
Fabryho choroba v dětském věku – přehled a kazuistika
Fabry disease in childhood – overview and a case report
Fabry disease is a rare lysosomal storage disorder leading to glycosphingolipid accumulation in most of tissues. Its singns and symptoms are quite distinctive, particulary in childhood. It is possible and so imperative to detect the disease at young age and start the enzyme replacement therapy to decrease the risk of late complication development in adulthood. The article offers a brief description of the disease and an illustrative case report of 12-years-old boy.
Keywords:
Fabry disease – childhood – acroparesthesia – angiokeratomas – cornea verticillata – agalsidase – ERT
Autoři:
Munzar Petr 1; Mazurová Stella 2; Dubská Zora 3
Působiště autorů:
Dětská neurologie Pardubice, s. r. o.
1; Klinika pediatrie a dědičných poruch metabolismu, Všeobecná fakultní nemocnice v Praze a 1. lékařská fakulta, Univerzita Karlova, Praha
2; Oční klinika, Všeobecná fakultní nemocnice v Praze a 1. lékařská fakulta, Univerzita Karlova, Praha
3
Vyšlo v časopise:
Čes-slov Pediat 2022; 77 (4): 219-225.
Kategorie:
Kazuistika
doi:
https://doi.org/10.55095/CSPediatrie2022/036
Souhrn
Fabryho choroba je vzácné dědičné metabolické onemocnění s progresivní lyzozomální akumulací glykosfingolipidu ve většině tělesných tkání. Má dobře definované klinické příznaky, které jsou zejména v dětství dosti specifické. Je velmi důležité je odhalit právě již v dětském věku a díky dostupné enzymové substituční terapii minimalizovat rozvoj závažných pozdních komplikací zvyšujících morbiditu i mortalitu v dospělosti. Článek přináší stručný popis onemocnění a názornou kazuistiku dětského pacienta.
Klíčová slova:
dětství – Fabryho choroba – akroparestezie – angiokeratomy – cornea verticillata – agalsidáza – ERT
Úvod
Fabryho choroba (FCh) je vzácné na X chromozom vázané metabolické onemocnění s progresivní lyzozomální akumulací glykosfingolipidů ve většině tělesných tkání, zejména endotelu cév a buňkách hladkého svalstva. Onemocnění známé též jako Andersonova–Fabryho choroba poprvé popsali nezávisle na sobě dermatolog J. Fabry (Německo, 1898 a 1930) a chirurg a anatom W. Anderson (Anglie, 1898) na konci devatenáctého století. Až v 70. letech 20. století byla zjištěna příčinná souvislost onemocnění s deficitem α - -galaktosidázy A.
Epidemiologie
Jedná se o druhé nejčastější lyzozomální střádavé onemocnění (po Gaucherově chorobě) s incidencí udávanou mezi 1 : 40 000 až 1 : 117 000 narozených chlapců, bez etnických vazeb.(1) Prevalence v ČR je u mužů 0,57 a u žen 0,77 případu na 100 000.(2) Prevalence na základě screeningu novorozenců v USA byla přibližně 1 : 3000 chlapců.(3,4) Důvodem této řádově vyšší incidence byl záchyt celého spektra fenotypů oproti původním studiím zachycujícím většinou jen fenotyp klasický.
Patogeneze
Příčinou onemocnění je mutace genu GLA, kódujícího α-galaktosidázu A. Gen obsahující 7 exonů (12 436 párů bazí) je lokalizován na dlouhém raménku chromozomu X (Xq22.1). Dosud bylo popsáno více než 1000 patogenních variant, z nichž většina je unikátních, vyskytujících se jen v dané rodině. Některé mutace (zejména missense) jsou spojované s pozdním nástupem onemocnění.(5,6) Jasná korelace mezi genotypem a fenotypem FCh však obecně není. I v rámci jednotlivých rodin se může fenotyp lišit v závislosti na pohlaví, věku nástupu onemocnění a dalších genetických i epigenetických faktorech. U žen přenašeček se navíc uplatňuje fenomén náhodné inaktivace X chromozomu (lyonizace), resp. nevyvážené inaktivace. Spektrum příznaků tedy sahá od asymptomatických (heterozygotek) po těžce nemocné s multiorgánovým postižením.
Dysfunkční enzym znemožňuje katabolismus α-galaktosylových částí glykolipidů, glykoproteinů a oligosacharidů buněčných membrán. Dochází tak k akumulaci glykosfingolipidů, především globotriaosylceramidu (Gb3), v lyzozomech buněk. Patofyziologickým korelátem orgánových manifestací je zejména deacylovaná forma globotriaosylceramidu – globotriaosylsfingosin (lyso-Gb3), jehož detekce slouží jako biomarker aktivity onemocnění.(7,8) Progresivní střádání glykolipidů v mnoha tělesných tkáních (zejména v endotelu, hladkém svalstvu, podocytech, kardiomyocytech, buňkách autonomního nervového systému) spouští kaskádu cytotoxických, zánětlivých, fibrotizujících procesů, vede k apoptóze buněk postižených tkání, působí vaskulární dysfunkci a protrombotický stav.(9,10) Vaskulární dysfunkce spolu s autonomní neuropatií vedou k poruše vazokonstrikce a vazodilatace. Tak je vysvětlována intolerance změn teploty. Autonomní neuropatie a chronická neuropatická bolest jsou způsobeny zejména depozicí Gb3 v autonomních gangliích a Schwannových buňkách. Podobným mechanismem vzniká senzorineurální porucha sluchu a tinnitus. Patogeneze renálního postižení je komplexní, předpokládá toxické působení Gb3 na všechny typy buněk.(3) Ještě před rozvojem snížení glomerulární filtrace je možno detekovat depozita Gb3 v téměř všech typech buněk ledvin (podocyty, tubulární i intersticiální buňky).
Klinické projevy
Klasická varianta Fabryho choroby (charakterizovaná nízkou aktivitou α-galaktosidázy A, často pod 1 %) začíná kolem 10. roku věku (u chlapců ve věku 9,5 ± 5,1; u dívek 9,7 ± 6,3 roku)(10) rozvojem akroparestetických krizí (epizodických neuropatických bolestí), hypohidrózy a angiokeratomů. Ve druhé dekádě se přidává hypertrofická kardiomyopatie a první projevy renálního postižení (mikroalbuminurie). Od třetí dekády progreduje postižení ledvin, srdce i CNS vedoucí k závažné morbiditě.(5,12)
Formy s pozdním nástupem (mezi 3. a 7. dekádou) mívají mírnější průběh s dominujícími orgánově specifickými projevy, a to díky částečně zachovalé aktivitě α-galaktosidázy A (2–30 %).
Stanovení diagnózy a zahájení léčby bývají významně opožděny. V mezinárodní studii zahrnující 151 účastníků dětského věku byla diagnóza stanovena průměrně za 2,8 roku od nástupu příznaků a léčba zahájena za další 2,6 roku.(13) V jiných souborech je běžné stanovení diagnózy za 10–12 let od prvních příznaků.(11)
Neléčená FCh zkracuje průměrnou očekávanou délku života o 15–20 let.(14)
Fabryovské krize (akroparestezie) a neuropatie tenkých vláken
Ataky minuty až dny trvajících bolestivých parestezií aker provokované námahou, horečkou, stresem nebo změnami teploty okolí jsou vysvětlovány zejména dysfunkcí tenkých myelinizovaných Aδ-vláken, C-vláken a poruchou vazomotoriky. Mohou se rozvíjet již od 3 let věku. Neuropatie tenkých vláken může působit i trvalé neuropatické bolesti, dysestezie, poruchy termického čití, pocení a gastrointestinální motility. Akroparestezie se s věkem zmírňují, nebo i vymizí. Neuropatická bolest může být trvalá.
Angiokeratomy
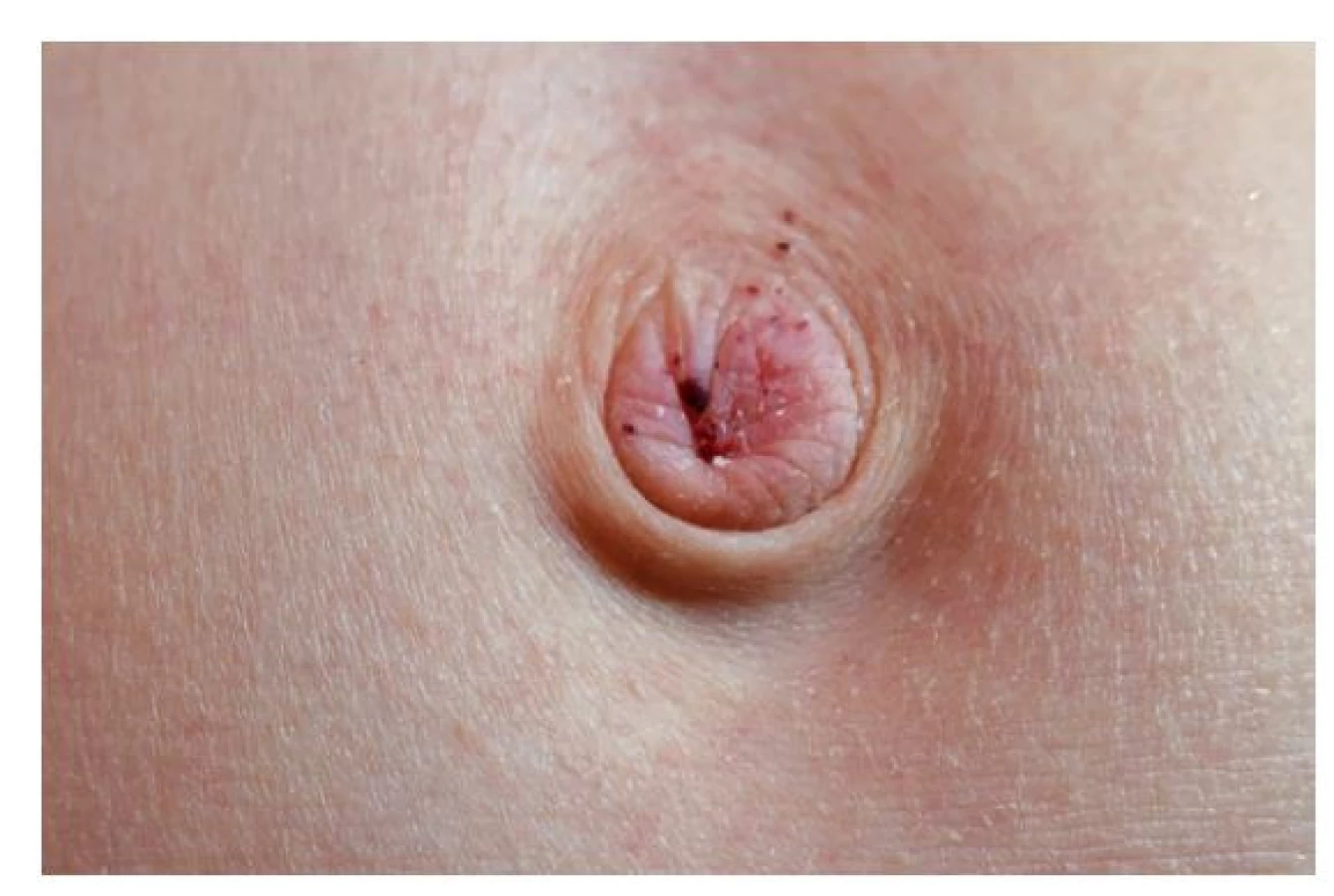
Jedná se o drobné tmavočervené kožní útvary (obr. 1 a 2) tvořené rozšířenými tenkostěnnými cévami dermis kryté hyperkeratotickou epidermis. Nejsou nálezem specifickým pro FCh. Můžeme se s nimi setkat i u několika dalších geneticky podmíněných onemocnění, ale i u pacientů léčených nízkomolekulárními hepariny.(15) Pro FCh je však relativně charakteristická jejich lokalizace v oblasti umbiliku, hýždí, hypogastria, třísel a genitálu; vzácněji v oblasti loktů, kolen a rukou.(16) Objevují se v první i druhé dekádě věku a jejich počet i velikost s věkem narůstá.

Hypohidróza
Snížená či chybějící potivost je popisována rodiči často již od 3 let věku, ústí v intoleranci tepla. Bývá spojena s recidivujícími subfebriliemi bez zjištěné příčiny. Je vysvětlována kombinací mechanické obstrukce potních žláz Gb3 a sympatické sudomotorické dysfunkce. Podstatně vzácněji je popisována i hyperhidróza.

Gastrointestinální obtíže
Autonomní neuropatie v kombinaci s akumulací Gb3 v hladké svalovině gastrointestinálního traktu se může projevit průjmem, zácpou, nauzeou, zvracením a bolestmi břicha. Na bolestech břicha se může podílet splenomegalie, méně často hepatomegalie. Známá je i dystrofizace a růstové opoždění v důsledku malabsorpce.(17)
Oční příznaky
Oční příznaky se u FCh objevují časně, a mohou dokonce na onemocnění upozornit. Charakteristickým nálezem je cornea verticillata (obr. 3). Jedná se o nahnědlá, případně našedlá depozita v epitelu rohovky a Bowmanovy membrány tvořící skvrny nebo linie podobající se víru či kočičím vousům. Přítomna bývá u téměř všech postižených mužů a u 70 % žen přenašeček. V dětském věku je nálezem relativně specifickým pro FCh, v dospělosti se s cornea verticillata setkáváme i u pacientů léčených amiodaronem, chlorochinem, indomethacinem, fenothiaziny a též po radiální keratotomii.(12) Dalšími příznaky mohou být vinuté cévy spojivky (obr. 4) i očního pozadí, změny v pigmentovém epitelu sítnice (detekovatelné pomocí autofluorescenčního filtru) a loukoťovité zákaly čočky (pod jejím předním i zadním pouzdrem).(18)


Postižení ledvin
Projevuje se úvodní glomerulární hyperfiltrací, následovanou albuminurií (ve 2. a 3. dekádě), snížením glomerulární filtrace a finální renální insuficiencí. Patologické hodnoty glomerulární filtrace je možno prokázat u 17 % pacientů do 18 let věku.(19)
Postižení myokardu
Hlavní komplikací je hypertrofie levé komory a zkrácení PQ intervalu.(16) Častěji (14,9 %) jsou v dětském věku popisovány i chlopenní vady.(11) V dospělosti se objevují komorové i supraventrikulární tachyarytmie, převodní poruchy a chlopenní insuficience. Kardiologické komplikace jsou hlavní příčinou předčasné smrti dospělých pacientů s FCh. 10–20 % pacientů vyžaduje v dospělosti trvalou kardiostimulaci.(16)
Cerebrovaskulární onemocnění
Dominantním projevem jsou ischemické cévní mozkové příhody (iCMP) a tranzitorní ischemické ataky (TIA), které jsou v dětském věku naštěstí vzácné.(6,20) I u dětí se však setkáváme s nespecifickými T2/FLAIR hypersignálními lézemi bílé hmoty. Mohou být izolované, mnohočetné i splývající; lokalizované subkortikálně, periventrikulárně i v hloubi bílé hmoty centrum semiovale.(19) Jsou často asymptomatické a s věkem jich přibývá. Data z amerických registrů FCh udávají výskyt CMP ve věkové kategorii 0–25 let 0,32 (u mužů), resp. 0,18 (u žen) případů na 1000 osob za rok. Pro srovnání ve věku 55–65 let se mrtvice vyskytovala s incidencí 22,05 u mužů, resp. 6,30 u žen případů na 1000 osob za rok.(21) Již u adolescentů se můžeme setkat s kognitivními poruchami s exekutivní dysfunkcí.
Diagnostika
Na základě anamnézy, klinických příznaků a laboratorních nálezů je možné pomýšlet na diagnózu Fabryho nemoci. V laboratoři mohou být přítomny známky střádání v retikuloendoteliálním systému: trombocytopenie, anemie, méně často leukopenie; dále elevace ferritinu, kyselé fosfatázy, laktátdehydrogenázy a zejména chitotriosidázy.
Specifickým biomarkerem je výše zmiňovaný lyso-Gb3, který je možné detekovat v plazmě i moči. Koncentrace Gb3 v plazmě je využívána k monitorování léčby.(6)
Podezření na Fabryho nemoc je možné potvrdit enzymologickým vyšetřením aktivity α-galaktosidázy v suché krevní kapce (u mužů) a dále v séru/plazmě či v izolovaných leukocytech periferní krve (u mužů i žen přenašeček). Definitivním potvrzením diagnózy je nález patogenní varianty v genu GLA.
K odhalení sekundárních komplikací využíváme dále EKG, echokardiografii, vyšetření moči (proteinurie, mikroalbuminurie, glomerulární filtrace), MRI a MRA mozku, oční vyšetření, ORL vyšetření včetně audiometrie, dermatologické vyšetření.(22,23) V některých zemích byl zaveden novorozenecký screening, jenž povede k časnější diagnóze a již nyní odhaluje podstatně vyšší incidenci FCh.(1)
Diferenciální diagnostika
Akroparestezie a jejich neobvyklé provokační mechanismy mohou lékařům připadat nevysvětlitelné, bizarní. Proto jsou často považovány za psychogenní příznaky v nejširším slova smyslu. U dětí jsou někdy tyto obtíže nesprávně hodnoceny jako růstové bolesti. Bez kontextu s klinikou je snadné zaměnit T2/FLAIR hyperintenzní léze na MRI za plaky roztroušené sklerózy. Diferenciální diagnostika angiokeratomů a cornea verticillata byla stručně zmíněna výše. Rovněž gastrointestinální a renální projevy je třeba klást do souvislosti s dalšími projevy systémového onemocnění a v případě nezjištěné příčiny uvažovat i o Fabryho chorobě. Tato problematika náleží specialistům jednotlivých oborů.
Léčba
Léčba Fabryho nemoci je založena především na enzymové substituční terapii (ERT – enzyme replacement therapy). Využívá rekombinantní agalsidázy α (Replagal®), případně agalsidázy β (Fabrazym®). Enzym je v doporučené dávce aplikován intravenózně ve dvoutýdenních intervalech. Již v prvních měsících po zahájení léčby je možné pozorovat snížení únavy a postupnou úpravu hematologických a dalších laboratorních parametrů, ustupuje hepatosplenomegalie, zlepšují se renální funkce, regreduje hypertrofie levé komory; po 3–4 letech léčby dochází ke stabilizaci neuropatické bolesti. Terapie zabraňuje dlouhodobé progresi orgánových komplikací (především srdečních a renálních), hůře ovlivňuje neurologické příznaky (zejména neuropatickou bolest). Intenzita i frekvence bolestivých krizí se po zahájení terapie obvykle snižují.(24) Nevýhodou ERT je nutnost její celoživotní aplikace a vysoká cena. Další možností terapie jsou tzv. chaperony, u FCh se užívá migalastat (Galafold®). Tato malá molekula se aktivně váže na enzym a pomáhá jeho stabilizaci a transportu do lyzozomu. Jedná se o perorální léčbu, kterou lze však využít jen pro nositele mutací způsobujících chybné sbalení (a tudíž nestabilitu) primárního proteinu. Takový genotyp může mít až 48 % pacientů.(24) Je schválena pro pacienty starší 16 let.(26,27) Ve fázi výzkumu je i cílená genová terapie.
Kvalitu života negativně ovlivňují ataky akroparestezií. Ve většině případů vede přerušení fyzické aktivity k rychlému ústupu těchto záchvatů. Pacienti se sami vyhýbají známým provokačním faktorům (expozice horku, slunci, intenzivní fyzické zátěži). Při nedostatečném efektu režimových opatření jsou v léčbě akutních krizí užívána analgetika-antipyretika a nesteroidní antiflogistika, méně často lidokainové či kapsaicinové náplasti a opioidy.
Již v dětském věku může být hendikepující rozvoj chronické neuropatické bolesti (perzistující či frekventně rekurentní). V léčbě je u dětí doporučován gabapentin, duloxetin, amitriptylin, karbamazepin, někdy i fenytoin.(1,12,17,23,28)
K ovlivnění těžké hypertenze či renálního postižení s mikroalbuminurií jsou podávány ACE inhibitory.
Gastrointestinálním příznakům mohou ulevit dietní opatření (menší a častější porce) a prevence zácpy.
Kazuistika
Dvanáctiletý mladík byl odeslán praktickou lékařkou pro děti a dorost do ambulance dětské neurologie pro opakující se epizody pálení plosek nohou a dlaní rukou.
Chlapec z gravidity po IVF, narozen ve 36. gestačním týdnu bez další perinatální zátěže. Jeho psychomotorický vývoj postupoval zcela fyziologickým tempem. V 7 letech podstoupil adenotomii. Od předškolního věku docházel na logopedii pro vývojovou dysfázii. Je sledován pedagogicko - psychologickou poradnou pro horší výkony ve škole. V 9,5 letech vyšetřován pro bolesti obou kyčlí, pro něž nemohl chodit: vyloučena Perthesova choroba; na základě MRI obrazu subchondrálního edému uzavíráno jako levostranná sakroileitida (zpětně hodnoceno jako obtíže nesouvisející s FCh). Ve 12,5 letech věku byl jednorázově vyšetřen endokrinologem pro malý vzrůst – shledán mírně opožděný kostní věk, bez prokázané endokrinopatie. V kojeneckém i batolecím věku dobře prospíval, ve školním věku již nikoli – je nejmenší ze třídy, hmotností i BMI pod 3. percentilem, výškou na 3. percentilu. Jinak zdráv.
Od 9 let si stěžuje na opakované pálivé parestezie, které popisuje i jako pocit vibrace lokalizovaný v oblasti plosek a prstů obou nohou, někdy též současně dlaní a prstů obou rukou. Frekvence epizod postupně stoupá, v letních měsících až ob den. Jejich trvání je proměnlivé od 1 hodiny až po dobu 2 dnů. Bezprostředním prodromem bývá pocit horka v oblasti hlavy a chladu v oblasti rukou. Objektivně jsou akra nejprve chladná a následně naopak horká, což bývá někdy provázeno vzestupem teploty (do 38 °C). Teploty klesají po podání paracetamolu nebo ibuprofenu. Na bolestivé parestezie však tyto léky vliv nemají. Ataky jsou pravidelně provokovány fyzickou aktivitou (běhání při tělocviku) nebo změnami okolní teploty, a to jak rychlým přechodem z chladu do horka, tak i naopak. Dokonce si povšiml, že epizody jsou provokovány nápadně častěji při pohybu za slunečného dne v černé obuvi. Trvání obtíží zkrátí a jistou úlevu v intenzitě přinese elevace končetin a jejich ochlazování proudem vzduchu (ventilátor). Udává hypohidrózu – téměř se nepotí. Chlapec vnímal velmi nepříjemně i skutečnost, že mu nikdo, kromě matky, jeho obtíže nevěřil. To komplikovalo vztahy s učiteli i spolužáky a kamarády. Současně (rovněž asi od 9 let) pozoruje tečkovitý červený exantém v oblasti skrota a umbiliku (obr. 1 a 2), který byl hodnocen jako petechie. Příčina jejich tvorby nebyla zjištěna.
Po prvním ambulantním vyšetření jsme doplnili následující pomocná vyšetření: UZ vyš. tepen dolních končetin s normálním nálezem; kardiologické vyšetření s nálezem akcidentálního šelestu při vazivové struně v levé komoře; laboratorní vyšetření (glukóza, kompletní mineralogram, Fe, ferritin, urea, kreatinin, transaminázy, CK, CRP, kys. močová, bilirubin, celk. bílkovina, FW, moč včetně sedimentu; krevní obraz; aPTT, INR, D-dimery) bez patologických hodnot. Dermatologické vyšetření potvrdilo podezření, že se jedná o angiokeratomy v oblasti umbiliku a skrota. Enzymologické vyšetření aktivity α-galaktosidázy A v suché krevní kapce a následně i v plazmě a leukocytech potvrdilo významné snížení aktivity tohoto enzymu; v plazmě chlapce 0,11 nmol/h/ml (norma 2,40–11,30). Dále bylo doplněno vyšetření molekulárně genetické, jež prokázalo hemizygotní patogenní variantu v GLA genu: GLA (NM_000169.2): c.[1133G>T];[0] Tato missense mutace byla již popsána,(28) není však nikterak častá.
K dalšímu vyšetření a péči byl pacient odeslán na Kliniku pediatrie a dědičných poruch metabolismu VFN v Praze. Tam doplněno UZ vyš. břicha s nálezem hraničně větší velikosti jater i sleziny, bez jednoznačných změn echogenity. Prokázána byla i zvýšená aktivita chitotriosidázy v séru 188,0 nmol/l (norma: 4,4–89,0) a mírná elevace kyselé fosfatázy – jakožto biomarkerů lyzozomálního střádání. Nebyla prokázána porucha renální funkce (albumin v moči, albumin/ kreatinin v moči; cystatin C).
Neurologický nález byl zcela normální s výjimkou poruchy pocení (hypohidróza). Bez průkazu poruchy termického/ algického čití. MRI mozku s normálním nálezem. Normální sluch podle audiometrie. Oftalmolog popsal jemně vinuté cévy spojivky i očního pozadí, bez obrazu cornea verticillata. U matky i sestry pacienta však tato charakteristická dystrofie rohovky byla přítomna (obr. 3 a 4).
Ke správné diagnóze nás navedla i rodinná anamnéza, neboť matka (42 let) sama trpěla ve věku od 10 do 17 let zcela shodnými paroxysmálními akroparesteziemi. V perigenitální oblasti má rovněž angiokeratomy. V adolescenci byla vyšetřována ve fakultní nemocnici, včetně provokačních chladových testů s odběry kapilární krve. Příčina obtíží nenalezena. Dle sdělení maminky uzavíráno jako „od srdce nebo od páteře“, nejspíše však psychogenní etiologie. Ke konci gravidity byla matka vyšetřována pro proteinurii. V dospělosti u ní došlo k rozvoji supraventrikulární arytmie, námahové dušnosti (od 41 let věku), hypohidrózy a oboustranného tinnitu (od 36 let věku). Aktivita α-galaktosidázy A v plazmě matky byla významně snížena: 0,74 nmol/h/ml (norma 2,40–11,30). Matka matky i jediná sestra matky jsou rovněž sledovány pro arytmie. Otec matčiny matky (pradědeček chlapce) zemřel v 60 letech na srdeční selhání. Mladší sestra našeho pacienta je sledována pro vývojovou dysfázii, jinak zdráva. Jiné sourozence nemá. U dalších členů rodiny se žádné suspektní příznaky nevyskytují.
Bylo potvrzeno přenašečství pro tutéž patologickou sekvenční variantu u matky, u níž byla dále diagnostikována kardiomyopatie a cerebrální vaskulopatie. I u ní byla zahájena substituční léčba. Přenašečství familiární varianty potvrzeno i u mladší sestry pacienta, která byla dosud bez subjektivních příznaků, oftalmolog však nově diagnostikoval cornea verticillata. Tatáž varianta v GLA genu byla nalezena i u matčiny matky (obr. 5). Můžeme se domnívat, že výše uvedený pradědeček chlapce zemřel na srdeční selhání podmíněné kardiomyopatií při FCh.

Diskuse
Vlastní stanovení diagnózy nám výrazně usnadnil sám pacient dokonalým vylíčením svých stesků, které v kontextu s rodinnou anamnézou umožnily na Fabryho chorobu pomýšlet již při prvním vyšetření. Vodítkem byla kombinace typických akroparestezií s angiokeratomy a současně shodné obtíže u matky (u níž se navíc projevily arytmie, proteinurie a tinnitus). Při podrobnějším rozboru rodinné anamnézy, který následoval v rámci dalších vyšetření, bylo možno vystopovat suspektní příznaky i u několika dalších příbuzných. X-vázaný vzorec dědičnosti může být užitečnou pomůckou v diagnostice.
Díky výborné spolupráci s Metabolickým centrem a KPDPM VFN v Praze byla indikována, schválena a do 10 týdnů od stanovení diagnózy zahájena enzymatická substituční terapie i.v. podávaným rekombinantním enzymem agalsidázou α (Replagal®) ve dvoutýdenních intervalech. Již po aplikaci prvních 4 dávek došlo ke dramatickému zlepšení kvality života chlapce. Frekvence epizod klesla na 1–2 příhody za měsíc, výrazně se snížila intenzita pálení, žádná další ataka netrvala déle než 1 hodinu. Subfebrilie se během atak rovněž již nevyskytují. Léčbu toleruje výborně. Na gastrointestinální obtíže si nikdy nestěžoval. Po zahájení léčby však začal podstatně více a s větší chutí jíst, přibral na váze, což je hodnoceno maminkou i pediatry velmi pozitivně. Očekáváme i akceleraci jeho růstu. Rodina i chlapec popisují celkově větší čilost, více životní energie, zlepšení školního prospěchu.
Vzhledem k širokému spektru fenotypů (od bezpříznakových průběhů po multiorgánové selhání) je nezbytné u každého pacienta s diagnostikovanou Fabryho chorobou vyšetřit a sledovat vždy funkce všech orgánů, které mohou být postiženy. Pomůckou mohou být markery tkáňové léze (tab. 2). Zahájení ERT v dětství a mladé dospělosti je prokazatelně sdruženo s výrazně pomalejší progresí postižení srdce i ledvin u Fabryho choroby.(4,13,17) U dospělých se doporučuje opakovat MRI mozku 1× za 3 roky (T1, T2/FLAIR, DWI). U mužů starších 21 let a žen starších 30 let je doporučována MR-angiografie.(20) Pro dětský věk zatím žádná doporučení k monitoraci centrální nervové soustavy MRI nebyla stanovena.

Závěr
Fabryho choroba je vzácné střádavé onemocnění s nepřehlédnutelnými příznaky, jejichž kombinace je vysoce specifická a měla by vést k diagnóze. V dětském věku, kdy jsou příznaky nejtypičtější, může na Fabryho chorobu upozornit kombinace recidivujících bolestivých akroparestezií provokovaných fyzickou aktivitou nebo změnou teploty okolí a angiokeratomů v kaudální polovině břicha a na genitálu. Podezření by měly vzbudit i recidivující gastrointestinální obtíže, hypohidróza či netolerance fyzické zátěže a typický oční nález (cornea verticillata). Každá nekardioembolizační ischemická cévní mozková příhoda v dětství či mladé dospělosti by rovněž měla být indikací k provedení screeningového testu v suché krevní kapce. V dospělosti se mohou k uvedeným příznakům přidat zejména chronická renální insuficience a hypertrofická kardiomyopatie.
Zásadní změnu v životech pacientů s tímto onemocněním přinesla možnost enzymatické substituční terapie, která dramaticky změnila prognózu i kvalitu života nemocných. Jedná se o mimořádně nákladnou centrovou léčbu dostupnou v naší zemi již od roku 2004.
V České republice bylo k srpnu 2021 sledováno 190 dospělých (hemizygotů i heterozygotek) s FCh. 90 z nich je léčeno intravenózní či perorální léčbou (II. interní klinika VFN, Praha). Dětských pacientů pak bylo diagnostikováno 20 (6 chlapců, 14 dívek). Čtyři chlapci jsou v současnosti léčeni intravenózní ERT (Klinika pediatrie a dědičných poruch metabolismu VFN, Praha).
Diagnóza bývá nejčastěji stanovena kardiologem, nefrologem a pediatrem, méně často neurologem, vzácně dermatologem, oftalmologem či jiným odborníkem.(29) Pacienti s Fabryho chorobou vyžadují interdisciplinární přístup s účastí pediatra/ internisty, neurologa, biochemika, kardiologa, nefrologa, oftalmologa, dermatologa, otorhinolaryngologa, genetika, psychologa, psychiatra, fyzioterapeuta i dalších odborníků.
Korespondenční adresa:
MUDr. Bc. Petr Munzar
Dětská neurologie Pardubice, s.r.o.
K. Šípka 282
53002 Pardubice
Zdroje
1. Dutra-Clarke M, et al. Variable clinical features of patients with Fabry disease and outcome of enzyme replacement therapy. Mol Gen Met Rep 2021; 26 : 1–11.
2. Poupětová H, et al. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33 : 387–396.
3. Olivera-González S, Josa-Laorden C, Torralba-Cabeza, MA. The pathophysiology of Fabry disease. Rev Clin Esp 2018; 218 (1):22–27.
4. Ellaway C. Paediatric Fabry disease. Transl Pediatr 2016; 5(1): 37–42.
5. Hirashio S, Kagawa R, Tajima G, Masaki T. A classic variant of Fabry disease in a family with the M2961 late-onset variant. CEN Case Rep 2021; 10(1): 106–110.
6. Michaud M, et al. When and how to diagnose Fabry disease in clinical practice. Am J Med Sci 2020; 360(6): 641–649.
7. Aerts JM, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. PNAS 2008; 105(8): 2812–2817.
8. Putko BN, et al. Anderson-Fabry cardiomyopathy: prevalence, pathophysiology, diagnosis and treatment. Heart Fail Rev 2015; 20 : 179–191.
9. Kok K, et al. Fabry Disease: Molecular Basis, Pathophysiology, Diagnostics and Potential Therapeutic Directions. Biomolecules 2021; 11(2): 271.
10. Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Molecular Genetics and Metabolism. 2017; 3(122): 19–27.
11. Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019; 96(2): 107–117.
12. Hewavitharana H, et al. Cornea verticillata in classical Fabry disease, first from Sri Lanka: a case report. BMC Pediatrics 2020; 20 : 338–341.
13. Parini R, et al. Analysis of renal and cardiac outcomes in male participants in the Fabry outcom survey starting agalsidase alfa enzyme replacement therapy before and after 18 years of age. Drug Des Devel Ther 2020; 14 : 2149–2158.
14. Špalek P. Orphan drugs v liečbe zriedkavých neuromuskulárních chorôb. Neurol Praxi 2021; 22(2): 104–112.
15. Lacina L, Kodet O, Štork J. Angiokeratom a stavy doprovázené jeho mnohočetným výskytem. Dermatol Praxi 2017; 11(3): 110–114.
16. Eng CM, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006; 8(9): 539 – 48.
17. Hopkin RJ, et al. The management and treatment of children with Fabry disease: A United States-based perspective. Mol Genet Metab 2015.
18. Dubská Z, Sedláková K, Becková J, et al. Cornea verticillata a význam jejího odhalení u Fabryho choroby. Acta Medicinae 2019; 15 : 77–78.
19. Tapia D, Kimonis VJ. Stroke and chronic Kidney disease in Fabry disease. J Stroke Cerebrovasc Dis. 2021; 30 : 105423.
20. Ortiz A, Germain DP, Desinch RJ, et al. Fabry disease revised: management and treatment recommondations for adult patients. Mol Genet Metab 2018; 123(4): 416–427.
21. Sims K, et al. Stroke in Fabry Disease Frequently Occurs Before Diagnosis and in the Absence of Other Clinical Events. Natural History Data From the Fabry Registry. Stroke 2009; 40 : 788–794.
22. Mena Rodríguez AL, et al. Histopathological findings in renal biopsies in Anderson–Fabry disease. Case series 2018; (81)4 : 243–247.
23. Rekova P, et al. Fabryho choroba, přehled problematiky a nejčastější neurologické projevy. Cesk Slov Neurol N 2018; 81(2): 156–163.
24. Nowak A, et al. Fabry disease genotype, phenotype, and migalastat amenability: Insights from a national cohort. J Inherit Metab Dis 2020; 43(2): 326–333.
25. Sasa H, Nagao M, Kino K. Safety and effectiveness of enzyme replacement therapy with agalsidase alfa in patients with Fabry disease: Post-marketing surveillance in Japan. Mol Genet Metab 2019; 126(4): 448–459.
26. McCafferty, et al. “Migalastat: A Review in Fabry Disease.” Drugs 2019; (79)5 : 543–554.
27. Goláň L. Migalastat v terapii Fabryho choroby. Interní Med 2017; 19(3): 167–170.
28. Sakuraba H, et al. Fabry disease in Japanese population – molecular and biochemical characteristics. Mol. Genet Metab Rep. 2018; 17 : 73 – 79.
29. Huges DA, Auiar P, Deegan PB, et al. Early indicators of disease progression in Fabry disease that may indicate the need for disease-specific treatment initiation: findings from the opinion-based PREDICT-FD modifies Delphi consensus initiative. BMJ Open 2020; 10.
Štítky
Neonatologie Pediatrie Praktické lékařství pro děti a dorostČlánek vyšel v časopise
Česko-slovenská pediatrie

2022 Číslo 4
- Rizikové období v léčbě růstovým hormonem: přechod mladých pacientů k lékařům pro dospělé
- Fexofenadin – nesedativní a imunomodulační antihistaminikum v léčbě alergických projevů
- Virová, nebo bakteriální etiologie? Stará otázka, nové metody...
Nejčtenější v tomto čísle
- Sepse u dětí
- Diferenciální diagnostika mikroskopické hematurie
- Hypertermie, její příčiny a rizika z pohledu patofyziologa
- Dystrofinopatie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy