
Hereditární amyloidózy – etiologie, klinický obraz a léčba
Hereditary amyloidosis – aetiology, clinical features and treatment options
Amyloidosis represents a heterogeneous group of diseases characterized by the deposition of pathological material of protein nature in target tissues and organs. Hereditary amyloidosis is a disease with autosomal dominant inheritance and considerable phenotypic variability and penetrance. It is caused by a germline mutation in the gene encoding amyloidogenic precursor protein. Development of hereditary amyloidosis is currently associated with mutations in the following amyloidogenic precursor proteins: transthyretin, apolipoprotein AI and AII, fibrinogen, gelsolin, lysozyme, cystatin C. Worldwide incidence of hereditary amyloidosis is highly variable with clearly visible differences between endemic areas and non-endemic areas. The incidence of hereditary amyloidosis in the Czech Republic remains unknown. As hereditary amyloidosis is a rare disease and awareness among physicians is low, diagnosis is often very difficult. Due to the hereditary nature of the disease, carefully acquired family medical history is a key from a diagnostic aspect. This review described the seven most common types of hereditary amyloidosis, their genetic background, clinical features and treatment options.
Key words:
hereditary amyloidosis, transthyretin, apolipoprotein, lysozyme, gelsolin, fibrinogen, cystatin C
Autoři:
Z. Kufová 1,2,3; T. Pika 4; T. Jelínek 1; F. Kryukov 1,2; R. Hájek 1,2,3
Působiště autorů:
Klinika hematoonkologie, Fakultní nemocnice Ostrava
1; Lékařská fakulta, Ostravská univerzita v Ostravě
2; Babákova myelomová skupina, Lékařská fakulta, Masarykova univerzita
3; Hemato-onkologická klinika, Lékařská fakulta Univerzity Palackého a Fakultní nemocnice Olomouc
4
Vyšlo v časopise:
Transfuze Hematol. dnes,21, 2015, No. 4, p. 184-192.
Kategorie:
Souhrnné práce, původní práce, kazuistiky
Amyloidózy tvoří různorodou skupinu onemocnění charakterizovanou tvorbou patologického bílkovinného materiálu s následným ukládáním v tkáních a orgánech. Příčinou hereditární amyloidózy je patologická germinální mutace v genu kódujícím některý z amyloidogenních prekurzorových proteinů. Hereditární amyloidóza patří mezi onemocnění s autozomálně dominantní dědičností a značnou fenotypovou variabilitou a penetrancí.
Souhrn
Amyloidózy tvoří různorodou skupinu onemocnění charakterizovanou tvorbou patologického bílkovinného materiálu s následným ukládáním v tkáních a orgánech. Příčinou hereditární amyloidózy je patologická germinální mutace v genu kódujícím některý z amyloidogenních prekurzorových proteinů. Hereditární amyloidóza patří mezi onemocnění s autozomálně dominantní dědičností a značnou fenotypovou variabilitou a penetrancí. Vznik hereditární amyloidózy je v současné době spojován s mutacemi proteinu transthyretinu, apolipoproteinu AI a AII, fibrinogenu, gelsolinu, lysozymu a cystatinu C. Světová incidence hereditární amyloidózy je velice variabilní s dobře patrnými rozdíly mezi endemickými a neendemickými oblastmi výskytu. Incidence v rámci České republiky je neznámá. Hereditární amyloidózy patří mezi raritní onemocnění a povědomí o nich mezi lékaři je nízké, diagnostika často velmi komplikovaná. Kvůli dědičnému charakteru onemocnění, patří důkladně odebraná rodinná anamnéza mezi stěžejní diagnostické aspekty. Toto souhrnné sdělení je zaměřeno na popis sedmi nejčastějších dosud známých typů hereditárních amyloidóz, jejich genetický podklad, klinický obraz a možnosti léčby.
Klíčová slova:
hereditární amyloidóza, transthyretin, apolipoprotein, lysozym, gelsolin, fibrinogen, cystatin C
ÚVOD
Amyloidózy tvoří velmi heterogenní skupinu onemocnění charakterizovanou tvorbou patologické bílkoviny fibrilárního charakteru a jejím ukládáním do tkání a orgánů. V současnosti je známo více než 30 prekurzorových amyloidogenních proteinů, na jejichž základě je založena současná nomenklatura [1]. Patologická konformace těchto proteinů vede ke ztrátě jejich rozpustnosti, což má za následek tvorbu extracelulárních depozit v různých cílových orgánech a jejich poškození [2]. Amyloidózy lze rozdělit do dvou základních skupin, a to na získané a dědičné (hereditární). Nejvýznamnější ze skupiny získaných je AL amyloidóza z volných lehkých řetězců imunoglobulinu, jejíž léčba je zásadně odlišná od hereditárních a podobá se terapii mnohočetného myelomu [3].
Hereditární amyloidózy patří mezi raritní onemocnění a incidence v rámci České republiky není známá. Na příkladu transthyretinové amyloidózy, nejznámější z hereditárních amyloidóz, lze názorně doložit, že incidence je celosvětově různorodá a pohybuje se od 1 případu na 538 obyvatel v endemické oblasti severního Portugalska [14] až po 1 případ na 100 000 obyvatel v americké populaci [5]. Všeobecné povědomí mezi lékaři je proto nízké a diagnostika často velmi komplikovaná zvláště v oblastech s nízkou incidencí. Klinické projevy se u většiny nemocných začínají objevovat až v dospělém věku a bývají různorodé v závislosti na typu amyloidogenního proteinu stejně jako na geografické lokalitě. Nejčastěji postiženými orgány jsou srdce, ledviny, periferní nervový systém a gastrointestinální trakt [6]. V rámci diagnostiky zůstává základním histologickým vyšetřením barvení kongo červení a průkaz dvojlomu (birefringence) v polarizovaném světle [7]. V diagnostice hereditárních amyloidóz hraje důležitou roli kromě imunohistochemie i molekulárně genetické vyšetření, pomocí kterého je možno konfirmovat přítomnost konkrétní genetické mutace zodpovědné za rozvoj onemocnění. Vzhledem k tomu, že nelze vyloučit současně probíhající monoklonální gamapatii nejasného významu (MGUS) a přítomnost hereditární amyloidózy, je vhodné u nejednoznačných případů suspektní AL amyloidózy provést toto genetické vyšetření k vyloučení hereditárních amyloidóz [8]. V současnosti neexistuje žádná specifická terapie, pomocí které by bylo možné odstranit již uložená depozita amyloidu, ať už je prekurzorový protein jakýkoliv. Všechny léčebné snahy vedou k redukci amyloidogenního proteinu přítomného v těle pacienta. Typickým příkladem je ortotopní transplantace jater u již zmíněné transthyretinové amyloidózy (ATTR), kdy dojde k odstranění zdroje patologického proteinu (jater) a zástavě progrese onemocnění [9]. U dalších typů hereditárních amyloidóz často nezbývá než provést transplantaci selhávajících cílových orgánů, a to nejčastěji ledvin nebo srdce, na rozdíl od zmíněné transplantace jater u ATTR však nejde o kauzální léčbu [10]. Toto souhrnné sdělení je zaměřeno na šest nejčastějších z dosud známých typů hereditárních amyloidóz, jejich genetický podklad, klinický obraz a možnosti léčby.
TRANSTHYRETIN (ATTR)
Transthyretinová amyloidóza (ATTR) je nejčastějším typem hereditární amyloidózy. Jedná se o autozomálně dominantní onemocnění způsobené mutací v genu pro transthyretin [11]. Protein transthyretin je syntetizován v játrech, v plexus choroideus v mozku a v retině a zastává roli transportního proteinu pro thyroxin a retinol [12]. Gen TTR kódující protein transthyretin je lokalizován na chromozomu 18 a skládá se ze čtyř exonů. Mutace v genu vedou k destabilizaci proteinu, který následně snadněji vytváří amyloidovou strukturu [13]. K dnešnímu dni bylo popsáno v TTR genu přibližně 120 mutací, všechny autozomálně dědičné, avšak s rozdílnou penetrancí. Jedná se o bodové mutace zahrnující jedno- nebo dvou-nukleotidové substituce, v raritních případech delece celého kodónu nebo mutace v genové oblasti UTR (untranslated region, nepřekládaná oblast). Nejčastěji vyskytující se mutací je jedno-nukleotidová substituce na pozici 30 způsobující záměnu valinu za methionin (Val30Met). Jednotlivé mutantní varianty se projevují klinicky značně odlišně a mají vliv na konkrétní postižení cílových orgánů, podle druhu mutace přítomné v genu. Komplikovanost onemocnění dále zvyšuje vysoká fenotypová heterogenita, která je ovlivňována řadou faktorů, např. přítomnost polymorfismů, geografický původ, pohlaví, věk aj. [14]. Stejně tak i prognóza nemocných je závislá na typu mutace, respektive dominantně postiženého orgánového systému. Hlavním klinickým příznakem ATTR je progredující axonální senzomotorická neuropatie, která bývá přítomna u 90 % nemocných. Typickou mutací TTR je Val30Met, která je podkladem pro familiární amyloidovou polyneuropatii I. typu (FAP). Věk klinické manifestace je však variabilní. Mutovaný protein, ačkoliv je exprimován od narození, vyvolává příznaky onemocnění projevující se po dvacátém roce života. U nemocných v Portugalsku dochází k rozvoji choroby mezi 20.–30. rokem s rychlou progresí, naopak u švédských nemocných je popisována klinická manifestace až mezi 60. a 70. rokem věku, ačkoliv obě populace nesou identickou mutaci. Tento fenotypový rozdíl je vysvětlován genetickými faktory a enviromentálními vlivy. Medián celkového přežití se pohybuje v rozpětí 5–15 let, v případě symptomatického srdečního postižení 3,4 roku. Ačkoliv se onemocnění klinicky manifestuje většinou postižením jednoho orgánu či orgánového systému, v pozdějších fázích dochází k postižení s klinickým projevem i u dalších orgánů [15]. Typická je vzestupná progredující ztráta citlivosti nejprve dolních a posléze i horních končetin, parestezie, bolest a svalová slabost s následnou atrofií. Syndrom karpálního tunelu bývá přítomen u poloviny nemocných. Postižení autonomního nervového systému je spojeno s ortostatickou hypotenzí, synkopami, poruchami motility gastrointestinálního traktu či impotencí. Některé práce popisují i postižení centrálního nervového systému ve formě leptomeningeální infiltrace vedoucí k ischemickým či krvácivým mozkovým příhodám či ataxii [12, 15, 16, 17].

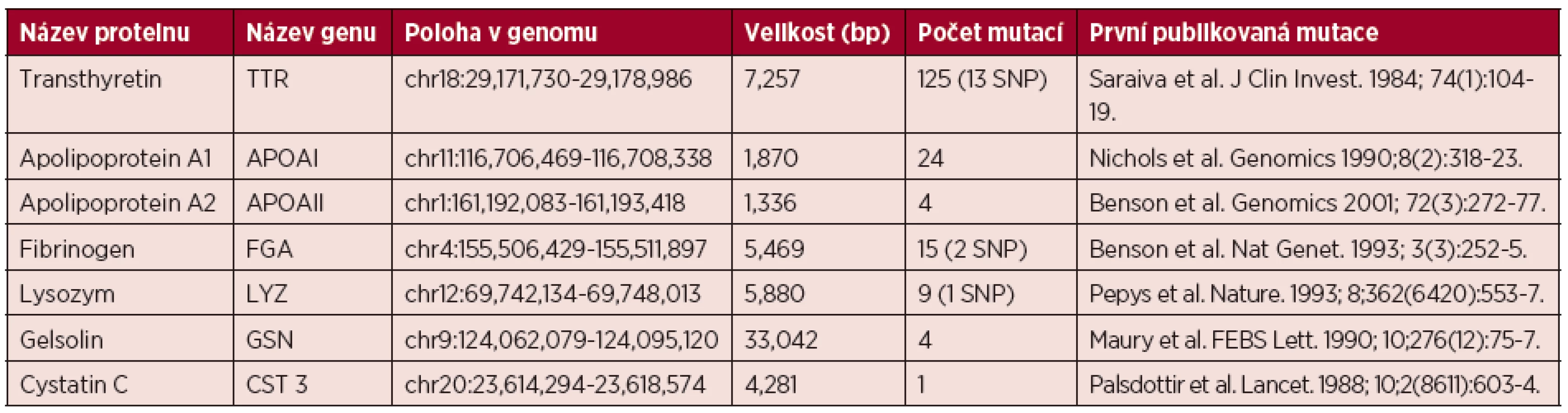
Srdeční postižení bývá pozorováno až u 62 % nemocných s rozvinutým onemocněním. V klinickém obraze dominuje městnavá srdeční slabost s dušností a otoky, časté jsou arytmie a síňokomorové blokády. Echokardiografický nález zahrnuje koncentrickou hypertrofii s rozšířením mezikomorového septa. V časném stadiu postižení bývá diastolická dysfunkce první známkou postižení. EKG známky a elevace srdečních biomarkerů rovněž může upozornit na srdeční postižení. Onemocnění s mutací TTR (Val122Ile, Gly47Val…) vedoucí k dominantnímu srdečnímu postižení jsou označovány jako familiární amyloidové kardiomyopatie (FAC). Mutace Val122Ile se vyskytuje u 3,5 % Afroameričanů [18]. Renální postižení – proteinurie a/nebo insuficience se objevuje přibližně u 1/3 nemocných. Plaky amyloidu (změněné proteinové shluky) se vyskytují nejen ve strukturách glomerulů a tubulů, ale bývají postiženy i cévy. Postižení ledvin je závislé na příčinné mutaci. Postižení gastrointestinálního traktu je přítomno přibližně u 50 % nemocných v době diagnózy onemocnění a závažnost příznaků se stupňuje s postupem onemocnění. Z klinických příznaků dominuje malabsorpce při infiltraci submukózy amyloidem či poruchách peristaltiky. Většinou nemocné postihuje průjem či střídání průjmu a zácpy. Malnutrice je jedním z nepříznivých prognostických faktorů [15]. Oční postižení ve smyslu zákalů sklivce a zvýšeného nitroočního tlaku často vyžadují operativní řešení [15, 17].
Transplantace jater, jako orgánu produkující defektní protein, představuje kauzální léčebnou modalitu pro většinu nemocných. Stejně tak lze uvažovat i o transplantaci srdce jako nejvíce postiženého orgánu v případě familiární amyloidové kardiomyopatie, eventuálně o duální transplantaci srdce a jater. V současné době je pro léčbu FAP registrován lék Vyndaqel (Tafamidis meglutime), stabilizující tetramery transthyretinu. Možné použití u jiných forem ATTR amyloidózy (FAC, senilní formy – wtTTR) je zatím předmětem klinických studií, stejně tak i jiné preparáty jsou zatím ve fázi testování (diflunisal, doxycyklin, antisense léčiva).

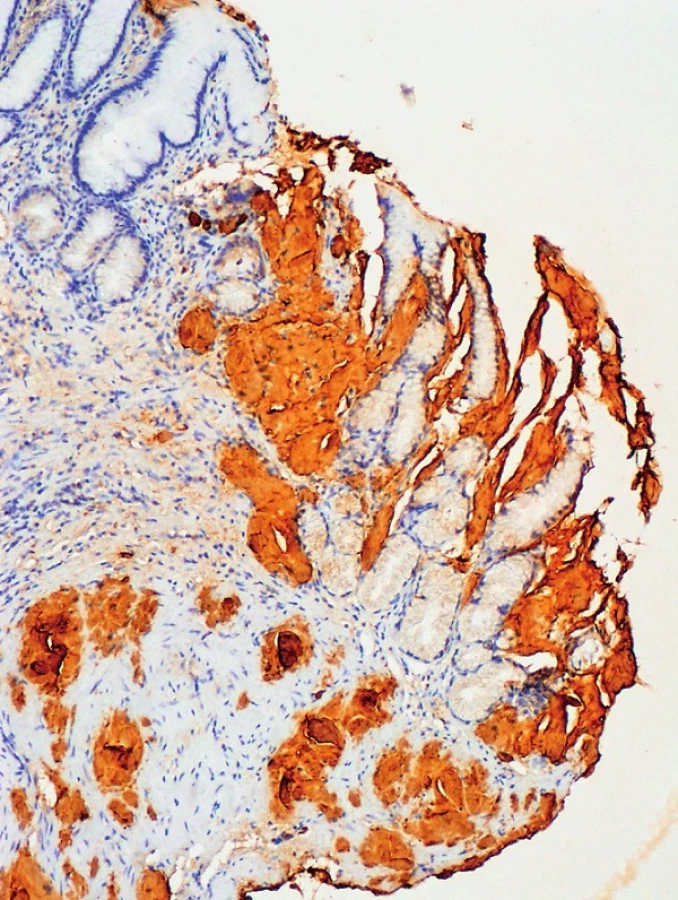
APOLIPOPROTEIN AI (AAPO AI)
Apolipoprotein AI (APO AI) amyloidóza je vzácné autozomálně dominantně dědičné onemocnění charakterizované progresivní akumulací amyloidních fibril v tkáních a orgánech vedoucích nejčastěji k renálnímu, jaternímu nebo srdečnímu poškození [19]. Apolipoprotein AI patří do skupiny tzv. vysokodenzitních proteinů (HDL – high density lipoproteins) a vyskytuje se v plazmě, kde se podílí na transportu lipidů, má kardioprotektivní účinek. Je také kofaktorem pro lecitin [20]. Gen APOAI se nachází na chromozomu 11, skládá se ze 4 exonů a kóduje protein apolipoprotein AI o 267 aminokyselinách, z nichž prvních 24 slouží jako signální peptid, molekulová hmotnost proteinu je 28 kDa a je syntetizován v játrech a tenkém střevě [21]. Patologická mutace v genu APOAI může být spojována se vznikem familiální hypercholesterolemie, kterou zapříčiňuje mutace způsobující ztrátu funkce genu (loss of function) nebo familiární systémové amyloidózy, kde naopak je patologická funkce mutací získána (gain of function) [20]. Celkem bylo u genu APOAI popsáno více než 50 mutací, z nichž 19 způsobuje vznik familiární systémové amyloidózy. Amyloidogenní mutace se nacházejí v exonech 3 a 4. Většinou se jedná o jednonukleotidové substituce, minoritní zastoupení tvoří mutace způsobující posun čtecího rámce (frameshift mutations) nebo inzerčně-deleční mutace (indely). Amyloidóza spojená s apolipoproteinem AI se může vyskytnout ve formě buď hereditární způsobené zárodečnou mutací v genu APOAI, nebo ve formě nedědičné jako depozita tzv. divokého proteinu (wild type protein), která jsou součástí aterosklerotických plaků [22]. Rozdíl mezi oběma formami je také v délce fragmentů, které vytváří fibrily. Zatímco u nehereditární formy se na vytváření fibril podílí protein o kompletní délce, u hereditárních forem dochází ke vzniku fibril pouze z N-terminálních konců proteinu konkrétně nejčastěji z peptidů 1–93. Manifestace onemocnění je udávána v třetí až páté dekádě života [12]. V nedávných studiích se ukázalo, že apolipoprotein AI působí jako antioxidant a má protizánětlivé vlastnosti, brání agregaci a neurotoxicitě amyloid β-peptidu, který je hlavním neurotoxinem při vzniku Alzheimerovy choroby [23].
Typ mutace má vliv na klinické příznaky, postižení orgánů, míru progrese, prognózu a věk, při kterém se nemoc začne projevovat. Různé druhy mutací mohou mít odlišné příznaky lišící se vzájemně věkem manifestace nemoci, klinickými příznaky. Významným rysem je také variace klinických příznaků u rodin se stejným typem mutace, nebo dokonce v rámci jedné rodiny [19]. Nejčastěji postiženým orgánem bývají u APO AI amyloidózy ledviny. Postižení ledvin je v tomto případě odlišné od poškození ledvin u AL nebo AA amyloidózy. Zatímco u AL nebo AA typu amyloidózy dochází ke vzniku silné proteinurie z důvodů ukládání amyloidních depozit v glomerulech, u APO AI amyloidózy dochází k depozici v cévách nebo v oblasti dřeně, nedochází k nárůstu proteinurie a nedochází ani k rozvoji nefrotického syndromu u takto postižených jedinců [12]. Dalším typem postižení je depozice amyloidu v srdci a s ní spojená kardiomyopatie. Typickým klinickým příznakem je klidová či ponámahová dušnost. Neléčená kardiomyopatie často vede k srdečnímu selhání [24]. Neuropatie je nejčastějším příznakem různých druhů systémové amyloidózy (spojené s mutací v genu pro transthyretin, gelsolin, fibrinogen…), u apolipoproteinu AI je však do dnešní doby popsána pouze jedna mutace způsobující právě neuropatii, proto se někteří autoři přiklání ke značení APO AI amyloidózy jako tzv. „non-neuropathy amyloidosis“ [12, 25]. Ukládání amyloidu může nastat také v játrech a způsobit hepatomegalii, popř. se může kombinovat s renálním poškozením, nebo v kůži, kde depozita způsobují kožní léze [25].
APOLIPOPROTEIN A II (AAPO AII)
Hereditární amyloidóza spojená s mutací v genu pro apolipoprotein A-II patří mezi autozomálně dědičná onemocnění. Příčinou vzniku tohoto onemocnění je mutace v stop kodónu genu APOA-II [26, 27]. Apolipoprotein A-II je druhým nejčastějším proteinem ze skupiny vysokodenzitních proteinů, vyskytuje se v plazmě jako monomer, homodimer nebo heterodimer spolu s apolipoproteinem D. Gen APOA-II se nachází na chromozomu 1 a skládá se ze 4 exonů. V současné době jsou známy 4 kauzální mutace v stop kodónu proteinu, které způsobí záměnu stop kodónu za kodón pro arginin, serin nebo glycin [28]. Amyloidní plaky se u osob postižených tímto druhem mutace nachází primárně v ledvinách, kde mohou způsobit poškození až selhání ledvin, ale mohou se vyskytovat i v jiných orgánech (srdci, slezině…) [27]. Nejčastější klinickou manifestací bývá nefropatie způsobená právě renálními amyloidními plaky, klinickými projevy se může podobat amyloidóze AFib (amyloidóza způsobená mutací v genu pro fibrinogen) nebo ALys (amyloidóza způsobená mutací v genu pro lysozym), ačkoliv u pacientů AFib bývá častější postižení jater a sleziny [28]. Prozatím jediným dostupným způsobem léčby je dialýza následovaná transplantací ledvin [26]. Defekt v genu APOA-II může být také příčinou Tangierovy choroby (A II deficience) nebo hypercholesterolemie [29].
FIBRINOGEN (AFib)
Mutace v genu kódujícím fibrinogen jsou spojovány se vznikem hereditární systémové amyloidózy označované jako AFib [30]. Jedná se o autozomálně dominantní onemocnění s variabilní penetrací, pozdním nástupem a typickou renální nefropatií vedoucí až k selhání ledvin. AFib je nejčastějším typem hereditární amyloidózy s renálním poškozením v Evropě a USA [31]. Fibrinogen je plazmatický protein s klíčovou rolí pro koagulační kaskádu a přeměnu na fibrin, je produkován výhradně játry. Protein fibrinogen se skládá ze dvou identických sad řetězců α, β, γ spojených disulfidickými můstky. Řetězce jsou kódovány geny FGA, FGB, FGG, všechny jsou lokalizovány na chromozomu 4. Největší z řetězců je α, skládá se z 610 aminokyselin a jeho molekulová hmotnost je 66 kDa [32]. Gen FGA kódující řetězec α se skládá z 6 exonů a jeho celkové velikost je 7kb. Mutace v tomto genu bývají vzácné, mohou způsobit dysfibrinogenemii, hypofibrinogenemii, afibrinogenemii nebo AFib amyloidózu. Všech 13 mutací fibrinogenu způsobující AFib amyloidózu je lokalizováno v exonu 5 genu FGA a postihují jednu nebo více aminokyselin na pozici od 517 do 555. Mezi mutacemi najdeme jak substituce, tak posunové nebo inzerčně-deleční mutace. Typ mutace může mít vliv na nástup nemoci, v případě inzerčně-delečních mutací se nástup nemoci projevuje už v dětství, zatímco u substitučních mutací nastupuje nemoc až ve středním věku. Nejčastější mutace Glu526Val (záměna kyseliny glutamové na pozici 526 za valin) je příčinou renální amyloidózy, typicky bývají postiženy glomeruly ledvinových kanálků, méně často již játra nebo slezina. Celých 5 % pacientů se systémovou AL amyloidózou je nositelem mutace Glu526Val v genu FGA [33]. Typickým znakem tohoto druhu amyloidózy je různorodý a komplexní fenotyp [31]. Průkaz depozice amyloidových mas s následnou typizací je obvykle proveden z renální biopsie, příčinná mutace je pak následně identifikována molekulárně genetickým vyšetřením. Genetické vyšetření by mělo být provedeno i u rodinných příslušníků spolu s posouzením renálních funkcí. Tento typ amyloidózy může způsobit viscerální, vaskulární, srdeční nebo neurologické postižení. Charakteristická je pomalá progrese onemocnění. Pro AFib je charakteristické postižení ledvin s typickou glomerulární masivní depozicí amyloidových mas bez výraznější infiltrace intersticia či cévních struktur. Kromě masivní proteinurie, jako dominantního laboratorního nálezu, zahrnují klinické příznaky edémy, arteriální hypertenzi (40–72 %) a progredující renální nedostatečnost (54 %) [16, 31, 36]. V případě nejběžnější mutace E526V představuje doba od nálezu vyšší proteinurie do doby nutnosti hemodialyzační terapie přibližně 10 let. Mezi další postižené orgány patří játra, avšak obvykle bez klinických atributů jen s obrazem laboratorní hepatopatie se zachováním funkcí. Infiltrace sleziny masami amyloidu je dominantně v trabekulární a subkapsulární lokalizaci, postižení sleziny může být spojeno s anémií/pancytopenií či rizikem ruptury, spontánní či perioperační v případě orgánové transplantace. Postižení sleziny a jater bývá výraznější u nemocných v hemodialyzační léčbě. Postižení autonomního nervového systému bývá spojeno s rytmickými srdečními poruchami (55 %) i ovlivněním motility gastrointestinálního traktu 68 % (průjem/obstipace) [31]. Naopak periferní nervový systém postižen není. Echokardiografické známky svědčící pro srdeční postižení bývají přítomny přibližně u poloviny nemocných. Velmi častá je incidence aterosklerotického postižení jak v systémovém (55 %), tak koronárním řečišti (68 %) [31]. Aterosklerotické pláty obsahují depozita amyloidu. V odborných publikacích můžeme najít zmínky o případech syndromu karpálního tunelu, způsobeného akumulací amyloidogenních proteinů, konkrétně interakcí fibrinogenu, transthyretinu a α-synukleinu, aktivní role fibrinogenu na vzniku neurodegenerativních onemocnění byla tímto prokázána. U těchto pacientů byla pozorována zvýšená hladina fibrinogenu [34]. Syndrom karpálního tunelu může být způsobený také změnami v genu kódujícím fibrinogen, a to díky polymorfismům vyskytujícím se v promotoru tohoto genu nebo mutacemi v genech, které se účastní regulace biosyntézy fibrinogenu (tzv. carpal tunel syndrome fibrinogen) [35]. Celkové příznaky zahrnují otoky, únavu, často i nevolnost. Léčba AFib je primárně transplantační, její vliv na proces amyloidogeneze není znám. Podle klinického stavu nemocného, pokročilosti renálního postižení a komorbidit lze zvážit renální (s rekurencí depozice amyloidu do 10 let), kombinovanou renální a jaterní či jaterní transplantaci před zahájením dialyzační léčby (tzv. preemptivní transplantaci), která by však vzhledem k nedostatečné kompletní penetranci mutace měla být provedena až po bioptickém průkazu depozice amyloidu [16, 31, 36].
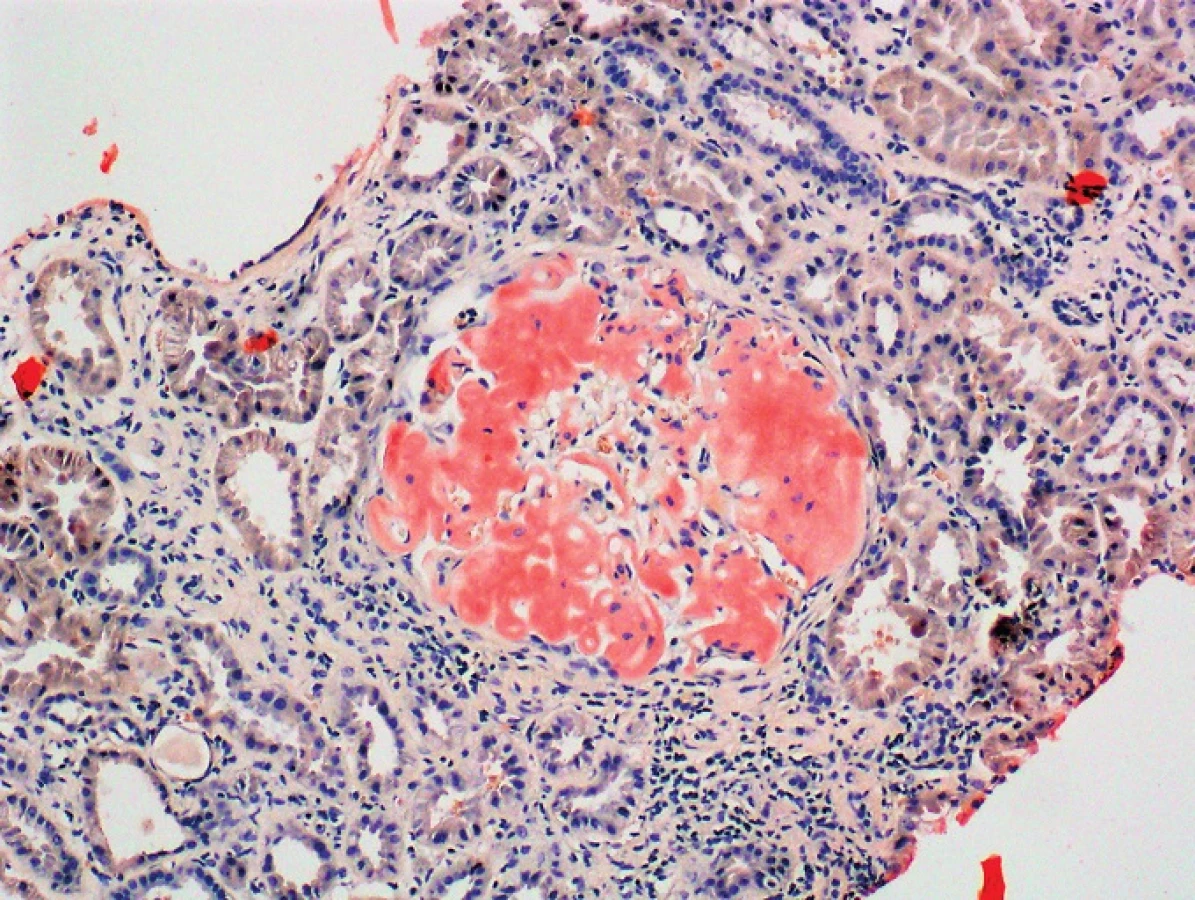
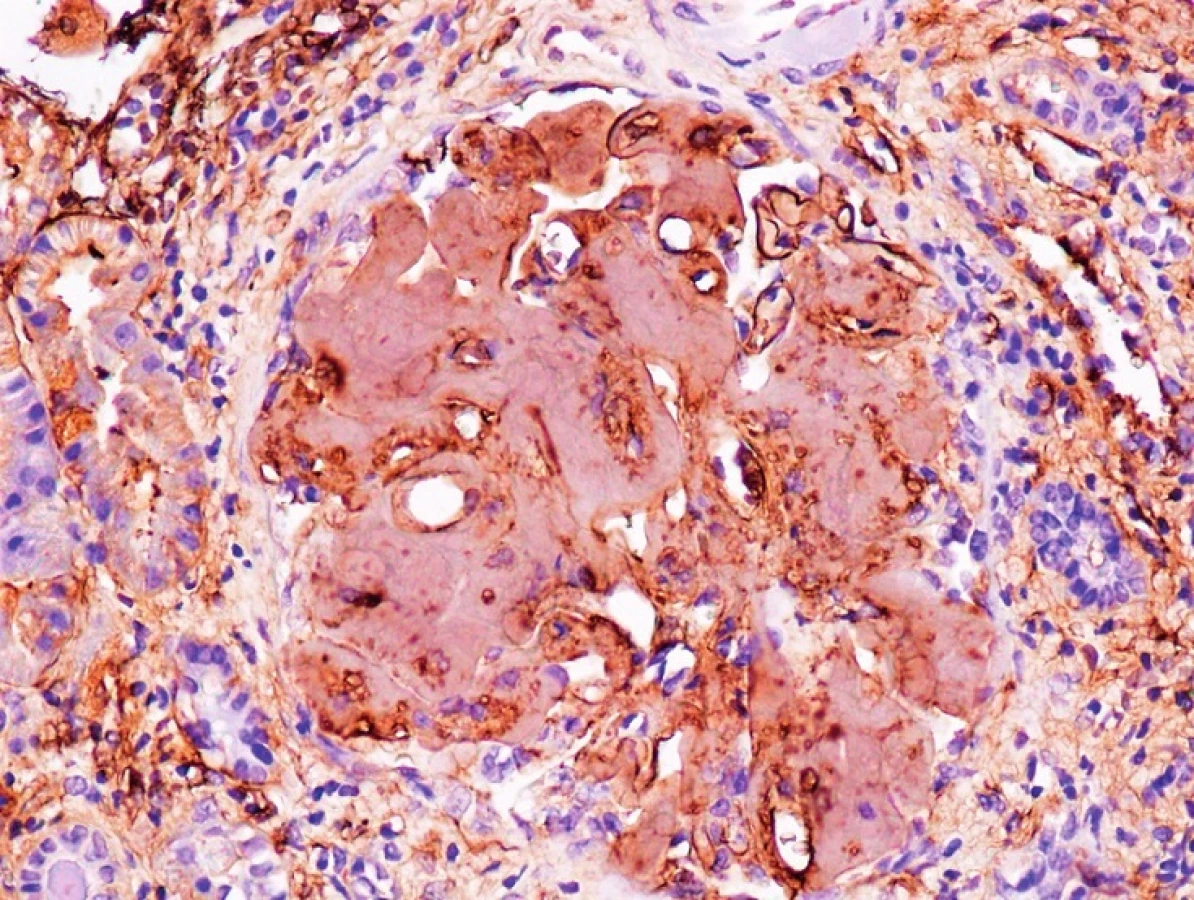
GELSOLIN (AGEL)
Gelsolinová amyloidóza (AGel) známá také pod názvem familiální amyloidóza finského typu (familial amyloidosis of Finnish type, FAF) je dědičné monogenně podmíněné onemocnění s autozomálně dominantním typem dědičnosti, které je způsobené mutací v genu GSN kódujícím protein gelsolin. AGel je onemocnění vyskytující se s vyšší frekvencí ve Finsku, kde také byla poprvé popsána, v jiných zemích je však vzácnější. Gelsolin je cytosolický kalcium senzitivní protein regulující gel-sol tranzici aktinu požadovaného pro pohyb makrofágů. V lidském metabolismu se vyskytuje ve dvou formách, jako intracelulární a jako sekretovaný. Obě formy vznikají alternativním sestřihem produktů jednoho genu označovaného GSN, nachází se na chromozomu 9 a skládá se ze 14 exonů. V dnešní době jsou známy 4 typy mutací vyskytujících se v GSN genu, jejichž následkem je vznik AGel, ve všech případech se jedná o substituce [37]. Nejčastější mutací a zároveň mutací vyskytující se především u finské populace je Asp187Asn, pro českou populaci je však typický výskyt jiné mutace konkrétně Asp187Tyr (tzv. amyloidóza dánského typu, Danish type gelsolin-related amyloidosis) s podobnými klinickými příznaky jako u předchozí záměny [38]. Ačkoliv v placích amyloidu AGel byly objeveny N terminální fragmenty gelsolinu, gelsolin o celkové délce může hrát hypotetickou roli při vazbě na jiné amyloidogenní prekurzory, např. amyloid β, který je hlavním prekurzorem vzniku Alzheimerovy choroby se sporadickým výskytem. Depozita amyloidu u AGel bývají nacházena v srdci, ledvinách a dalších viscerálních orgánech. Postižení jedinci trpí často dystrofií rohovky (mřížkovitá dystrofie), což se nezřídka řeší její transplantací. Obvyklé je neuropatické postižení kraniálních nervů dominantně s obrazem obrny lícního nervu spojeným s ptózou víček s následnou nutností blefaroplastiky [36]. Podkladem neuropatického postižení je zřejmě mikroangiopatie s ischemií [39]. Angiopatie CNS je zřejmě také příčinou jemných neuropsychiatrických změn, které jsou u nemocných sledovány [40]. Dalším příznakem je cutis laxa (granulomatózní volná kůže) se zvýšenou cévní fragilitou. Gelsolin spolu s amyloidem β, transthyretinem, cystatinem C, prionovým proteinem a ABri /ADan řadíme také mezi geny spojené s hereditární formou mozkové amyloidní angiopatie [41]. Další funkce gelsolinu ovlivňují viskozitu krve a zánětlivou odpověď organismu [37]. AGel se může vyskytnout také jako sporadická forma, a to v asociaci s AA amyloidózou, mnohočetným myelomem, AL amyloidózou nebo DM 2. typu (diabetes mellitus), někdy také může dojít k jeho mylné záměně za Sjögrenův syndrom [42]. Obvykle AGel není spojena s kratším přežitím, ačkoliv u pacientů, kteří jsou homozygotní v mutaci Asp187Asn, je akcelerováno renální postižení [36].
LYSOZYM (ALYS)
ALys je označení pro formu hereditární systémové amyloidózy s autozomálně dominantním typem dědičnosti způsobené výskytem patologické mutace v genu kódujícím protein lysozym. Ve srovnání s ATTR se jedná o vzácný typ hereditární amyloidózy. Amyloidogenní prekurzorový protein lysozym je hydrolytický enzym s ubikvitinovou aktivitou, který je produkován v gastrointestinálním traktu hepatocyty a makrofágy. Jeho fyziologickou úlohou je katalýza rozkladu polysacharidových řetězců v buněčných stěnách bakterií. Gen LYZ kódující protein lysozym se nachází na chromozomu 12 a skládá se ze 4 exonů. Celkem bylo u genu LYZ popsáno devět mutací způsobující ALys a ve všech případech se jedná o substituce v exonu 2 nebo 4. V literatuře najdeme vzácnou zmínku o výskytu dvou typů mutací v rámci jedné alely u jednoho pacienta, tyto dvě mutace se musí doplňovat, neboli musí být komplementární [43]. Jiná zmínka v literatuře referuje o ALys ve spojení s MGUS (monoklonální gamapatie nejasného významu), která byla zpočátku mylně diagnostikována jako AL amyloidóza [44]. Fenotypový projev této nemoci je variabilní a závisí na penetranci, věku a postiženém orgánu. Fenotypový projev se může lišit u nemocných se stejným typem mutace, v rámci rodiny však bývá podobný. ALys bývá vzhledem k podobnému klinickému obrazu (dominantní postižení ledvin) občas mylně zaměňována za AL amyloidózu, proto je velice důležité dbát na precizní a přesnou typizaci amyloidu pomocí histologických metod. Nebyla nalezena přímá korelace mezi mutací a následnými symptomy, ale např. u pacientů s mutací Trp64Arg bylo pozorováno časté poškození ledvin spojené se sicca syndromem nebo u Asp67His zase spontánní hepatická hemoragie [45]. Společným klinickým rysem ALys je postižení ledvin s progredující renální nedostatečností, u většiny nemocných s proteinurií až nefrotického typu. Přítomnost arteriální hypertenze však není pravidlem. Míra afekce dalších jiných orgánů je variabilní, nebývá postiženo srdce, ale zřídka bývá postižený periferní nervový systém [46]. Orgánové poškození při ALys zahrnuje také postižení gastrointestinálního traktu, jater, sleziny, slinných žláz s rozvojem sicca syndromu či kožní petechie [36, 45]. Extenzivní depozita amyloidu bývají nezřídka příčinou fatálních krvácivých komplikací (ruptura jater, sleziny, krvácení z gastrointestinálního traktu) [45, 46]. Specifická terapie není k dispozici, ani transplantace jater není přínosem, jelikož lysozym je syntetizován na více místech v organismu [46]. Transplantace ledvin, jako nejzávažněji postiženého orgánu, představuje u řady nemocných léčebnou modalitu s dobrou funkcí štěpu často přesahující 15 let [36].
CYSTATIN C (ACYS)
Hereditární amyloidóza spojená s mutací v genu pro cystatin C neboli hereditární angiopatie islandského typu je onemocnění s autozomálně dominantní dědičností a vysokou mírou penetrance. Protein cystatin C je zástupcem rodiny inhibitorů cysteinových proteáz II. třídy a je produkován mnoha typy buněk v organismu včetně neuronů. Cystatin C je kódován genem CST3, který se nachází na chromozomu 20. V současnosti je známá jediná mutace spojená se vznikem ACys, a to Leu68Gln, která se nachází v exonu 2 a byla popsána roku 1987 na Islandu [47, 48]. Depozice vzniklého amyloidu nastává především v mozkových plenách, cerebrálním kortexu, v bazálních gangliích, mozkovém kmeni a v mozečku. Plaky lze také najít v periferních tkáních např. v lymfoidních orgánech, kůži, slinných žlázách nebo ve varlatech. Jedná se o onemocnění často s fatálním průběhem, časným krvácením do mozku popř. nástupem demence, průměrný věk nástupu nemoci je 30 let. Cystatin C může hrát roli také v patogenezi jiných druhů amyloidóz (např. Aβ amyloidózy), zatímco polymorfismy v genu CST3 mohou zvyšovat riziko vzniku Alzheimerovy choroby [49]. Pro ACys neexistuje specifická terapie. Předpokládá se, že zvýšená teplota potencuje formaci amyloidových fibril, proto se doporučuje předcházet a časně léčit febrilní stavy [50].
ZÁVĚR
Hereditární amyloidózy představují heterogenní skupinu onemocnění s rozdílnou etiologií, klinickým obrazem i obdobím manifestace. Výskyt v našich podmínkách je spíše výjimečný, proto je stanovení správné diagnózy obvykle pozdní a obtížné, vyžadující spolupráci řady medicínských odborníků. Vzhledem k dědičnému charakteru onemocnění, patří důkladně odebraná rodinná anamnéza mezi stěžejní diagnostické aspekty. Genetické vyšetření rodinných příslušníků s identifikací postižených jedinců nesoucích patogenní mutaci, je zásadní pro určení vhodného medicínského přístupu. Vzhledem k raritnímu výskytu platí, že v případě klinického podezření je vhodné nemocného odeslat k dalšímu došetření a léčbě do specializovaného centra.
Podíl autorů na přípravě rukopisu
ZK – sepsání rukopisu, finální verze
TP – sepsání rukopisu, finální verze
TJ – sepsání rukopisu
FK – připomínkování rukopisu
RH – připomínkování rukopisu, finální verze
Poděkování
Práce byla realizována za podpory Dotačního program Moravskoslezského kraje „Podpora vědy a výzkumu v MS kraji 2014“ pod grantovým číslem MSK 02680/2014/RRC, za podpory Specifického VŠ výzkumu Lékařské fakulty, Ostravské univerzity v Ostravě „Screening mutačních stavů hereditární amyloidózy v České republice“ pod grantovým číslem SGS02/LF/2014 a za podpory projektu Interní grantové agentury IGA MZ ČR NT 14400.
Čestné prohlášení autora
Autor práce prohlašuje, že v souvislosti s tématem, vznikem a publikací tohoto článku není ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou. Toto prohlášení se týká i všech spoluautorů.
Doručeno do redakce: 16. 7. 2015
Přijato po recenzi: 24. 9. 2015
prof. MUDr. Roman Hájek, CSc.
Klinika hematoonkologie
Fakultní nemocnice Ostrava
17. listopadu 1790
708 52 Ostrava-Poruba
e-mail: roman.hajek@fno.cz
Zdroje
1. Sipe JD, Benson MD, Buxbaum JN, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid 2014; 21: 221–224.
2. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. New Engl J Med 1997; 337: 898–909.
3. Mahmood S, Palladini G, Sanchorawala V, Wechalekar A. Update on treatment of light chain amyloidosis. Haematologica 2014; 99: 209–221.
4. Conceição I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late- and early-onset cases in Portugal. Muscle Nerve 2007; 35: 116–118.
5. Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis 2013; 8: 31.
6. Gertz MA, Kyle RA, Thibodeau SN. Familial amyloidosis: a study of 52 North American-born patients examined during a 30-year period. Mayo Clin Proc 1992; 67: 428–440.
7. Steensma DP. ‘Congo’ red: out of Africa? Arch Path Lab Med 2001; 125: 250–252.
8. Comenzo RL, Zhou P, Fleisher M, et al. Seeking confidence in the diagnosis of systemic AL (Ig light-chain) amyloidosis: patients can have both monoclonal gammopathies and hereditary amyloid proteins. Blood 2006; 107: 3489–3491.
9. Herlenius G, Wilczek HE, Larsson M, et al. Familial Amyloidotic Polyneuropathy World Transplant Registry (2004). Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation 2004; 77: 64–71.
10. Pinney JH, Lachmann HJ, Sattianayagam PT, et al. Renal transplantation in systemic amyloidosis-importance of amyloid fibril type and precursor protein abundance. Am J Transplant 2013; 13: 433–441.
11. Sipe JD, Benson MD, Buxbaum JN, et al. Amyloid fibril protein nomenclature: recommendations from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2012; 19: 167–170.
12. Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 2007; 36: 411–423.
13. Lastovičková J. Hereditární amyloidóza s defektem transthyretinu a její neurologické projevy. Neurologie pro praxi 2011; 12(2): 142–144.
14. Hazenberg BPC. Amyloidosis: A clinical overview. Rheumat Dis Clin North Amer 2013; 39: 323–345.
15. Zeldenrust SR. ATTR: Diagnosis, prognosis, and treatment. In: Gertz MA, Rajkumar SV. Amyloidosis: Diagnosis and treatment. Humana Press, Springer, New York 2010.
16. Ryšavá R. Systémové amyloidózy a jejich léčba. Maxdorf, Praha 2013.
17. Wixner J, Karling P, Rydh A, et al. Gastric emptying in hereditary transthyretin amyloidosis: the impact of autonomic neuropathy. Neurogastroenterol Motility 2012; 24: 1111- e568.
18. Fikrle M, Palecek T, Kuchynka P, et al. Cardiac amyloidosis: A comprehensive review. Cor et Vasa 2013; 55: 60–75.
19. Traynor CA, Tighe D, O’Brien FJ, et al. Clinical and pathologic characteristics of hereditary apolipoprotein A-I amyloidosis in Ireland. Nephrology 2013; 18: 549–554.
20. Raimondi S, Guglielmi F, Giorgetti S, et al. Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of apolipoprotein A-I. J Mol Biol 2011; 407: 465–476.
21. Haase CL, Frikke-Schmidt R, Nordestgaard BG, et al. Population--based resequencing of APOA1 in 10,330 individuals: Spectrum of genetic variation, phenotype, and comparison with extreme phenotype approach. PLoS Genetics 2012; 8(11): e1003063.
22. Eriksson M, Schonland S, Yumlu S, et al. Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimen. J Mol Diagn 2009; 3: 257–262.
23. Ramella NA, Rimoldi OJ, Prieto ED, et al. Human apolipoprotein A-I--derived amyloid: its association with atherosclerosis. PLoS ONE 2011; 6(7): e22532.
24. Obici L, Bellotti V, Mangione P, et al. The new apolipoprotein A-I variant Leu174Ser causes hereditary cardiac amyloidosis, and the amyloid fibrils are constituted by the 93-residue N-terminal polypeptide. Am J Pathol 1999; 155(3): 695–702.
25. Testro AG, Brennan SO, Macdonell RAL, et al. Hereditary amyloidosis with progressive peripheral neuropathy associated with apolipoprotein AI Gly26Arg: Outcome of hepatorenal transplantation. Liver Transplant 2007; 13: 1028–1031.
26. Gursky O. Hot spots in apolipoprotein A-II misfolding and amyloidosis in mice and men. FEBS Letters 2014; 18; 588(6): 845–850.
27. Benson MD, Liepnieks JJ, Yazaki M, et al. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics 2001; 72: 272–277.
28. Yazaki M, Liepnieks JJ, Barats MS, et al. Hereditary systemic amyloidosis associated with a new apolipoprotein AII stop codon sta-tion Stop78Arg. Kidney Int 2003; 64(1): 11–16.
29. Sawashita J, Kametani F, Hasegawa K, et al. Amyloid fibrils formed by selective N-, C-terminal sequences of mouse apolipoprotein A-II. Biochim Biophys Acta 2009; 1794(10): 1517–1529.
30. Picken MM. Fibrinogen amyloidosis: the clot thickens! Blood 2010; 115(15): 2985–2986.
31. Stangou AJ, Banner NR, Hendry BM, et al. Hereditary fibrinogenA - chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood 2010; 115(15): 2998–3007.
32. Tennent GA, Brennan SO, Stangou AJ, et al. Human plasma fibrinogen is synthetized in the liver. Blood 2007; 109(5): 1971–1974.
33. Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. New Engl J Med 2002; 346: 1786–1791.
34. Barreiros AP, Galle PR, Otto G. Familial amyloid polyneuropathy. Digest Dis 2013; 311:170–174.
35. Utrobičić I, Novak I, Marinović-Terzic I, et al. Carpal tunnel syndrome is associated with high fibrinogen and fibrinogen deposits. Neurosurgery 2014; 75(3): 276–285.
36. Benson MD. Other systemic form of amyloidosis. In: Gertz MA, Rajkumar SV. Amyloidosis: Diagnosis and treatment. Humana Press, Springer, New York 2010.
37. Solomon JP, Page LJ, Balch WE, Kelly JW. Gelsolin amyloidosis: genetics, biochemistry, pathology and possible strategies for therapeutic intervention. Crit Rev Bioch Mol Biol 2012; 47(3): 282–296.
38. Taira M, Ishiura H, Mitsui J, et al. Clinical features and haplotype analysis of newly identified Japanese patients with gelsolin-related familial amyloidosis of Finnish type. Neurogenetics 2012; 13(3): 237–243.
39. Kiuru-Enari S, Somer H, Seppalainen AM, et al. Neuromuscular pathology in hereditary gelsolin amyloidosis. J Neuropathol Exp Neurol 2002; 61: 565–571.
40. Kantanen M, Kiuru-Enari S, Salonen O, et al. Subtle neuropsychiatric and neurocognitive changes in hereditary gelsolin amyloidosis (AGel amyloidosis). PeerJ 2014; 2:e493.
41. Yamada M. Cerebral amyloid angiopathy and gene polymorphisms. J Neurol Sci 2004; 15; 226(1-2): 41–44.
42. Juusela P, Tanskanen M, Nieminen A, et al. Hereditary gelsolin amyloidosis mimicking Sjögren’s syndrome. Clin Rheumatol 2009; 28(11): 1351–1354.
43. Röcken Ch, Becker K, Fändrich M, et al. ALys amyloidosis caused by compound heterozygosity in exon 2 (Thr70Asn) and exon 4 (Trp112Arg) of the lysozyme gene. Human Mut 2006; 27(1): 119–120.
44. Granel B, Serratrice J, Disdier P, et al. Underdiagnosed amyloidosis: Amyloidosis of lysozyme variant. Am J Med 2005; 118:321–322.
45. Granel B, Valleix S, Serratrice J, et al. Lysozyme amyloidosis, Report of 4 cases and a review of the literature. Medicine 2006; 85: 66–73.
46. Gillmore JD, Booth DR, Madhoo S, et al. Hereditary renal amyloidosis associated with variant lysozyme in large English family. Nephrol Dialysis Transplant 1999; 14: 2639–2644.
47. Revesz T, Holton JL, Lashley T, et al. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 2009; 118(1): 115–130.
48. Palsdottir A, Helgason A, Palsson S, et al. A drastic reduction in the life span of cystatin C L68Q carriers due to life-style changes during the last two centuries. PLoS Genetics 2008; 20; 4(6): e1000099.
49. Revesz T, Ghiso J, Lashley T, et al. Cerebral amyloid angiopathies: a pathologic, biochemical, and genetic view. J Neuropathol Exp Neurol 2003; 62(9): 885–898.
50. Abrahamson M, Grubb A. Increased body temperature accelerates aggregation of the Leu68 Gln mutant cystatin C, the amyloid-forming protein in hereditary cystatin C amyloid angiopathy. Proc Nat Acad Sci 1994; 91: 1416–1420.
Štítky
Hematologie a transfuzní lékařství Interní lékařství Neurologie OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2015 Číslo 4
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- Metamizol jako analgetikum první volby: kdy, pro koho, jak a proč?
- MINISERIÁL: Když ženám stoupá tlak...
- Specifika v komunikaci s pacienty s ránou – laická doporučení
- Deficience vitaminu B12 navozená metforminem − aktuální poznatky a tipy pro klinickou praxi
Nejčtenější v tomto čísle
-
Doporučení Společnosti pro transfuzní lékařství ČLS JEP
č. STL2015_12 ze dne 01. 09. 2015 verze 1
Doporučené postupy pro podání transfuzních přípravků - Metabolismus železa u dárců krve
- Hereditární amyloidózy – etiologie, klinický obraz a léčba
- Cytomorfologie a imunofenotyp lymfomu z plášťových buněk