné pooperační adjuvantní léčby, jejíž intenzita je vedena histologickou odpovědí na neoadjuvantní léčbu. Osteosarkom je špatně citlivý vůči radioterapii. Neoadjuvantní léčba má za cíl zmenšit velikost nádoru a zvýšit úspěšnost operace, která je klíčovou léčebnou modalitou. Pacienti s nádorem sahajícím do okrajů excize a s méně než 90% nekrózou nádorové tkáně mají 30% výskyt lokálních recidiv. U osteosarkomů končetin jsou v současnosti dvě třetiny operačních zákroků záchovných, jedna třetina je amputací. K záchovným zákrokům se používá protéz kovových či z jiných materiálů (keramické), autologních kostních štěpů (fibula), alogenních kostních štěpů. Komplikacemi jsou infekce, fraktury, nespojení kostí. Problémem je pokračující růst dětských kostí, který resekce zasahující do epifýzy poškozuje. Funkční výsledky distálních amputací se neliší od operací záchovných, čím je amputace více proximálně, tím je ale funkční rozdíl větší. Problémem amputovaných končetin jsou fantomové bolesti. Děti se po adekvátní psychologické přípravě dokáží s mutilujícími operacemi vyrovnat. Zkušené ortopedické centrum je pro tyto typy operací nutností.
Před érou chemoterapie se dařilo vyléčit pouhou operací 15 % dětí s osteosarkomy. Metastázy lokální a vzdálené byly pravidlem. Z cytostatik jsou u osteosarkomu nejúčinnější cisplatina, doxorubicin, ifosfamid, vysokodávkovaný metotrexát. Plicní metastázy při diagnóze či progresi nemoci je nutné odstranit chirurgicky, i když jsou mnohočetné a recidivující. Jiná naděje na jejich vyléčení není. Metastázy do dalších orgánů a tkání jsou spojeny s extrémně nepříznivou prognózou.
3.2 Ewingův sarkom
Ewingův sarkom (ES) je druhým nejčastějším zhoubným kostním nádorem s incidencí 2,1 na milion dětí. Vrchol výskytu je v druhé dekádě života, 20–30 % případů se vyskytuje u dětí mladších 10 let, 20 % u pacientů starších 20 let.
Etiopatogeneze
Klasický Ewingův sarkom je nádor z malých tmavých buněk, vykazujících v imunohistochemickém vyšetření diferenciaci do nervové tkáně. Stejně se chová i diferencovanější varianta ES, periferní primitivní neuroektodermální nádor (PNET). Nádory skupiny ES/PNET jsou charakterizovány chromozomálními přestavbami, které zahrnují gen EWS na 22. chromozomu (22q12) a geny ets skupiny transkripčních faktorů (FLI1, FEV, ERG). Nejčastější specifickou translokací je t(11;22)(q22;q12), která se nachází u 85 % pacientů. Vytvořený fúzní gen EWS-FLI1 lze detekovat metodou PCR nebo FISH a stanovit jeho průkazem v kostní dřeni rozsah minimální diseminované nemoci při diagnóze či v průběhu léčby.
Klinický obraz
Prvním projevem nádoru je lokální bolest, která mění svou intenzitu, ale úplně neustoupí ani v noci. Často je přičítána předchozímu úrazu. Trvá-li bez zřetelného lokálního nálezu déle než měsíc, měla by být důvodem vyšetření zobrazovacími metodami. Na bolest navazuje vznik lokální rezistence, což ovšem u nádorů pánve či hrudní stěny může trvat velmi dlouho. Průměrná doba mezi začátkem potíží a stanovením diagnózy trvá 3 až 9 měsíců. Onemocnění může být považováno za zánět šlach, kyčelního kloubu, osteomyelitidu (je často provázeno horečkami, což podezření na osteomyelitidu zvyšuje).
Postižena může být jakákoliv kost. Nejčastěji se jedná o pánevní kosti (26 %), femur (20 %) a hrudní stěnu (žebra, obratle). Ewingův sarkom může vycházet i z měkkých tkání bez postižení kosti (extraskeletální ES). Až 25 % pacientů má primárně metastatickou nemoc, plíce jsou postiženy v 10 %, kosti a kostní dřeň rovněž v 10 %, kombinovaná postižení tvoří 5 % případů.
Diagnostika
Nativní snímek prokáže osteolýzu, periostální reakci, zduření měkkých tkání. Na rozdíl od osteosarkomu (postižení metafýz) vychází ES z diafýzy dlouhých kostí. MRI určí rozsah lokálního postižení. CT plic, scintigrafie skeletu a aspirace kostní dřeně slouží k průkazu metastáz. Pozitronová emisní tomografie je vysoce účinná v průkazu kostních metastáz. Diagnózu určí histochemické vyšetření biopsie nádoru a potvrdí molekulární vyšetření translokace.
Terapie a prognóza
Prognosticky nepříznivým faktorem je přítomnost metastáz. Pacienti s plicními metastázami mají o něco lepší prognózu (přežití 30 %) než děti s metastázami v kostech či kostní dřeni (přežití méně než 20 %). Děti mladší 10 let mají lepší výsledky léčby než děti starší. Velké nádory (většinou se jedná o nádory pánve) mají horší prognózu. Důležitým prognostickým znakem je také odpověď na iniciální léčbu.
Před érou chemoterapie se dařilo vyléčit 10 % pacientů. Současná léčba kombinuje neoadjuvantní (předoperační) chemoterapii, operaci, pooperační adjuvantní chemoterapii, event. radioterapii. Iniciální léčba zmenšuje nádor a činí ho operabilním. Současně léčí mikrometastázy. Operace je většinou záchovná, se stejnými přístupy i komplikacemi jako u osteosarkomu. Ewingův sarkom je vysoce radiosenzitivní nádor. Radioterapií je možné nahradit operaci, pokud není možná, před operací zmenšit nádor nebo po operaci s mikroskopickým přesahem okrajů resekátu nádorem snížit riziko lokální recidivy. Nejúčinnějšími cytostatiky jsou vinkristin, ifosfamid, doxorubicin, etoposid. Šance na vyléčení pacientů s lokalizovaným onemocněním je 70%. Riziko relapsu je 30%, větší u pacientů s metastatickým onemocněním. Šance na vyléčení relapsu je 20% v závislosti na intervalu od iniciální diagnózy (prognóza časných relapsů v prvních dvou letech je infaustní).
Pozdní následky jsou kombinací ortopedických problémů po operaci, důsledků radioterapie na růst kostí, svalů, postradiační malignity (osteosarkomy) a komplikací chemoterapie (2 % pacientů onemocní sekundární akutní leukemií do 3 let od léčby). Kvalita života většiny vyléčených dětí je velmi dobrá.
4.0 Sarkomy měkkých tkání
Sarkomy měkkých tkání (maligní mezenchymální nádory) vznikají maligní transformací primitivního mezodermu, který normálně vyzrává do skeletálních svalů, hladkých svalů, tuku, fibrózní tkáně, kostí a chrupavek. Jedná se o pátou nejčastější malignitu v dětském věku. Dělí se na dvě velké skupiny – rabdomyosarkomy a nonrabdomyosarkomy. Rabdomyosarkomy (RMS) tvoří 40 % sarkomů měkkých tkání, u dětí mladších 5 let 60 %. Non-rabdomyosarkomy (NRSTS) tvoří tři čtvrtiny sarkomů u dospívajících mezi 15–19 lety.
4.1 Rabdomyosarkom
Rabdomyosarkom (RMS) je s incidencí 4,3 případů na milion dětí třetím nejčastějším extrakraniálním nádorem u dětí po neuroblastomu a nefroblastomu. Dvě třetiny nádorů jsou diagnostikovány u dětí mladších 6 let, druhý menší vrchol výskytu je v dospívání. Histologická klasifikace dělí RMS na dvě kategorie: 80 % tvoří embryonální RMS, alveolární varianta 20–30 %. Vzdálené metastázy jsou nalézány u 15 % dětí.
Etiopatogeneze
Zvýšený výskyt RMS je u některých familiárních genetických syndromů, jako jsou neurofibromatóza I. typu a syndrom Li-Fraumeni se zárodečnou mutací tumor supresorového genu p53. RMS se v těchto rodinách spojuje s adrenokortikálním karcinomem a časným vznikem karcinomu prsu.
Osmdesát procent pacientů s alveolárním RMS má v nádorových buňkách translokaci t(2;13) s fúzním genem PAX3-FOXO1a (FKHR), pro embryonální RMS je charakteristická ztráta heterozygozity lokusu 11p15. Obě tyto změny mají za následek zvýšenou produkci růstového faktoru IGF-II (insulin growth factor), stimulujícího růst nádorových buněk. Padesát procent případů RMS má mutaci v dráze tumor supresorového genu p53, třetina pacientů má mutace onkogenů RAS.
Klinický obraz
Sarkomy zpočátku nepůsobí větší klinické obtíže, a proto je stanovení jejich diagnózy často pozdní se záchytem lokálně pokročilých forem. Různorodost nádorových příznaků vyplývá z lokalizace primárního nádoru. Obvykle nádor imponuje jako tuhá, nebolestivá, progresivně rostoucí rezistence v měkkých tkáních, případně na nádorové onemocnění upozorní obtíže z útlaku či tahu okolních struktur. Přibližně 40 % RMS pochází z oblasti hlavy a krku (tab. 2). Nádory očnice se manifestují protruzí a poruchou pohyblivosti očního bulbu spolu s jeho deviací, ztrátou vidění a bolestí oka. Chronická sekrece z nosu, opakovaná epistaxe, porucha nosní průchodnosti a dysfagie mohou upozornit na tumor lokalizovaný v oblasti nosu, vedlejších nosních dutin a nosohltanu. Bolest ucha s chronickou sekrecí ze zvukovodu upozorňují na nádor zevního zvukovodu a středouší. Až 25 % nádorů pochází z urogenitálního ústrojí a prezentují se poruchou vyprazdňování moči, dysurií, hematurií či tuhým zvětšováním varlete. Necelých 20 % nádorů postihuje končetiny, z ostatních lokalizací je nejčastěji postižen trup (10 %).
![Rabdomyosarkom – lokalizace primárního nádoru a přežití.
[Upraveno podle: Stevens MCG. Treatment of childhood rhabdomyosarcoma: the cost of cure. Lancet Oncol 2005; 6: 77–84.]](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image/ca15d31ea6c93f2f314f637cc4650267.png)
Nejčastějším místem vzdálených metastáz jsou plíce (50 %), kostní dřeň (20–30 %), kosti (10 %), polovina pacientů má postižen pouze jeden orgán (nejčastěji plíce). Do mízních uzlin metastazuje 20 % nádorů.
Diagnostika
Ultrazvukové vyšetření, CT, MRI, PET stanoví lokalizaci a velikost primárního nádoru a metastáz. Kostní scintigrafie a aspirace kostní dřeně slouží k vyloučení metastáz. Diagnózu potvrdí histochemické a molekulární vyšetření tkáně nádoru odebrané biopsií.
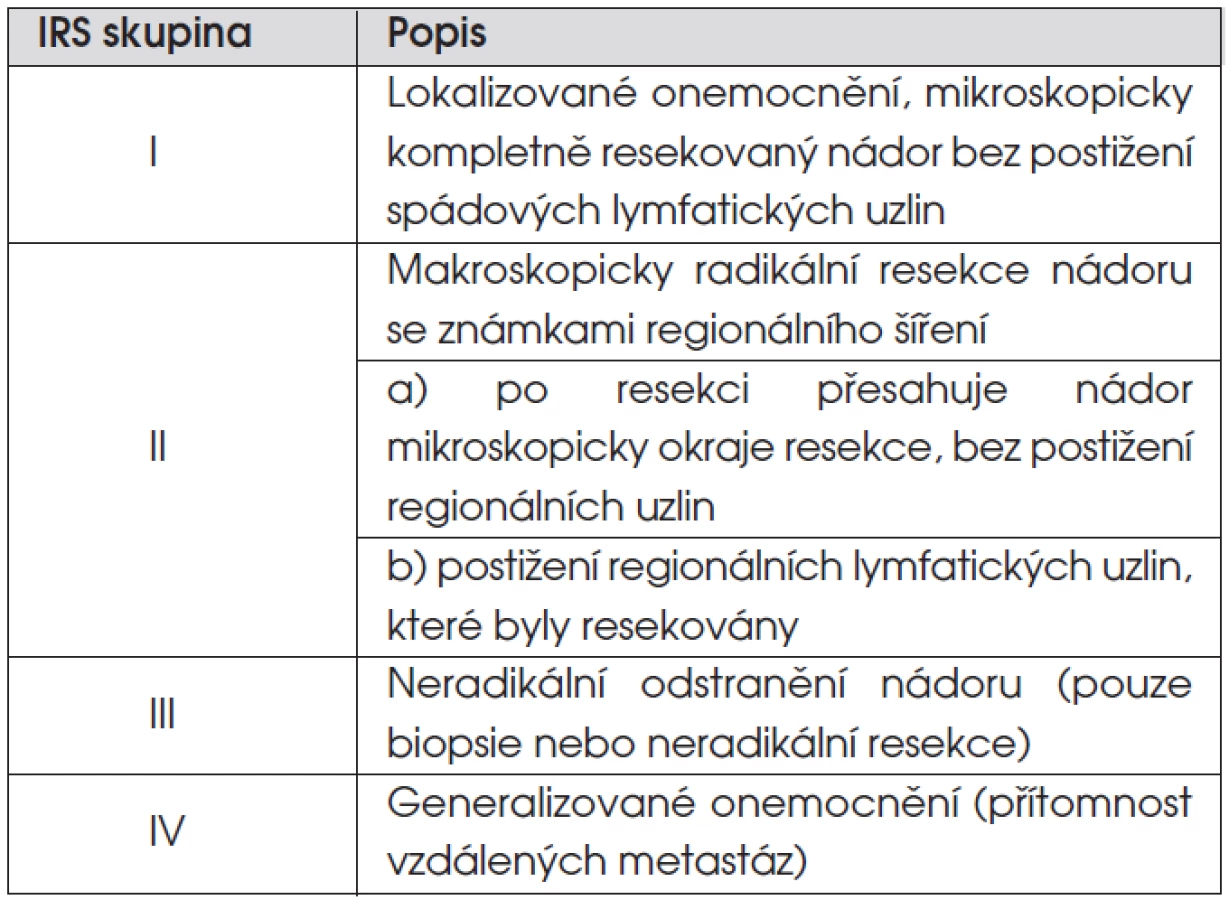
Z klasifikací řadících onemocnění do klinických stadií jsou nepoužívanější dvě. SIOP TNM předoperační staging řadí nádory do čtyř stadií: stadium 1 – lokalizovaný nádor omezený na orgán či tkáň svého původu; stadium 2 – lokální nádor extenzivně se šířící; stadium 3 – postižení regionálních lymfatických uzlin; stadium 4 – vzdálené metastázy. Pooperační staging severoamerické pracovní skupiny IRS (Intergroup Rhabdomyosarcoma Study) dělí pacienty do skupin podle rozsahu reziduálního nádoru po iniciální operaci (tab. 3).
Terapie a prognóza
Rabdomyosarkom je chemosenzitivní nádor a více než 70 % pacientů s lokalizovaným onemocněním bez vzdálených metastáz může být vyléčeno. Léčba je multimodální a skládá se z operace, chemoterapie a radioterapie. Optimální volba léčebného schématu záleží na přítomnosti rizikových faktorů.
Nejdůležitějšími prognostickými faktory, ovlivňujícími léčebné výsledky, jsou stadium nemoci, lokalizace nádoru, patologický podtyp a věk pacienta. Většina pacientů (74 %) bez metastatické nemoci má po iniciálním chirurgickém zákroku měřitelnou reziduální nemoc (IRS skupina III), mikroskopickou reziduální nemoc (IRS skupina II) má 12 % dětí a primární kompletní resekce nádoru (IRS skupina I) je možná pouze u 14 % dětí s RMS. Důvodem neúspěchu iniciální operace je lokalizace nádoru u dětí. Cílem operace je vyhnout se mutilaci pacienta. Lokalizace nádoru v oblasti hlavy, krku a urogenitálního traktu u 65 % dětí s RMS většinou radikální operační zákrok neumožňuje.
Lokalizace nádoru je významným rizikovým faktorem (tab. 2). Riziko metastáz je v některých lokalizacích zvýšené stejně jako existence nepříznivé (alveolární) histologie. Metastázy se vyskytují pouze u 4 % nádorů orbity, ale u 40 % sarkomů končetin. Nejnižší výskyt nepříznivého alveolárního podtypu je u nádorů orbity (8 %), naopak nejvyšší u nádorů končetin (70 %). Pacienti s parameningeálními nádory mají zvýšené riziko přímého šíření nádoru na mozkomíšní pleny a do centrálního nervového systému. Cytologické vyšetření mozkomíšního moku je u nich nutností.
Pacienti s alveolárním histologickým typem nádoru jsou většinou starší, mají častěji nádory končetin a mají vysoké riziko vzniku metastáz ve srovnání s RMS embryonálním. Věk nad 10 let a pod 1 rok jsou považovány za nepříznivé. Roli hraje i velikost nádoru. Pacienti vysokého rizika jsou děti starší 10 let s velikostí nádoru 5 cm a více.
Současné léčebné postupy stratifikují intenzitu léčby podle stupně rizika za účelem nepřeléčovat děti s dobrou prognózou a zlepšit osud pacientů s prognózou špatnou. To se nedaří u dětí se vzdálenými metastázami při diagnóze, jejichž prognóza je velmi špatná bez ohledu na volbu léčebného schématu.
Pacienti IRS skupiny I po kompletní resekci jsou léčeni pouze chemoterapií v situaci embryonálního nádoru, pro zvýšené riziko lokální recidivy je u alveolárních nádorů využívána i radioterapie. Chemoterapie dokáže u 35 % dětí s reziduálním nádorem po iniciální operaci/biopsii (IRS skupina III) dosáhnout kompletní remise. Rozhodnutí o zařazení radioterapie u nich stejně jako u dětí IRS skupiny II (mikroskopické reziduum) závisí na přítomnosti rizikových faktorů. Chemoterapie umožňuje zmenšením nádoru radikální operaci u řady pacientů. V situaci, kdy operace není možná nebo přetrvává reziduum i po druhé operaci, je na místě zařadit radioterapii. Z cytostatik jsou nejúčinnější vinkristin, aktinomycin, ifosfamid, cyklofosfamid, antracykliny.
Zařazení radioterapie do léčebného plánu snižuje riziko lokálních recidiv, zvyšuje ale výskyt pozdních následků, které závisí na lokalizaci nádoru. Ozáření parameningeálních nádorů má u malých dětí riziko postižení mozku. Bez ozáření se nejspíše obejdou děti mladší 10 let s malým primárním nádorem a embryonální histologií. Radioterapii lze u primárně neozářených dětí zařadit do léčebného plánu v případě recidivy.
4.2 Non-rabdomyosarkomy měkkých tkání
Non-rabdomyosarkomy měkkých tkání (NRSTS) jsou heterogenní skupinou nádorů, obvykle vycházející z končetin. Jsou charakterizovány lokálním agresivním růstem. Jejich sklon metastazovat závisí na histologickém stupni malignity (grade). Všeobecně jsou považovány za středně citlivé vůči chemoterapii i ozáření. Pacienti s nádory vysokého stupně malignity těží z adjuvantní chemoterapie. NRSTS se dělí na nádory malých dětí, které jsou biologicky méně agresivní než nádory dospělého typu, které se vyskytují u dospívajících.
Infantilní fibrosarkom
Infantilní fibrosarkom postihuje velmi malé děti do dvou let věku a je charakterizován nálezem translokace t(12;15). Léčbou první volby je operace, nádor nemá tendenci metastazovat, prognóza je velmi dobrá (přežití 90%).
NRSTS dospívajících
Nejčastějším nádorem je v této věkové skupině synovialosarkom, který tvoří 30 % nádorů, postihuje především dolní končetiny a nese translokaci t(X;18). Druhým nejčastějším nádorem je maligní nádor z obalů periferních nervů (MPNST), který tvoří 10 % nádorů dospívajících a v 20–50 % případů se spojuje s neurofibromatózou 1. typu. U 2–13 % pacientů s neurofibromatózou 1. typu se vyvine během života MPNST z plexiformního neurofibromu. Léčba sarkomů dospívajících závisí na lokalizaci a velikosti nádoru a stupni malignity. Většinou se kombinuje operace, chemoterapie i radioterapie. Kompletní resekce lokalizovaného nádoru dává 70% šanci na vyléčení.
5.0 Pozdní následky onkologické léčby
Komplexní onkologickou léčbou lze vyléčit více než 75 % dětí s nádory. V současné době je jeden z 900 dospělých mladších 45 let vyléčeným ze zhoubného nádoru v dětství. K zásadnímu zlepšení výsledků léčby došlo v dětské onkologii v 70. a 80. letech. Šedesát procent vyléčených pacientů z té doby má alespoň jedno chronické postižení jako důsledek předchozí léčby. Je velmi pravděpodobné, že počet vyléčených s dlouhodobými následky mezi dětmi léčenými pro zhoubný nádor v 90. letech a v současnosti klesne, protože současná léčba více zohledňuje riziko pozdních následků a je z tohoto pohledu méně toxická. Preventivní ozáření mozku bylo dříve aplikováno u všech dětí s akutní lymfoblastickou leukemií, v současnosti se využívá pouze u 15 % pacientů s nejvyšším rizikem vzniku leukemické infiltrace mozku. Děti s nádory mozku mladší 3–5 let se v první linii léčby neozařují na kraniospinální osu. Tato léčba se odkládá až na progresi nemoci. Současná léčebná schémata kontrolují kumulativní dávky antracyklinů a alkylačních agens a snižují tak pozdní kardiotoxicitu i riziko vzniku sekundárních leukemií. Radioterapie lymfatického systému se u Hodgkinova lymfomu provádí na redukované pole nebo úplně vynechává a tím se snižuje výskyt nádorů štítné žlázy a prsu, které vznikaly desítky let po úspěšném vyléčení z této nemoci.
Četnost výskytu pozdních následků onkologické léčby vedla k vzniku specializovaných ambulancí, kde je dětský onkolog koordinátorem komplexní diagnostické a léčebné péče o vyléčené onkologické pacienty i v dospělosti.
Poruchy růstu
Děti léčené ozářením krania mají nejvyšší riziko vývoje poruch růstu v důsledku poškození hypofýzy a snížené produkce růstového hormonu. Nejvyšší riziko opožděného růstu mají děti mladší 5 let. Tito pacienti mohou těžit z léčby růstovým hormonem. Ozáření páteře snižuje růst obratlů a tím výšku ve stoje, či způsobuje skoliózu. Poruchu růstu ale může způsobit i chemoterapie. Sledování růstové křivky po skončení onkologické léčby je nutností.
Endokrinní komplikace
Hypotyreóza je častá u dětí po celotělovém ozáření, ozáření krania a ozáření krku. Obvyklá doba do jejího vzniku je 12 měsíců, nicméně může trvat i několik let. Zvýšený TSH a normální hladina tyroxinu jsou pravidlem. Substituční léčba je nutná, protože hrozí riziko vzniku nádorů štítné žlázy při její trvalé stimulaci TSH.
Předčasná puberta, opožděná puberta a neplodnost jsou potenciálními důsledky protinádorové léčby. Předčasnou pubertu způsobuje aktivace hypotalamo-hypofyzární osy ozářením krania a je častější u dívek. Poškození gonád u chlapců způsobuje ozáření varlat při celotělovém ozáření, léčbě testikulárního relapsu leukemie, léčbě Hodgkinovy nemoci. Důsledkem je azoospermie, méně často i snížená produkce testosteronu, protože Leydigovy buňky jsou radiorezistentní. Azoospermii vyvolávají i některá cytostatika, jako například alkylační agens cyklofosfamid či prokarbazin. Všem dospívajícím chlapcům musí být před zahájením léčby provedena kryokonzervace spermatu. Ovaria poškozuje ozáření i některé léky (busulfan). Hypergonadotropní hypogonadismus, porucha menstruace, neplodnost, předčasná menopauza jsou pravidlem po transplantaci kostní dřeně s myeloablativní přípravou, hrozí ale i při ozáření břicha pro nádor či při léčbě Hodgkinovy nemoci. Hormonální blokáda gonád s jejich uvedením do předpubertálního klidového stavu je částečně úspěšným řešením.
Žádná studie neprokázala zvýšený výskyt potratů, předčasných porodů, vrozených malformací či genetických onemocnění u dětí narozených ženám a mužům léčeným v dětství pro zhoubný nádor.
Kardiopulmonální komplikace
Bleomycin, deriváty nitrosourey, busulfan, celotělové ozáření mohou poškozovat plíce a způsobovat restriktivní plicní poruchu a plicní fibrózu. Kardiální poškození je nejčastěji důsledkem vysoké kumulativní dávky antracyklinových cytostatik (daunorubcin, doxorubicin, mitoxantron), která poškozují buňky srdečního svalu a mohou vést až k srdečnímu selhání. Překročení kumulativní dávky antracyklinů 550 mg/m2 způsobuje poruchu srdeční funkce u minimálně 5 % takto léčených pacientů 6–19 let po léčbě. Vyšší riziko poškození mají malé děti. Ozáření mediastina, jako je tomu u Hodgkinovy nemoci, může poškodit koronární arterie. Pravidelné monitorování zátěžové echokardiografie a EKG je po léčbě antracykliny nutností.
Renální komplikace
Cisplatina, ifosfamid a cyklofosfamid mohou způsobit chronické poškození ledvin, projevující se snížením clearance kreatininu. Alkylační agens vyvolávají obávanou hemoragickou cystitidu. Ifosfamid může způsobit Fanconiho syndrom, který při neadekvátní substituční léčbě hrozí vznikem křivice. Ozáření pánve může poškodit funkci močového měchýře.
Neuropsychologické komplikace
Neuropsychologické pozdní následky jsou nejčastěji důsledkem ozáření mozku v léčbě mozkových nádorů a leukemie. Projevem jsou poruchy pozornosti, neschopnost soustředění, porucha krátkodobé paměti, logického myšlení, řešení matematických úloh. Důsledkem pak jsou školní problémy, poruchy chování, omezené možnosti pracovního zařazení. Riziko vzniku poškození mozku je větší při léčbě u dětí mladších 8 let, kdy je vyvíjející se mozek nejzranitelnější.
Sekundární malignity
Sekundární zhoubné nádory postihují 20 let po léčbě primárního nádoru 3–12 % dětí. Rizikovými faktory pro jejich vznik jsou některá cytostatika (etoposid, cyklofosfamid, antracykliny), radioterapie, primární diagnóza Hodgkinova lymfomu a retinoblastomu, dědičné genetické syndromy zvýšeného výskytu nádorů, jako je Li-Fraumeni syndrom či neurofibromatóza 1. typu. Alkylační agens a etoposid indukují vznik sekundární akutní myeloidní leukemie. Radioterapie vede ke vzniku sarkomů, karcinomů a mozkových nádorů v ozařovacím poli. Pacienti s Hodgkinovým lymfomem mají v 15 letech od diagnózy 8% riziko vzniku sekundárních nádorů. Nejčastěji je postihuje karcinom prsu a karcinom štítné žlázy.
6.0 Transplantace krvetvorných buněk
Transplantace krvetvorných buněk (HSCT = hematopoietic stem cell transplantation), pro kterou je rovněž často užíván termín transplantace kostní dřeně, je používána v léčbě krevních malignit, aplastické anemie, některých vrozených krevních onemocnění, primárních těžkých imunodeficiencí, dědičných poruch metabolismu a několika solidních nádorů. Indikací HSCT u těchto onemocnění je selhání nebo malá úspěšnost jiné léčby (chemoterapie u maligních onemocnění) nebo skutečnost, že jiná účinná léčba dosud chybí (imunodeficience, dědičné poruchy metabolismu).
Alogenní HSCT využívá krvetvorné kmenové buňky kostní dřeně, periferní krve nebo placentární krve zdravého rodinného nebo nepříbuzného dárce, shodného s pacientem v základních znacích HLA systému. HLA antigeny I. třídy (HLA-A, B, C) a II. třídy (HLA-DR, DQ) hrají klíčovou roli v rejekci štěpu a rozvoji reakce štěpu proti hostiteli. Každé dítě dědí jednu sadu HLA antigenů od matky a jednu od otce. Pravděpodobnost shody v antigenech I. a II. třídy je mezi sourozenci 25%. Autologní transplantace využívá pacientovy vlastní kmenové buňky kostní dřeně nebo periferní krve.
Principem alogenní HSCT je podání vysoké dávky chemoterapie, často v kombinaci s radioterapií v předtransplantační přípravě za účelem zničit vlastní normální a abnormální krvetvorbu (myeloablace), vytvořit „prostor“ v mikroprostředí kostní dřeně, umožňující dárcovým kmenovým buňkám proliferovat a dosažení účinné imunosuprese (lymfoablace), umožňující přihojení štěpu. Takto intenzivní předtransplantační příprava má za následek zvýšené riziko výskytu bakteriálních, virových a plísňových infekčních komplikací v potransplantačním období. Dalšími významnými komplikacemi alogenní transplantace jsou rejekce štěpu, reakce štěpu proti hostiteli – graft-versus-host disease (GVHD) a relaps nemoci po transplantaci. Morbidita a mortalita způsobené potransplantačními komplikacemi limitují využití alogenní HSCT na onemocnění neslučitelná s dlouhodobým přežitím. Pokroky v HLA typizaci, předtransplantační přípravě a podpůrné léčbě umožňují rozšíření indikačního spektra HSCT. Příkladem je rozvoj transplantací s redukovanou intenzitou předtransplantační přípravy, které jsou vhodné zejména pro nemaligní onemocnění a vyznačují se sníženou potransplantační morbiditou i mortalitou.
V posledních deseti letech došlo k prudkému rozvoji transplantace od nepříbuzných dárců díky zavedení molekulárních metod do vyšetření HLA systému, což umožňuje dosáhnout lepší shody v HLA mezi příjemcem a dárcem a snížit tak potransplantační komplikace, zejména výskyt GVHD. V současné době je ve světových registrech dárců kostní dřeně registrováno více než 10 milionů dobrovolníků. Dalším významným zdrojem krvetvorných buněk je zejména pro dětské pacienty placentární krev, odebraná z pupečníkových cév po porodu placenty, která obsahuje velké množství nezralých imunokompetentních buněk, schopných krvetvorby v příjemcově dřeni. Tyto transplantace jsou zatíženy menším rizikem GVH reakce a umožňují použít placentární štěp i při neúplné shodě v HLA. Nevýhodou je pozdější přihojení a možnost použití pouze pro dětské pacienty pro malý obsah krvetvorných buněk.
Předtransplantační příprava
Samostatné použití celotělového ozáření nebo cyklofosfamidu mělo za následek vysoký výskyt relapsů leukemie po transplantaci. Kombinace obou léčebných modalit toto riziko významně snížila. Alternativou této přípravy je od 70. let busulfan. Předtransplantační příprava by měla splnit 3 podmínky: 1) ablace příjemcovy kostní dřeně s vytvořením prostoru pro proliferaci dárcových krvetvorných buněk, 2) imunosuprese příjemce za účelem prevence rejekce štěpu, 3) eradikace nádorových buněk. Intenzita předtransplantační přípravy závisí na typu základního onemocnění. Pacienti s aplastickou anemií a těžkými kombinovanými imunodeficiencemi mohou být léčeni méně intenzivně s dobrým efektem transplantace. Cyklofosfamid zajistí u aplastické anemie účinnou imunosupresi bez nutnosti ablace již aplastické dřeně. Pacienti s kombinovanou těžkou imunodeficiencí mají snížené riziko nepřihojení štěpu a mohou dokonce úspěšně podstoupit transplantaci bez předtransplantační přípravy. Naopak pacienti s pokročilými typy myelodysplastického syndromu (RAEB, RAEBt) těží z intenzivní předtransplantační přípravy, například s použitím 3 alkylačních agens – busulfanu, cyklofosfamidu a melfalanu. Transplantace pro ALL je spojena s méně intenzivním GVL efektem (efekt štěpu proti leukemii) ve srovnání s CML i AML. Intenzivní předtransplantační příprava je u ní nutností.
Předtransplantační příprava se sníženou intenzitou nabývá na významu pro starší pacienty. Nadějná ale může být svou redukcí rizika pozdních následků i pro děti. Velmi často je využíván fludarabin, který má antileukemický efekt a je účinným imunosupresivem. Je kombinován s melfalanem či nízkou dávkou busulfanu. Tento typ předtransplantační přípravy je schopen zajistit přihojení štěpu a generuje efekt štěpu proti nádoru.
Předtransplantační příprava u autologní tranplantace nemusí zajistit imunosupresi pacienta. Byla vyvinuta řada typů předtransplantační přípravy, např. kombinace BCNU, etoposid, cytosin-arabinosid, melfalan (BEAM), cyklofosfamid, BCNU, cisplatina (CBP) a další.
Závěrem lze konstatovat, že dosud nebylo dosaženo konsensu o optimální předtransplantační přípravě u alogenní i autologní transplantace. Žádný režim nedosáhl jednoznačně lepších výsledků v celkovém přežití nebo v přežití bez relapsu základního onemocnění.
Alogenní HSCT
Optimálním a nejčastějším dárcem krvetvorných buněk pro alogenní transplantaci je HLA identický sourozenec. Se zlepšením metod detekce HLA antigenů, imunosuprese a prevence GVHD jsou stále více využíváni alternativní dárci. Jsou jimi zejména HLA shodní nepříbuzní dárci krvetvorných buněk z registrů dobrovolných dárců kostní dřeně. Jejich HLA znaky jsou vyšetřeny a uloženy v databance národních registrů. Jednotlivé národní registry jsou propojeny počítačovou sítí Bone Marrow Donors Worldwide (BMDW) s centrálou v nizozemském Leidenu. Zadáním HLA znaků pacienta tak lze v krátké době zjistit počet potenciálních dárců krvetvorných buněk v národních i zahraničních registrech. Banky placentární krve vznikají od 90. let. Placentární krev je odebrána po porodu placenty a kryokonzervována. Před tím je provedeno vyšetření na infekční agens a vyšetření HLA znaků. Banka placentární krve existuje i v České republice.
Volba vhodného dárce z registrů se řídí stupněm shody v HLA systému mezi pacientem a dárcem. Čím je větší neshoda, tím je větší riziko rejekce štěpu a reakce štěpu proti hostiteli. V případě shody HLA dárce a příjemce závisí výběr optimálního dárce na pohlaví, počtu těhotenství dárce, věku dárce a nosičství CMV viru. Riziko GVH reakce je zvýšené při volbě dárce ženy, která prodělala více těhotenství, a při vyšším věku dárce. Je-li pacient sérologicky CMV negativní, je dávána přednost CMV negativním dárcům. Šance na nalezení HLA identického nepříbuzného dárce závisí na etnickém původu a složení registrů dárců. Pro bělocha původem ze střední Evropy dosahuje pravděpodobnost nalezení vhodného dárce v registrech 70 %. Není-li nalezen dárce shodný s pacientem minimálně v 8 z 10 hodnocených znaků HLA systému, je vhodnou alternativou použití štěpu placentární krve, kde není pro úspěšnou transplantaci vyžadována úplná shoda mezi dárcem a příjemcem.
Odběr a zpracování štěpu
Krvetvorné buňky kostní dřeně jsou odebírány opakovanými vpichy z pánevních kostí. Optimální množství odebrané dřeňové krve záleží na váhovém poměru mezi příjemcem a dárcem a činí 15–20 ml/kg příjemce. Výkon je pro dárce spojen s nutností celkové anestezie, pooperační bolestí a nutností transfuze krve. Alternativou, která nabývá na významu nejen pro příbuzné dárce, je odběr periferních krvetvorných buněk jejich mobilizací z kostní dřeně dárce růstovými faktory, nejčastěji G-CSF. Odběr periferních buněk je prováděn leukaferézou na separátoru krevních elementů. Podání růstových faktorů dárci může způsobit bolest kostí, leukaferéza způsobuje trombocytopenii. Obě komplikace jsou přechodné, dlouhodobé následky podání růstových faktorů zdravému dárci nebyly pozorovány.
Prevence reakce štěpu proti hostiteli a efekt štěpu proti leukemii, potransplantační komplikace
Nejužívanějším schématem prevence GVHD je v současné době kombinace cyklosporinu A (CsA) s krátkým kurzem metotrexátu. Délka léčby CsA závisí na výskytu GVH reakce a stadiu nemoci. V případě výskytu GVHD léčba CsA pokračuje do jejího zvládnutí. U nepříbuzných transplantací se v prevenci GVHD úspěšně uplatňuje antitymocytární globulin podávaný jako součást předtransplantační přípravy.
Reakce štěpu proti hostiteli se i přes účinnou profylaxi rozvíjí u 20–70 % pacientů po alogenní HSCT. Její výskyt ovlivňují HLA shoda mezi dárcem a příjemcem, typ štěpu, věk pacienta a dárce, pohlaví dárce. Akutní GVHD vzniká v prvních 100 dnech po transplantaci. Prvním projevem je typicky makulopapulózní kožní exantém. Postižení střev vede k sekrečním průjmům, postižení jater k cholestatickému ikteru. Chronická GVHD se objevuje déle než 100 dní po transplantaci a může postihnout řadu orgánů. Skleróza kůže, malabsorpce, hubnutí, mukositida, chronické postižení plic, cholestatický ikterus jsou častými projevy. Léčbou GVHD jsou kortikoidy a další imunosupresiva.
Efekt štěpu proti leukemii (GvL) nebo štěpu proti tumoru (GvT) v případě solidních nádorů je vyvolán aloreaktivitou T lymfocytů dárce, které ničí zbylé nádorové buňky v těle příjemce. V současné době ho nelze oddělit od GVH reakce. Pacienti se silnou akutní a zejména s chronickou GVH nemocí mají nižší výskyt relapsu leukemie po transplantaci než pacienti, u nichž je GVH reakce slabá nebo se vůbec nevyskytne.
V prvních měsících po transplantaci ohrožují imunosuprimovaného pacienta infekční komplikace bakteriální (sepse) a plísňové (aspergillus, candida). Obávané virové infekce vznikají přenosem od dárce nebo reaktivací vlastní latentní infekce. Jedná se zejména o cytomegalovirus, který může být příčinou fatálních pneumonií, a EBV, který hrozí vznikem potransplantační EBV lymfoproliferace. Riziko rozvoje těchto infekcí snižuje preemptivní léčba založená na monitorování hladin viru v krvi molekulárními metodami a včasném zahájení antivirové léčby (ganciklovir u CMV, rituximab u EBV). Profylakticky podávaný acyklovir snižuje výskyt infekcí herpes simplex, cotrimoxazol pak riziko pneumonie vyvolané Pneumocystis jiroveci. Po šestém měsíci od transplantace jsou pacienti ohroženi pneumokokovými infekcemi, infekcemi vyvolanými virem varicella-zoster. Pacienti s chronickou GVHD reakcí mají zvýšený výskyt život ohrožujících infekčních komplikací.
Onemocnění indikovaná k alogenní HSCT
Tři čtvrtiny alogenních transplantací u dětí jsou prováděny u pacientů s krevním maligním onemocněním. HSCT je léčebnou metodou první volby u pacientů s myelodysplastickým syndromem a juvenilní myelomonocytární leukemií. U CML je HSCT i v éře imatinibu indikována v situaci nalezení HLA identického dárce v rodině či v registrech. V situaci pokročilé nemoci či selhání léčby inhibitory tyrosikináz je indikována HSCT i od neidentických dárců. ALL a AML v první remisi je indikací k HSCT výjimečně u pacientů nejvyššího rizika. Naopak relaps ALL a AML je nejčastější indikací HSCT u dětí.
Neonkologická onemocnění tvoří 25 % indikací k alogenní HSCT. Nejčastěji se jedná o získanou aplastickou anemii, méně často o vrozená selhání kostní dřeně, jako je Fanconiho anemie, Diamondova-Blackfanova anemie či dyskeratosis congenita. Talasémie major a těžké formy srpkovité anemie jsou vyléčitelné transplantací kostní dřeně. U těžkých vrozených imunodeficiencí obnoví dodané kmenové buňky normální imunitu. Nejčastějšími onemocněními indikovanými k transplantaci jsou těžká kombinovaná imunodeficience (SCID), Wiskottův-Aldrichův syndrom, primární hemofagocytující lymfohistiocytóza, nepříznivě probíhající chronická granulomatóza. U maligní osteopetrózy dodá dárcovská kostní dřeň funkční osteoklasty a nadměrně tvořená kost je resorbována. Včas provedená transplantace dokáže postiženým dětem i zachránit zrak. HSCT je účinnou léčbou některých lyzozomálních a peroxyzomálních onemocnění. Chybějící enzym je vnesen do organismu buňkami monocytárně-makrofágového systému, které se vyvíjejí z kmenových buněk dárce a nahrazují příjemcovy buňky v játrech, slezině, plicích, ale i v mozku. Progrese onemocnění se zastaví a dochází k reparaci poškozených orgánů. Podmínkou úspěchu je včasné provedení transplantace před rozvojem ireverzibilního poškození mozku. Nejčastějšími nemocemi indikovanými k transplantaci v této skupině jsou mukopolysacharidóza I. typu – nemoc Hurlerové a adrenoleukodystrofie se začátkem v dětském věku.
Autologní HSCT
Hypotéza podporující provádění autologní transplantace předpokládá, že nádorové buňky jsou účinně ničeny vysokou dávkou cytotoxických léků. Protože hlavním limitem výše dávky cytostatika je toxicita pro kostní dřeň v podobě dřeňové aplazie, lze ji oslabit podáním autologních krvetvorných buněk odebraných z kostní dřeně nebo periferní krve před zahájením vysokodávkované terapie. Dostatečně buněčný štěp periferních krvetvorných buněk je získáván mobilizací pacienta podáním vysokodávkovaného cyklofosfamidu s následnou aplikací G-CSF. Alternativou je sběr krvetvorných buněk z krve po bloku intenzivní chemoterapie základního onemocnění s následným podáním růstového faktoru. Sběr buněk je prováděn leukaferézou. Progenitory periferní krve se přihojují rychleji než kostní dřeň.
Studie s využitím genového značení prokázaly, že nádorové buňky obsažené v autologním štěpu jsou schopny vyvolat relaps nemoci po transplantaci. Další příčinou potransplantačního relapsu jsou nádorové buňky v těle pacienta, které předtransplantační příprava nedokázala zničit. Nevýhodou autologní transplantace je chybění GvL efektu, a proto je její přínos v léčbě leukemie diskutabilní.
Autologní transplantace přináší méně komplikací v potransplantačním období než transplantace alogenní. Důvodem je chybění GVH reakce a tím pádem nutnost méně intenzivní imunosuprese, což má za následek nižší výskyt infekčních komplikací.
Indikace k autologní transplantaci jsou v současné době v dětské onkologii omezené. Využívá se v léčbě neuroblastomu vysokého rizika, pokročilých forem Ewingova sarkomu, některých nádorů mozku, časných relapsů Hodgkinova lymfomu a nehodgkinských lymfomů.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2011 Číslo 3
- Horní limit denní dávky vitaminu D: Jaké množství je ještě bezpečné?
- Diagnostický algoritmus při podezření na syndrom periodické horečky
- Isoprinosin je bezpečný a účinný v léčbě pacientů s akutní respirační virovou infekcí
Nejčtenější v tomto čísle
- Skríning kritických vrodených chýb srdca u novorodencov pulznou oxymetriou v regióne severného Slovenska
- Poruchy príjmu potravy z pohľadu pedopsychiatra
- Vybrané kapitoly z dětské onkologie a transplantace kostní dřeně
- Periodické horečky a další syndromy s poruchou regulace zánětlivé odpovědi