
Genetika monogénových foriem diabetu
Genetics of monogenic forms of diabetes
Monogenic diabetes mellitus is a type of diabetes, where genetics without any other factors is strong enough to cause the disease. According to the clinical features monogenic diabetes can be divided to the mild familial early onset diabetes, familial fasting hyperglycemia, diabetes with extrapancreatic features and neonatal diabetes mellitus. During the last several years the number of genes causing monogenic diabetes has continuously increased. The clinical picture of the monogenic diabetes is very heterogeneous, thus DNA analysis is required for identification of the diabetes etiology, which influences also the choice of treatment. This article is an overview of current knowledge on monogenic diabetes, focusing at the clinically and epidemiologically most important forms.
Key words:
monogenic diabetes mellitus – MODY – neonatal diabetes – DNA analysis
Autoři:
J. Staníkihash3ihash6 1, 2 1, 4 1, 4
Působiště autorů:
DIABGENE & Laboratórium diabetu a porúch metabolizmu, Ústav experimentálnej endokrinológie SAV Bratislava, Slovenská republika, riaditeľ prof. MUDr. Iwar Klimeš, DrSc.
1; Detské diabetologické centrum SR pri I. detskej klinike LF UK a DFNsP Bratislava, Slovenská republika, prednostka doc. MUDr. Oľga Červeňová, CSc.
2; I. detská klinika LF UK a DFNsP Bratislava, Slovenská republika, prednostka doc. MUDr. Oľga Červeňová, CSc.
3; Molekulárno-medicínske centrum SAV Bratislava, Slovenská republika, riaditeľ MUDr. Richard Imrich, CSc.
4
Vyšlo v časopise:
Vnitř Lék 2011; 57(11): 937-945
Kategorie:
80. narozeniny prof. MUDr. Jaroslava Rybky, DrSc.
Souhrn
Monogénový diabetes je typom cukrovky, kde genetická porucha je rozhodujúca pre vznik ochorenia. Podľa klinických príznakov sa rozdeľuje na cukrovku so skorým začiatkom a rodinným výskytom, familiárnu hyperglykémiu nalačno, diabetes s extrapankreatickými príznakmi a neonatálny diabetes. V posledných rokoch kontinuálne rastie počet génov, ktorých mutácie spôsobujú monogénový diabetes. Ich klinický obraz je veľmi heterogénny, preto je nutná DNA analýza na identifikáciu príčiny ochorenia, od ktorej sa odvíja aj liečba. Náš článok prináša prehľad súčasným poznatkov o monogénovej cukrovke so zameraním na klinicky a epidemiologicky najzávažnejšie formy.
Kľúčové slová:
monogénový diabetes mellitus – MODY – neonatálny diabetes – DNA analýza
Diabetes mellitus je jednou z častí medicíny, kde je v posledných rokoch jasný prienik metód molekulárnej genetiky, čo začína ovplyvňovať jeho diagnostiku, ako aj liečbu. Presná znalosť etiológie ochorenia na úrovni DNA umožňuje voliť liečebný postup „šitý na mieru“ pacienta, čo je hlavnou myšlienkou, v súčasnosti veľmi vyzdvihovanej personalizovanej medicíny. Prvou skupinou, kde sa v diabetológii uplatnila DNA analýza, boli rôzne formy monogénovej cukrovky. V posledných 5–6 rokoch však veľmi pokročila aj znalosť genetického pozadia polygénových foriem diabetu. Tento článok sa venuje ťažiskovo najnovším poznatkom z oblasti genetiky monogénových diabetov.
Monogénový diabetes mellitus je rôznorodá skupina ochorení, líšiacich sa etiológiou, patogenézou, liečbou a prognózou. Spoločnou črtou je chronická hyperglykémia, ktorú vyvoláva mutácia génu, zasahujúceho do vylučovania alebo účinku inzulínu [1].
Monogénový diabetes tvorí približne 1–2 % spomedzi všetkých pacientov s cukrovkou, pričom sa v minulosti považoval za diabetes 1. alebo 2. typu [1]. Najskôr sa spomedzi polygénových diabetov vyčlenil typ MODY (Maturity Onset Diabetes of the Young), so začiatkom cukrovky do 25. roku života, rodinným výskytom v minimálne 2 generáciách bez prerušenia a merateľným C-peptidom minimálne 3 roky po manifestácii [2], neskôr neonatálny diabetes so začiatkom hyperglykémie v prvých 6 mesiacoch života a mitochondriálny diabetes.
Aktuálna klasifikácia podľa Murphyovej rozdeľuje monogénovú cukrovku podľa etiológie a klinického priebehu na 4 základné formy [3]:
- diabetes s rodinným výskytom a skorým začiatkom,
- hyperglykémiu nalačno s rodinným výskytom,
- diabetes s extrapankreatickými príznakmi a
- neonatálny diabetes mellitus.
Výhodou tejto klasifikácie je lepšie zohľadnenie klinickej a etiologickej charakteristiky ochorenia. Zameriava sa najmä na nosnú klinickú črtu ochorenia – vek začiatku (do 6 mesiacov – neonatálny diabetes), hyperglykémiu nalačno, rodinný výskyt bez iných prídavných príznakov alebo extrapankreatické príznaky. V praxi ešte veľmi rozšírený termín MODY táto klasifikácia už nepozná a jednotlivé jeho podtypy sú rozdelené vo viacerých skupinách (ako je diabetes s rodinným výskytom, familiárna hyperglykémia nalačno alebo diabetes s extrapankreatickými príznakmi).
Spoločnou črtou všetkých skupín monogénovej cukrovky je neustále rastúci počet nových génov, ktorých mutácie spôsobujú toto ochorenie [4]. Náš prehľad sa zameriava na klinicky najvýznamnejšie typy monogénovej cukrovky, ako aj na jej najnovšie identifikované gény.
Diabetes s rodinným výskytom a skorým začiatkom
Toto označenie sa používa v spojitosti s monogénovou cukrovkou, ktorá vzniká na základe mutácií génov pre transkripčné faktory a vekom sa hyperglykémia zhoršuje. Toto ochorenie patrí do skupiny MODY diabetov [3–4].
Ako prvé sa identifikovali mutácie génov HNF4A (MODY-1) a HNF1A (MODY-3), a to začiatkom 90. rokov minulého storočia. Ostatné gény, ako sú PDX1 (MODY-4), NEUROD1 (MODY-6), PAX4 (MODY-7) a KLF11 (MODY-9) sa objavili neskôr. Prehľad génov je v tab. 1.
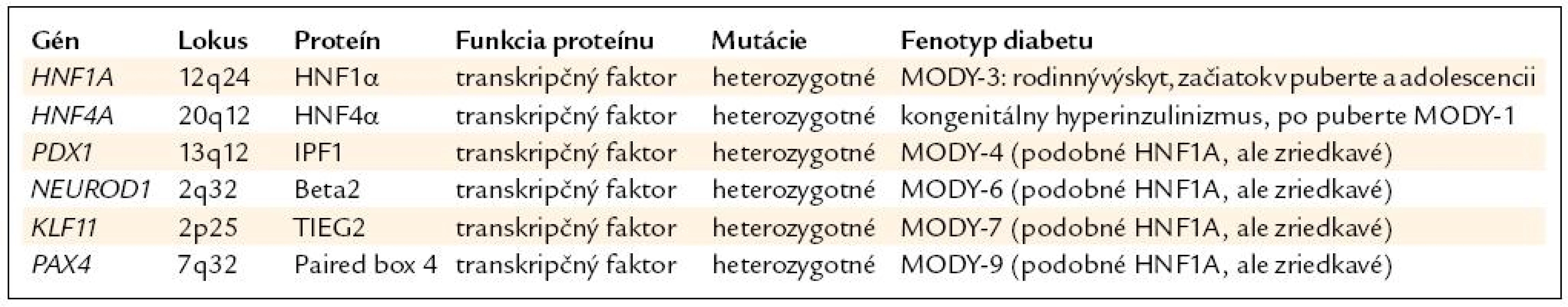
Najčastejším ochorením z tejto skupiny je HNF1A-diabetes (MODY-3), ktorý tvorí približne 0,5 % spomedzi všetkých typov diabetu [1] a približne 10–20 % spomedzi pacientov s MODY v Českej republike a na Slovensku [5], (Staník, Gašperíková, Klimeš – nepublikované). Mutácie génu pre HNF4A tvoria asi 1–5 % pacientov s MODY, ostatné typy sú omnoho zriedkavejšie [6].
Mutácie HNF1A (MODY-3) a HNF4A (MODY-1)
Genetika. HNF1A-diabetes vzniká na podklade heterozygotných mutácií génu pre hepatálny nukleárny faktor 1 α (HNF1A), ktorý sa nachádza na chromozóme 12, má 10 exónov a doposiaľ sa našlo viac ako 190 mutácií (v exónoch a exón-intrónových spojeniach) [7]. Mutácie majú najčastejšie charakter zámeny nukleotidov, čo spôsobuje zámenu aminokyselín v proteíne (missense mutácie), alebo vznik stop-kodónu (nonsense) s predčasným ukončením prepisu DNA. Vyskytujú sa aj inzercie či delécie nukleotidov s posunom ostatných nukleotidov (frame-shift) a vznikom nezmyselného proteínu. Ochorenie sa dedí z generácie na generáciu s autozómovo dominantným typom dedičnosti, de-novo mutácie (vznik ochorenia priamo u pacienta) sú veľmi zriedkavé [2]. Gén pre HNF4A (chromozóm 20) má rovnako 10 exónov, mutácií bolo ale identifikovaných len viac ako 45 [7].
Patogenéza. Gény tejto skupiny ochorení kódujú proteíny s funkciou transkripčného faktora, ktorý ovplyvňuje aktivitu ostatných génov v bunke. V B-bunkách ovplyvňujú schopnosť produkovať a vylučovať inzulín [8]. Transkripčných faktorov je v B-bunke niekoľko desiatok, pričom chýbanie jedného (pri poškodení mutáciou) dokážu ostatné kompenzovať aj niekoľko rokov. Nedostatok sa prejaví až pri zvýšených nárokoch na produkciu inzulínu, teda v období puberty, adolescencie, alebo aj neskôr, pričom porucha vyučovania inzulínu sa vekom zhoršuje.
Klinický obraz pri HNF1A-diabete sa začína glykozúriou pri normálnej glykémii v priebehu detstva. Hyperglykémia sa manifestuje najčastejšie počas puberty a adolescencie, miernymi alebo žiadnymi príznakmi [9]. Nikdy sa nevyskytuje ketoacidóza [10]. Glykémia je zvýšená najskôr po záťaži, až neskôr nalačno [11]. Chronické mikrovaskulárne komplikácie sú pri zlej kompenzácii diabetu časté. Mutácie HNF4A sa prejavujú veľmi podobne, rozdielom býva vysoká pôrodná hmotnosť (nad 4 000 g) a niekedy kongenitálny hyperinzulinizmus s hypoglykémiami. Hypoglykémie v 1. roku života ustúpia a v adolescencii sa objaví diabetes.
Diagnostika je postavená na splnení diagnostických kritérií MODY diabetu. Indikuje sa zvyčajne najskôr DNA analýza génu HNF1A priamym sekvenovaním, pri negativite analýza HNF4A. Výnimkou je vysoká pôrodná hmotnosť pacienta, kedy sa gén HNF4A analyzuje ako prvý [12], lebo aj keď už boli opísané makrozomické plody aj pri HNF1A-diabete, ide skôr o zriedkavý nález [13].
Liečba závisí od veku a HbA1c. Pri manifestácii a HbA1c pod 6,5 % (DCCT) sa môže prechodne ordinovať diéta. Pri vzostupe HbA1c sú liekom voľby deriváty sulfonylurey [14], v nízkych dávkach, nakoľko sú na ne títo pacienti zvýšene citliví (pre ich pomalšie metabolizovanie) [14]. Aj u pacientov liečených spočiatku inzulínom môže byť liečba zmenená na derivát sulfonylmočoviny, dokonca so znížením glykemických výkyvov a poklese hodnoty glykovaného hemoglobínu [15]. Táto liečba má účinnosť aj niekoľko desaťročí, avšak pri veľkom poklese produkcie inzulínu B-bunkami je potrebné v niektorých prípadoch opätovne prejsť na liečbu inzulínom [14].
Mutácie génov pre PDX1 a NEUROD1 sa našli len u niekoľkých rodín (mutácie NEUROD1 sa našli vo vyššom percente v Českej republike) [16]. Klinický obraz bol miernejší ako v prípade HNF1A-diabetu. Mutácie v homozygotnej forme spôsobujú neonatálny diabetes (v prípade PDX1 ide o závažnú agenézu pankreasu).
Mutácie génov pre KLF11 (MODY-7) a PAX4 (MODY-9)
Ide o novoidentifikované gény, kódujúce transkripčné faktory. V roku 2005 sa opísali 3 rodiny s 2 rôznymi mutáciami génu KLF11 (Kruppel-like Factor 11) s fenotypom diabetu so začiatkom od 17 do 56 rokov na liečbe perorálnymi antidiabetikami alebo inzulínom [17]. Mutácie v géne PAX4 (Paired Box Gene 4) sa našli v roku 2007 [18] v 2 rodinách z Thajska s vekom začiatku diabetu od 13 do 50 rokov na liečbe diétou alebo perorálnymi antidiabetikami.
Hyperglykémia nalačno s rodinným výskytom
Ochorenie vzniká na podklade mutácie génu pre glukokinázu (nazýva sa aj GCK-diabetes alebo MODY-2), začína do 30. roku života, má rodinný výskyt a veľmi dobrú prognózu.
Odhaduje sa, že GCK-diabetes tvorí približne 0,5 % spomedzi všetkých ľudí s diabetom [19].
Genetika. Ochorenie vzniká na podklade heterozygotnej inaktivačnej mutácie génu pre glukokinázu (GCK), ktorý má 10 exónov a 2 promótory. Mutácie (spolu viac ako 600) [20] charakteru missense, nonsense alebo frame-shift sa našli vo všetkých exónoch, priľahlých intrónových častiach a najnovšie aj v panktreatickom promótore. Táto promótorová mutácia (–71G > C) má na Slovensku vysokú prevalenciu [21]. Dedičnosť GCK-diabetu je autozómovo dominantná – t. j. riziko prenosu ochorenia na potomkov je 50 %. Typický je rodinný výskyt v niekoľkých generáciách po sebe.
Patogenéza. Glukokináza je enzým, ktorý fosforyluje glukózu a umožňuje jej tak vstúpiť do metabolizmu, na konci ktorého je na energiu bohatá molekula ATP. Pri poškodení enzýmu glukokinázy mutáciou vzniká v B-bunke málo ATP, ktorý normálne stimuluje vylučovanie inzulínu do krvi [22]. Kým nalačno je glukóza hlavným regulátorom produkcie inzulínu, po najedení prichádzajú aj iné stimuly, ako sú aminokyseliny a hormóny vylučované črevom. Preto mutácie glukokinázy spôsobujú hyperglykémiu nalačno, pričom po záťaži (jedle, oGTT) je glykémia normálna alebo zvýšená len mierne [5].
Klinický obraz. MODY-2 sa prejavuje bezpríznakovou hyperglykémiou nalačno, zvyčajne medzi 5,5 a 8,5 mmol/l [23–25]. Po jedle alebo pri orálnom glukózovo-tolerančnom teste býva zvyčajne len malý vzostup glykémie, typicky do 4 mmol/l [26]. Hyperglykémia nalačno je stacionárna a vekom sa nezhoršuje. Riziko vzniku chronických komplikácií diabetu je pri MODY-2 veľmi nízke [25].
Diagnostika je postavená na kritériách MODY s opakovanou hyperglykémiou nalačno a HbA1c do 7,5 % (DCCT) [27]. Potvrdenie diagnózy prináša DNA analýza génu pre glukokinázu.
Liečba MODY-2. U detí nie je indikovaná žiadna medikamentózna liečba, pretože len znižuje telu vlastnú produkciu inzulínu a hyperglykémiu nalačno nezmierni [1]. Efekt prísnej diabetickej diéty je rovnako diskutabilný; odporúča sa preto len racionálna strava. U dospelých sa odporúča racionálna strava, v prípade vyšších hodnôt glykémií sa odporúča obmedzenie množstva voľných monosacharidov v strave, pri vyšších hodnotách HbA1c (býva veľmi zriedka) deriváty sulfonylurey [26]. V prípade gravidity žien s MODY-2 je pri rýchlejšom raste plodu v dôsledku hyperglykémie u matky indikovaná liečba inzulínom (ktorá sa po pôrode ukončí).
Inaktivačné mutácie GCK v homozygotnom stave sa prejavujú neonatálnym diabetom, ktorý vyžaduje liečbu inzulínom [20]. Dôležité je preto genetické poradenstvo, nakoľko homozygoti majú najčastejšie oboch rodičov s mutáciou v glukokinázovom géne.
Glukokinázový regulačný proteín
V roku 2010 sa identifikovali 3 japonské rodiny s mutáciami v géne pre glukokinázový regulačný proteín (GCKR). Klinicky sa títo pacienti prejavovali ako pacienti s MODY-2 diabetom [28].
Diabetes s extrapankreatickými príznakmi
Diabetes asociovaný s inými príznakmi má často monogénový základ [1]. Sprievodné ochorenie je preto často výborným vodidlom k správnej diagnóze. Prehľad génov a ochorení je v tab. 2.
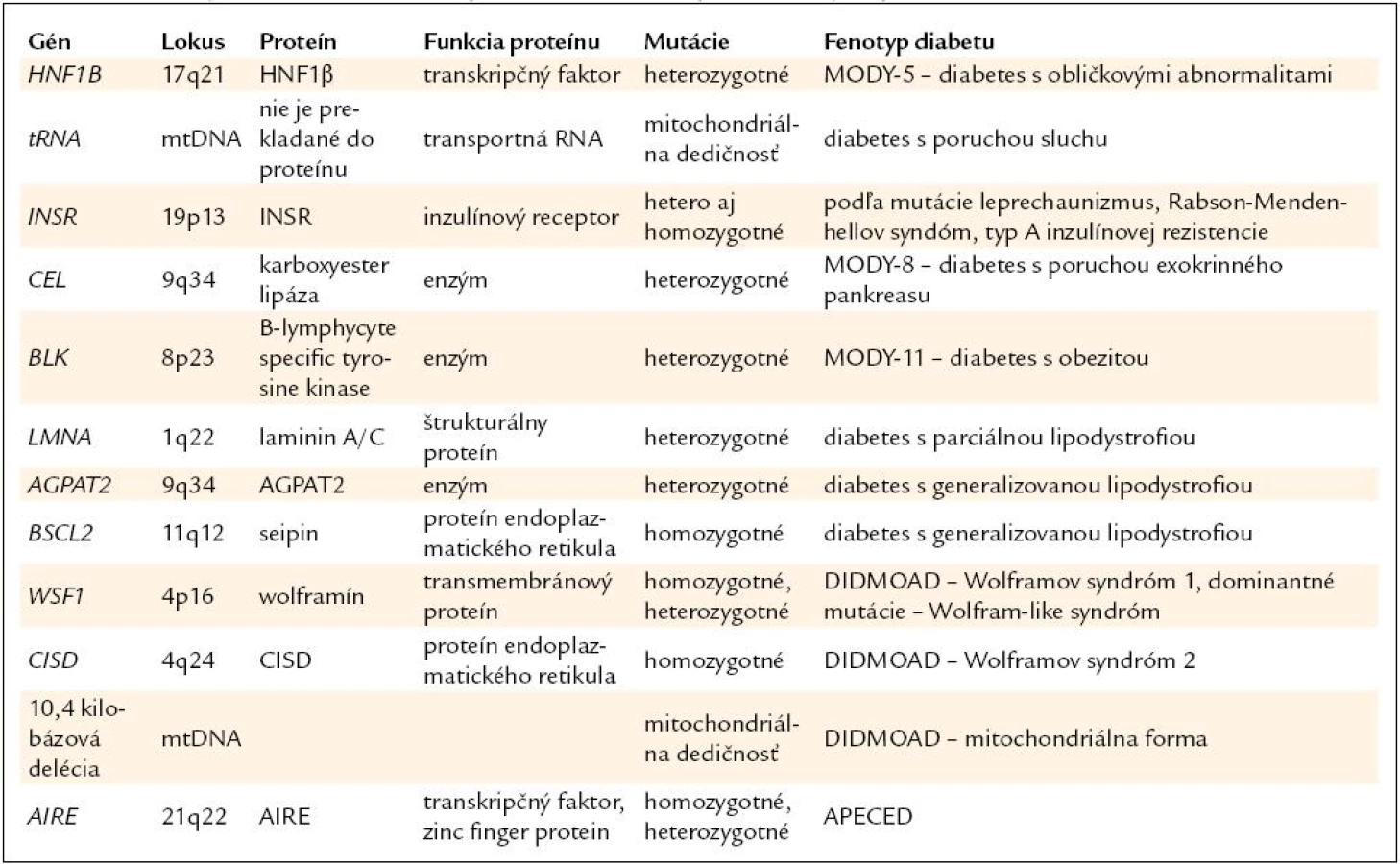
Diabetes a obličkové cysty
Monogénová cukrovka asociovaná s obličkovými cystami (nazývaná aj HNF1B-diabetes alebo MODY-5) spája obličkové cysty zachytené zvyčajne v priebehu detstva s diabetom, ktorý má začiatok do 25. roku života, rodinný výskyt a má tendenciu sa vekom zhoršovať [29].
HNF1B-diabetes tvorí menej ako 0,1 % všetkých pacientov s diabetom [2].
Genetika. Ochorenie vzniká na podklade mutácie génu pre hepatálny nukleárny faktor 1 β (HNF1B) a má autozómovo-dominantný charakter s rodinným výskytom [2]. Gén pre HNF1B sa nachádza na chromozóme 17, obsahuje 9 exónov. Mutácie môžu byť bodové (substitúcie, inzercie, delécie), ale častejšie ide o veľké delécie zasahujúce niekoľko exónov alebo aj celú alelu génu. Táto skutočnosť je veľmi dôležitá pri DNA analýze, kde okrem priameho sekvenovania je potrebné vykonať aj vyhľadávanie veľkých delécií metódou MLPA.
Patogenéza. Gén pre hepatálny nukleárny faktor 1 β (HNF1B) kóduje proteín-transkripčný faktor, ktorý sa podieľa v B-bunkách na sekrécii inzulínu a v obličkách na vývoji nefrónov. Kým sa porucha v obličkách prejaví už počas ich vývoja, v B-bunke zastúpením chýbajúceho transkripčného faktora inými, sa diabetes manifestuje až po puberte [29].
Klinický obraz. Postihnutie urogenitálneho systému má najčastejšie charakter obličkových cýst, menej často ide o malformácie genitálu a maternice [29]. Diabetes má miernejší priebeh ako cukrovka 1. typu, aj keď sa väčšina pacientov lieči inzulínom. Chronické komplikácie diabetu závisia od kompenzácie ochorenia, ale sú menej časté ako pri MODY-3.
Diagnostika je založená na kombinácii diabetu s obličkovými cystami a na ich rodinnom výskyte [2]. DNA analýza potvrdí diagnózu HNF1B-diabetu.
Liečba diabetu je takmer výlučne inzulínom. Obličkové malformácie vyliečiť nemožno, je však dôležitá prevencia sekundárneho poškodenia nefrónov v dôsledku hyperglykémií a diabetickej nefropatie [29]. Dobrá kompenzácia diabetu je preto veľmi dôležitá.
Diabetes a porucha sluchu
Percepčná porucha sluchu, začínajúca do adolescencie a diabetes mellitus manifestujúci sa v 3. a 4. decéniu sú charakteristickými črtami syndrómu MIDD (Mitochondrial Diabetes and Deafness) [3].
Epidemiológia. Odhaduje sa, že MIDD tvorí približne 1 % spomedzi všetkých typov diabetu.
Etiológia. Ochorenie vzniká na podklade mutácie A3243G mitochondriálnej DNA a má matrilineárny typ dedičnosti: dedí sa výlučne od matky [30]. Výrazne zriedkavejšia je mutácia T14709C s podobným fenotypom ako pri A3243G.
Patogenéza. V každej bunke je niekoľko desiatok až stoviek mitochondrií a každá z nich má niekoľko desiatok kópií mitochondriálnej DNA. Nie všetky kópie mávajú mutáciu, preto klinický obraz závisí od percenta DNA poškodenej mutáciou – čím je mutovanej DNA viac, tým sú príznaky závažnejšie. Poškodené mitochondrie nie sú schopné adekvátne pracovať – produkovať energiu vo forme ATP. Porucha sa preto prejaví v tkanivách a orgánoch náročných na energiu, v prípade MIDD je to vnútorné ucho a B-bunky pankreasu (napr. pri syndróme MELAS je to aj sval a mozog). Vekom sa funkcia mitochondrií zhoršuje, preto sa zvýrazňujú aj príznaky MIDD [3].
Klinický obraz. Percepčná porucha sluchu zvyčajne predchádza vznik diabetu, obe sa objavujú vo veku do 40 rokov. Diabetes spočiatku nevyžaduje liečbu inzulínom, neskôr však u časti pacientov je exogénne podávanie inzulínu nevyhnutné [30]. Chronické komplikácie diabetu závisia od kompenzácie diabetu.
Diagnostika sa zakladá na kombinácii diabetu s poruchou sluchu so začiatkom do 40. (max. 50.) roku života. Vek je podmienkou diagnostiky, pretože diabetes vo vyššom veku môže byť sprevádzaný stareckou poruchou sluchu, ktorá nemá mitochondriálny základ [30].
Liečba v prípade diabetu je spočiatku orálnymi antidiabetikami, neskôr s progresiou ochorenia je potrebná liečba inzulínom. Nakoľko poškodením mitochondrií je poškodená oxidatívna fosforylácia a je riziko zvýšenia laktátu, liečba metformínom pre veľké riziko laktátovej acidózy je kontraindikovaná [3].
Dedičné syndrómy inzulínovej rezistencie
Ide o menej časté ochorenia s výraznou inzulínovou rezistenciou, ktoré vznikajú na podklade mutácií génu pre inzulínový receptor (ISNR) [31].
Inzulínový receptor má 22 exónov a jeho mutácie môžu mať recesívny alebo dominantný charakter. Poškodenie inzulínového receptora spôsobuje inzulínovú rezistenciu s následnou hyperinzulinémiou a zníženým clearensom inzulínu z plazmy. Pre nedostatok receptorov pretrváva inzulín v plazme dlhšie, čo zvyšuje pomer k C-peptidu (ten sa odbúrava iným spôsobom ako cez inzulínový receptor). Stupeň inzulínovej rezistencie závisí od typu mutácie [31,32].
Homozygotné mutácie sa prejavujú závažnými syndrómami už vo včasnom veku. Pri leprechaunizme spôsobuje nadprodukcia inzulínu v dôsledku vysokej inzulínovej rezistencie už prenatálne závažné hypoglykémie v novorodeneckom období, často fatálne v prvých mesiacoch života. Naopak, pri Rabson-Mendenhallovom syndróme pacienti väčšinou novorodenecké hypoglykémie prežijú a postupne prejdú do hyperglykémií. Pacienti zomierajú v detstve v inzulín-rezistentnej ketoacidóze [33].
Heterozygotné autozómovo-dominantné mutácie sa prejavujú u žien ako inzulínová rezistencia typu A (našli sa približne v 1 % tohto ochorenia) [33].
V dôsledku poškodenia INSR chromozómovým zlomom sa opísal aj intermediálny fenotyp s hypoglykémiami v detstve a striedaním hypo- a hyperglykémií v dospelosti [32].
Diabetes s poruchou exokrinného pankreasu
Aj keď sa exokrinná porucha pankreasu opisuje aj v určitom percente diabetu 1. a 2. typu, je opísaná aj monogénová forma týchto ochorení. Vzniká na podklade delécií VNTR (variabilný počet tandemových opakovaní) karboxylesterlipázy a zaraďuje sa medzi diabetes typu MODY (MODY-8) [34]. Porucha bola opísaná v roku 2006 u 2 rodín z Nórska [35].
Ochorenie sa prejavuje typicky kombináciou diabetu a poruchy trávenia v dôsledku exokrinnej pankreatickej insuficiencie. Intenzita príznakov môže kolísať aj v rámci jednej rodiny, pričom u každého jedinca môže byť v popredí iný príznak (diabetes alebo maldigescia). Diabetes sa manifestoval do 25. roku života u viacerých členov opísaných rodín [35].
Diagnostika sa opiera o prítomnosť exokrinnej pankreatickej poruchy (deficit elastázy v stolici) u pacientov spĺňajúcich kritériá MODY. DNA analýza CEL génu potvrdí klinické podozrenie [36].
Diabetes s obezitou
Mutácie génu BLK spôsobujú MODY diabetes asociovaný s obezitou [37].
Genetika. Gén BLK sa nachádza na chromozóme 8, pričom sa identifikovalo jeho 5 rôznych mutácií v piatich rodinách v USA. Gén kóduje jednu z tyrozínkináz a exprimuje sa okrem B-lymfocytov aj v pankrease.
Klinický obraz. Penetrancia mutácií je pomerne nízka, len 30 % nosičov má klinické prejavy diabetu. Vyšší výskyt diabetu sa asociuje s obezitou u pacientov [37].
Lipoatrofický diabetes
Ide o skupinu ochorení, kde je primárna porucha tukového tkaniva, spôsobujúca abnormálnu distribúciu tuku (parciálnu alebo generalizovanú) s inzulínorezistentným diabetom s acanthosis nigricans a dyslipidémiou [38].
Parciálne formy s atrofiou tuku na končatinách a trupe a hromadením na krku a na tvári spôsobujú mutácie génu pre laminin A/C (LMNA) [39].
Generalizované formy s atrofiou podkožného tuku na celom tele vznikajú na podklade génov pre 1-acylglycerol-3-phosphate O-acyltransferase-2 (AGPAT2) a seipin (BSCL2) [40]. Typ diabetu je pri všetkých formách podobný.
Diagnostika lipoatrofického diabetu je postavená na kombinácii diabetu s abnormálnou distribúciou (chýbaním) podkožného tuku a DNA analýze príslušného génu [38].
Wolframov syndróm (DIDMOAD)
Syndróm DIDMOAD charakterizuje postihnutie viacerých orgánov, pričom typická je kombinácia Diabetes Insipidus, Diabetes Mellitus, Optickej Atrofie a hluchoty (Deafness), z ktorej je aj odvodený názov. Ide o ochorenie, ktoré podmieňujú mutácia vo viacerých génoch. Najčastejšie ide o mutácie génu pre wolframín (WSF1) [41], menej často génu CISD2 (WSF2) a ojedinele ho spôsobujú mutácie mitochondriálnej DNA [42].
Diagnostika sa opiera o kombináciu diabetes mellitus a atrofie optického nervu (ostatné príznaky sú menej konštantné) a dôkaz mutácií DNA analýzou.
Syndróm APECED
Ide o monogénový autoimunitný typ ochorenia s autozómovo-recesívnym alebo dominantným typom dedičnosti, podľa typu mutácie. Vzniká na podklade mutácií AIRE (AutoImmune Regulator) génu, ktorý je zodpovedný za navodzovanie tolerancie počas zrenia T-lymfocytov v thymuse. APECED charakterizujú autoimunitné polyendokrinopatie, candidóza a ektodermálna dystrofia. Diabetes nepatrí do klinického obrazu konštantne, pričom má charakter diabetu 1. typu a začína vo včasnom detstve [43].
Diagnostika je postavená na kombinácii 2 ochorení z Adisonovej choroby, hypoparatyreózy a mukokutánnej kandidózy a na dôkaze mutácie v AIRE géne [43].
Neonatálny diabetes mellitus
Väčšina cukrovky vzniknutej v 1. polroku života má monogénový charakter a prípady diabetu 1. typu sú v tejto skupine raritné. Neonatálny diabetes, ktorý vymizne do niekoľkých týždňov až mesiacov od vzniku, sa nazýva tranzientný neonatálny diabetes (TNDM). Diabetes bez remisie má charakter permanentného neonatálneho diabetu (PNDM) [3,44]. Najčastejšou príčinou novorodeneckej cukrovky sú poruchy imprintingu (majú charakter výlučne TNDM) [45,46] a mutácie génov pre Kir6.2 (Kalium inward rectifier 6.2) a SUR1 (Sulfonylurea Receptor 1) podjednotky draslíkového kanála B-buniek (podľa typu mutácie sa prejavia ako TNDM alebo PNDM) [45]. Prehľad génov neonatálneho diabetu je v tab. 3.

Epidemiológia. Incidencia permanentného neonatálneho diabetu bola na Slovensku, ako v prvej krajine na svete, presne určená na základe informácií z Národného registra detí s diabetom a je 1 prípad na 214 000 živo narodených detí [47]. Výskyt tranzientného NDM je približne rovnaký [48].
Poruchy imprintingu
Genetika. V lokuse 6q24 sa nachádza gén PLAGL1, ktorého prepis do proteínovej formy je regulovaný, pretože ovplyvňuje vylučovanie inzulínu v B-bunke [49]. Zabezpečuje sa to vyradením alely získanej od matky tzv. metyláciou. Iba alela získaná od otca je funkčná. Ochorenie vzniká pri zvýšenom množstve proteínu, čo spôsobuje porucha metylácie alely od matky, alebo duplikovanie aktívnej alely od otca (formou mikroduplikácií alebo uniparentálnou paternálnou izodizómiou, t. j. zdedením oboch chromozómov 6 od otca). Zrením B-buniek sa dokáže nadprodukcia proteínu dočasne vykompenzovať, neskôr v priebehu života sa opäť prejaví [50,51].
V roku 2008 sa identifikovali mutácie génu ZFP57 (chromozóm 6p) ako ďalšia príčina hypometylácie pri TNDM. Na rozdiel od výlučnej hypometylácie 6q24 mali títo pacienti aj akcesórne príznaky, ako je zaostávanie v psychomotorickom vývoji a hypoplázia corpus callosum (aj keď niektorí mali aj normálny vývoj CNS a intelektu) [52].
Patogenéza. Zvýšený prepis PLAGL1 v lokuse 6q24 spôsobuje prechodné zvýšenie vylučovania inzulínu [51]. Pri mutáciách ZFP57 ide o porušenú metyláciu na rôznych miestach vrátane PLAGL1, preto diabetes vzniká rovnakým mechanizmom.
Klinický obraz. Zvýšené hladiny glukózy sa objavujú zvyčajne už 1. týždeň života a vyžadujú liečbu inzulínom. Sprievodnými znakmi bývajú nízka pôrodná hmotnosť, makroglosia a umbilikálna hernia. Do 6. mesiaca života sa glykémie upravia (pacienti sú vtedy bez liečby), pričom u asi 50 % sa diabetes vráti v priebehu dospelosti [1].
Diagnostika TNDM je postavená na zistení neonatálneho diabetu a jeho remisii v 1. roku života.
Liečba je v neonatálnej fáze výlučne inzulínom, v prípade relapsu sú alternatívou inzulínu deriváty sulfonylmočoviny [1].
Mutácie génov pre Kir6.2 a SUR1 podjednotky draslíkového kanála
Mutácie génov pre Kir6.2 a SUR1 sú najčastejšou príčinou permanentného neonatálneho diabetu, ale ich klinický obraz môže byť veľmi rôznorodý, v závislosti od typu mutácie. Pacienti vo väčšine prípadov dobre odpovedajú na liečbu derivátmi sulfonylurey.
Genetika. Gény KCNJ11 (kóduje Kir6.2) a ABCC8 (kóduje SUR1) sa nachádzajú na chromozóme 11. Kým KCNJ11 má len 1 exón, ABCC8 ich má 39 [53]. Táto skutočnosť výrazne ovplyvňuje diagnostický postup. Mutácie sa našli takmer vo všetkých častiach kódujúcich úsekov, ako aj priľahlých intronických častiach. Oba gény sú ale veľmi polymorfné s veľkým množstvom polymorfizmov, ako aj rôznych typov mutácií, čo značne komplikuje DNA diagnostiku a najmä interpretáciu výsledku [50]. Ochorenie má autozómovo-dominantný charakter dedičnosti, ale väčšina mutácií vzniká de-novo – priamo u pacienta. Len asi 20 % detí mutáciu zdedí po jednom z rodičov [47].
Patogenéza. Podjednotky Kir6.2 a SUR1 formujú od ATP závislý draslíkový kanál B-buniek [54]. Takéto kanály sú v membráne bunky a regulujú jej napätie. Ak je kanál otvorený, draslíkové ióny voľne prechádzajú, čo udržuje membránu hyperpolarizovanú a neschopnú prepustiť sekrečné granuly s inzulínom. Keď stúpne koncentrácia ATP v bunke (napr. po najedení sa), kanál sa pôsobením ATP uzavrie, membrána sa depolarizuje a uvoľní inzulín do cirkulácie. Pri poškodení podjednotiek aktivačnými mutáciami je kanál stále otvorený (alebo len pootvorený), inzulín sa nemôže dostať do cirkulácie a vzniká diabetes [54]. Od zvyškovej funkcie kanála závisí klinický obraz [55].
Klinický obraz je závislý od typu mutácie. Mutácie, ktoré len mierne ovplyvňujú funkčnosť kanála, sa prejavujú podobne ako MODY s manifestáciou diabetu až v puberte alebo dospelosti. Čím je porucha kanálu závažnejšia, tých skôr sa ochorenie prejaví – preto môže mať charakter TNDM s relapsom v detstve, permanentného neonatálneho diabetu (najčastejší klinický obraz) a v prípade úplného vyradenia funkcie kanála syndrómu DEND (Developmental delay, Epilepsy, Neonatal Diabetes) s epilepsiou a zaostávaním vo vývoji [56]. Prechodné formy existujú medzi všetkými typmi. Chronické komplikácie diabetu sa vyskytujú v závislosti od kompenzácie ochorenia [55].
Diagnostika sa zakladá na čase vzniku diabetu. Pri permanentnom neonatálnom diabete a syndróme DEND je najčastejšou príčinou mutácia génu KCNJ11, preto sa tento gén analyzuje vždy ako prvý. Pri TNDM je častejšou príčinou mutácia ABCC8, ale pre veľkosť tohto génu (39 exónov) sa analyzuje až po KCNJ11 (má len 1 exón). Pri MODY sa analýza KCNJ11 a ABCC8 robí zvyčajne až pri negatívnom výsledku analýzy častejších príčin (gény pre GCK, HNF1A, HNF4A) [45].
Liečbou voľby sú vo väčšine prípadov deriváty sulfonylmočoviny [28], ktoré sa nadväzujú cez SUR1 podjednotku na kanál a uzatvárajú ho. Umožňujú tak depolarizáciu a vylúčenie inzulínu. Sekréciu inzulínu nestimulujú len priamo, ale B-bunka sa stáva vnímavá na fyziologické stimuly (ako napr. gastrointestinálne neuropeptidy a iné) a vylučuje inzulín podľa potreby tela [47]. Výnimku tvoria pacienti so syndrómom DEND a niektorými mutáciami, výrazne ovplyvňujúcimi funkciu kanála.
Mutácie génu pre inzulín
Mutácie inzulínového génu sú druhou najčastejšou príčinou permanentného diabetu (po mutáciách KCNJ11), ale môžu sa prejavovať aj ako MODY diabetes (v závislosti od typu mutácie) [45].
Genetika. Inzulínový gén má 3 exóny a boli opísané viaceré polymorfizmy a bodové mutácie. Mutácie spôsobujúce PNDM môžu byť autozómovo-dominantné alebo recesívne v homozygotnom stave. MODY spôsobujú výlučne dominantné mutácie v heterozygotnom stave (označuje sa ako MODY-10).
Patogenéza. Najzávažnejšie dominantné mutácie spôsobujú apoptózu B-buniek, ostatné dominantné a recesívne mutácie vedú k tvorbe inzulínu, ktorý je rôzne poškodený (od úplnej afunkčnosti bez vylúčenia do cirkulácie pri PNDM až po miernejšie formy s určitou hladinou inzulínu v sére pri MODY).
Klinický obraz závisí od typu mutácie. Pri PNDM je nulová inzulinémia, často s ketoacidózou. Pri MODY je klinický obraz miernejší: začiatok od detstva až do vyššieho veku, miernejšie príznaky [45].
Diagnostika sa pri PNDM zakladá na DNA analýze – inzulínový gén sa zvyčajne analyzuje, ak sa v KCNJ11 nenájdu žiadne mutácie. V rámci MODY ide o zriedkavú príčinu, preto sa DNA analýza robí až po vylúčení majoritných MODY génov (HNF1A, HNF4A a GCK).
Liečba je pri PNDM výlučne inzulínom, pri MODY sú to nízke dávky inzulínu alebo deriváty sulfonylurey [45].
Menej časté formy neonatálneho diabetu
Ide o celé spektrum génov, ktorých mutácie sa prejavujú ako permanentný neonatálny diabetes s rôznymi vývojovými anomáliami alebo extrapankreatickými príznakmi. Patria sem homozygotné mutácie PDX1 spôsobujúce agenézu pankreasu (heterozygoti majú fenotyp MODY-4), homozygotné mutácie glukokinázy (heterozygoti majú MODY-2), mutácie WRS spôsobujúce Wolcott-Rallisonov syndróm s epifyzeálnymi dyspláziami, mutácie PTF1A s hypopláziou pankreasu a mozočka, mutácie GLIS3 spájané s kongenitálnou hypotyreózou a mutácie Foxp3 prejavujúce sa ako syndróm IPEX s autoimunitným diabetom a ďalším autoimunitnými endokrinopatiami [57].
Homozygotné mutácie HNF1B (heterozygoti majú MODY-5) sa prejavia ako tranzientný neonatálny diabetes s renálnymi abnormalitami [57].
Záver
Z uvedeného prehľadu vyplýva, že počet diagnóz spadajúcich pod monogénový diabetes neustále pribúda. Na ich presnú identifikáciu a následne cielenú liečbu sú nutné metódy DNA analýzy. V Českej republike je viacero pracovísk, ktoré sa zaoberajú DNA diagnostikou monogénových foriem diabetu, na Slovensku je to laboratórium DIABGENE sídliace v Ústave experimentálnej endokrinológie v Bratislave (www.diabgene.sk).
prof. MUDr. Iwar Klimeš, DrSc.
www.endo.sav.sk
e-mail: iwar.klimes@savba.sk
Doručeno do redakce: 24. 9. 2011
Zdroje
1. Hattersley A, Bruining J, Shield J et al. The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes 2009; 10 (Suppl 12): 33–42.
2. Ellard S, Bellanné-Chantelot C, Hattersley AT. European Molecular Genetics Quality Network (EMQN) MODY group. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia 2008; 51: 546–553.
3. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab 2008; 4: 200–213.
4. Vaxillaire MDP, Bonnefond A, Froguel P. Breakthroughs in monogenic diabetes genetics: from pediatric forms to young adulthood diabetes. Pediatr Endocrinol Rev 2009; 6: 405–417.
5. Pruhova S, Dusatkova P, Sumnik Z et al. Glucokinase diabetes in 103 families from a country-based study in the Czech Republic: geographically restricted distribution of two prevalent GCK mutations. Pediatr Diabetes 2010; 11: 529–535.
6. Dusatkova P, Vesela K, Pruhova S et al. Lack of PAX4 mutations in 53 Czech MODYX families. Diabet Med 2010; 27: 1459–1460.
7. Ellard S, Colclough K. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young. Hum Mutat 2006; 27: 854–869.
8. Vesterhus M, Haldorsen IS, Raeder H et al. Reduced pancreatic volume in hepatocyte nuclear factor 1A-maturity-onset diabetes of the young. J Clin Endocrinol Metab 2008; 93: 3505–3509.
9. Katra B, Klupa T, Skupien J et al. Dipeptidyl peptidase-IV inhibitors are efficient adjunct therapy in HNF1A maturity-onset diabetes of the young patients – report of two cases. Diabetes Technol Ther 2010; 12: 313–316.
10. Nyunt O, Wu JY, McGown IN et al. Investigating maturity onset diabetes of the young. Clin Biochem Rev 2009; 30: 67–74.
11. Pinés Corrales PJ, López Garrido MP, Aznar Rodríguez S et al. Clinical differences between patients with MODY-3, MODY-2 and type 2 diabetes mellitus with I27L polymorphism in the HNF1alpha gene. Endocrinol Nutr 2010; 57: 4–8.
12. Pearson ER, Boj SF, Steele AM et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 2007; 4: e118.
13. Dusatkova P, Pruhova S, Sumnik Z et al. HNF1A mutation presenting with fetal macrosomia and hypoglycemia in childhood prior to onset of overt diabetes. J Pediatr Endocrinol Metab 2011; 24: 377–379.
14. Shepherd M, Shields B, Ellard S et al. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med 2009; 26: 437–441.
15. Bazalova Z, Rypackova B, Broz J et al. Three novel mutations in MODY and its phenotype in three different Czech families. Diabetes Res Clin Pract 2010; 88: 132–138.
16. Gonsorcikova L, Pruhova S, Cinek O et al. Autosomal inheritance of diabetes in two families characterized by obesity and a novel H241Q mutation in NEUROD1. Pediatr Diabetes 2008; 9: 367–372.
17. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci USA 2005; 102: 4807–4812.
18. Plengvidhya N, Kooptiwut S, Songtawee N et al. PAX4 mutations in Thais with maturity onset diabetes of the young. J Clin Endocrinol Metab 2007; 92: 2821–2826.
19. Shields BM, Hicks S, Shepherd MH et al. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 2010; 53: 2504–2508.
20. Osbak KK, Colclough K, Saint-Martin C et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat 2009; 30: 1512–1526.
21. Gasperikova D, Tribble ND, Stanik J et al. Identification of a novel beta-cell glucokinase (GCK) promoter mutation (–71G>C) that modulates GCK gene expression through loss of allele-specific Sp1 binding causing mild fasting hyperglycemia in humans. Diabetes 2009; 58: 1929–1935.
22. Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci 2009; 66: 27–42.
23. Gloyn AL, van de Bunt M, Stratton IM et al. Prevalence of GCK mutations in individuals screened for fasting hyperglycaemia. Diabetologia 2009; 52: 172–174.
24. Codner E, Rocha A, Deng L et al. Mild fasting hyperglycemia in children: high rate of glucokinase mutations and some risk of developing type 1 diabetes mellitus. Pediatr Diabetes 2009; 10: 382–388.
25. Martin D, Bellanné-Chantelot C, Deschamps I et al. Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care 2008; 31: 1321–1323.
26. Cuesta-Muñoz AL, Tuomi T, Cobo--Vuilleumier N et al. Clinical heterogeneity in monogenic diabetes caused by mutations in the glucokinase gene (GCK-MODY). Diabetes Care 2010; 33: 290–292.
27. Feigerlova E, Pruhova S, Dittertova L et al. Aetiological heterogeneity of asymptomatic hyperglycaemia in children and adolescents. Eur J Pediatr 2006; 165: 446–452.
28. Tanaka D, Nagashima K, Sasaki M et al. GCKR mutations in Japanese families with clustered type 2 diabetes. Mol Genet Metab 2011; 102: 453–460.
29. Oram RA, Edghill EL, Blackman J et al. Mutations in the hepatocyte nuclear factor-1beta (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am J Obstet Gynecol 2010; 203: 364 e1–e5.
30. Laloi-Michelin M, Meas T, Ambonville C et al. Mitochondrial Diabetes French Study Group. The clinical variability of maternally inherited diabetes and deafness is associated with the degree of heteroplasmy in blood leukocytes. J Clin Endocrinol Metab 2009; 94: 3025–3030.
31. Vincent-Desplanques D, Faivre-Defrance F, Wémeau JL et al. Monogenic severe insulin resistance syndromes. Rev Med Interne 2005; 26: 866–873.
32. Suliman SG, Stanik J, McCulloch LJ et al. Severe insulin resistance and intrauterine growth deficiency associated with haploinsufficiency for INSR and CHN2: new insights into synergistic pathways involved in growth and metabolism. Diabetes 2009; 58: 2954–2961.
33. Musso C, Cochran E, Moran SA et al. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore) 2004; 83: 209–222.
34. Johansson BB, Torsvik J, Bjørkhaug L et al. Diabetes and Pancreatic Exocrine Dysfunction Due to Mutations in the Carboxyl Ester Lipase Gene-Maturity Onset Diabetes of the Young (CEL-MODY): a protein misfolding disease. J Biol Chem 2011; 286: 34593–34605.
35. Raeder H, Johansson S, Holm PI et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet 2006; 38: 54–62.
36. Torsvik J, Johansson S, Johansen A et al. Mutations in the VNTR of the carboxyl-ester lipase gene (CEL) are a rare cause of monogenic diabetes. Hum Genet 2010; 127: 55–64.
37. Borowiec M, Liew CW, Thompson R et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and beta-cell dysfunction. Proc Natl Acad Sci USA 2009; 106: 14460–14465.
38. Garg A. Acquired and inherited lipodystrophies. N Engl J Med 2004; 350: 1220–1234.
39. Garg A, Misra A. Lipodystrophies: rare disorders causing metabolic syndrome. Endocrinol Metab Clin North Am 2004; 33: 305–331.
40. Gomes KB, Fernandes AP, Ferreira AC et al. Mutations in the seipin and AGPAT2 genes clustering in consanguineous families with Berardinelli-Seip congenital lipodystrophy from two separate geographical regions of Brazil. J Clin Endocrinol Metab 2004; 89: 357–361.
41. d‘Annunzio G, Minuto N, D‘Amato E et al. Wolfram syndrome (diabetes insipidus, diabetes, optic atrophy, and deafness): clinical and genetic study. Diabetes Care 2008; 31: 1743–1745.
42. Domènech E, Gómez-Zaera M, Nunes V. Study of the WFS1 gene and mitochondrial DNA in Spanish Wolfram syndrome families. Clin Genet 2004; 65: 463–469.
43. Zaidi G, Sahu RP, Zhang L et al. Two novel AIRE mutations in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) among Indians. Clin Genet 2009; 76: 441–448.
44. Polak M, Shield J. Neonatal Diabetes Mellitus – genetic aspects 2004. Pediatr Endocrinol Rev 2004; 2: 193–198.
45. Edghill EL, Flanagan SE, Patch AM et al. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008; 57: 1034–1042.
46. Polak M. Neonatal diabetes mellitus: insights for more common forms of diabetes. J Clin Endocrinol Metab 2007; 92: 3774–3776.
47. Stanik J, Gasperikova D, Paskova M et al. Prevalence of permanent neonatal diabetes in Slovakia and successful replacement of insulin with sulfonylurea therapy in KCNJ11 and ABCC8 mutation carriers. J Clin Endocrinol Metab 2007; 92: 1276–1282.
48. Shield JP, Temple IK, Sabin M et al. An assessment of pancreatic endocrine function and insulin sensitivity in patients with transient neonatal diabetes in remission. Arch Dis Child Fetal Neonatal Ed 2004; 89: F341–F343.
49. Mackay DJ, Boonen SE, Clayton--Smith J et al. A maternal hypomethylation syndrome presenting as transient neonatal diabetes mellitus. Hum Genet 2006; 120: 262–269.
50. Stanik J, Lethby M, Flanagan SE et al. Coincidence of a novel KCNJ11 missense variant R365H with a paternally inherited 6q24 duplication in a patient with transient neonatal diabetes. Diabetes Care 2008; 31: 1736–1737.
51. Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet 2002; 39: 872–875.
52. Mackay DJ, Callaway JL, Marks SM et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet 2008; 40: 949–951.
53. Bonnefond A, Durand E, Sand O et al. Molecular diagnosis of neonatal diabetes mellitus using next-generation sequencing of the whole exome. PLoS One 2010; 5: e13630.
54. Männikkö R, Flanagan SE, Sim X et al. Mutations of the Same Conserved Glutamate Residue in NBD2 of the Sulfonylurea Receptor 1 Subunit of the KATP Channel Can Result in Either Hyperinsulinism or Neonatal Diabetes. Diabetes 2011; 60: 1813–1822.
55. Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes 2005; 54: 2503–2513.
56. Flanagan SE, Patch AM, Mackay DJ et al. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007; 56: 1930–1937.
57. Bonnefond A, Froguel P, Vaxillaire M. The emerging genetics of type 2 diabetes. Trends Mol Med 2010; 16: 407–416.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2011 Číslo 11
- MINISERIÁL: Když ženám stoupá tlak...
- Není statin jako statin aneb praktický přehled rozdílů jednotlivých molekul
- Specifika v komunikaci s pacienty s ránou – laická doporučení
- Moje zkušenosti s Magnosolvem podávaným pacientům jako profylaxe migrény a u pacientů s diagnostikovanou spazmofilní tetanií i při normomagnezémii - MUDr. Dana Pecharová, neurolog
Nejčtenější v tomto čísle
- Antibiotická léčba akutních bakteriálních infekcí
- Terapie obezity – postupy, účinnost a perspektivy
- Moderní technologie v diabetologii. CSII (kontinuální subkutánní infuze inzulinu) a CGM (kontinuální glykemický monitoring) v klinické praxi
- Genetika monogénových foriem diabetu