
Holoprosencefalie
Authors:
A. Nogolová 1; N. Filáková 1; J. Všetička 2
Authors‘ workplace:
Oddělení dětského lékařství, Městská nemocnice Ostrava, p. o.
1; Genetika Ostrava s. r. o.
2
Published in:
Čes-slov Pediat 2017; 72 (6): 345-350.
Category:
Case Report
Overview
Vrozené vývojové vady centrálního nervového systému zahrnují široké spektrum anomálií, které vznikají v průběhu ontogeneze mozku a míchy. Etiologie vad je značně heterogenní, na jejich vzniku se podílejí faktory genetické, exogenní či endogenní noxy. V 60 % případů zůstává etiologie nejasná. Vyskytují se zhruba u 0,5–0,7 % novorozenců, stojí často za úmrtím fétu. Postnatálně se podílejí na vysoké morbiditě i mortalitě dětí, jsou častou příčinou psychomotorické retardace, smyslového postižení, epilepsie atd. Klinický obraz je závislý na typu a rozsahu vady. Terapie bývá většinou jen symptomatická. Diagnostika je možná i prenatálně, zvláště přínosná jsou strukturální zobrazení – sonografie, MRI. Důležitá je péče genetika a multioborová spolupráce.
KLÍČOVÁ SLOVA:
malformace centrální nervové soustavy, holoprosencefalie, hypernatremie
ÚVOD
Vrozené vývojové vady centrálního nervového systému zahrnují široké spektrum anomálií, které vznikají v průběhu ontogeneze mozku a míchy. Celkem je popsáno více než 2000 vývojových vad [1]. Jednotlivé struktury nervového systému se vyvíjejí podle daného časového a prostorového plánu. Neurální ploténka se zakládá kolem 18. dne gestace. Z ploténky se vytváří neurální roura, která se kaudálně uzavírá během 3. a 4. týdne. Do 5. gestačního týdne probíhá primární segmentace mozku na prosencefalon, mezencefalon a rombencefalon, vytváří se základy chorioideálního plexu a poté mozečku. Komorový systém se formuje v 8. týdnu , v 10. týdnu corpus callosum. K migraci a proliferaci buněk dochází hlavně mezi 7.–25. týdnem. Gyrifikaci pozorujeme od 22.–24. týdne, končí kolem 40. týdne gestace. Synaptogeneze je patrná zhruba od 4. měsíce vývoje a pokračuje po celý život. Myelinizace začíná kolem 20. gestačního týdne ve hřbetní míše, postupuje kraniálně a pokračuje i postnatálně [1–3].
Malformace CNS, které vznikají v časných fázích vývoje, bývají většinou podmíněny genetickými faktory. Z exogenních či endogenních nox se uplatňuje maternální infekce, diabetes mellitus, radiace, medikace, působení toxinů atd. V pozdějším údobí gestace dominují léze destruktivního charakteru nebo poruchy dozrávání již vytvořených struktur CNS. V 60 % případů zůstává etiologie nejasná [2].
Malformace CNS se vyskytují zhruba u 0,5–0,7 % novorozenců, u potracených plodů až ve 3 %. Jsou příčinou cca 75 % úmrtí fétu [2]. Postnatálně se podílejí na vysoké morbiditě i mortalitě dětí, jsou častou příčinou psychomotorické retardace, smyslového postižení, epilepsie atd. Klinický obraz je závislý na typu a rozsahu vady. Malformace CNS můžeme dělit podle mnoha hledisek – např. podle období vzniku, narušeného vývojového procesu, z hlediska molekulární genetiky atd.
KAZUISTIKA
Rodinná anamnéza pacientky je negativní. Matka pravidelně docházela do prenatální poradny. V 8. týdnu gravidity susp. prodělala parotitidu (nepotvrzeno laboratorně ). Je kuřačka. Dítě se rodí z první gravidity, porod byl indukován ve 40. týdnu gestace pro růstovou stagnaci plodu. Novorozenec ženského pohlaví byl porozen záhlavím, porodní hmotnost byla 2800 g, délka 46 cm. Bez poruch časné adaptace. Ihned po narození zjištěna mikrocefalie, OH byl 29 cm, kraniofaciální stigmatizace. Podle sonografického vyšetření mozku byla verifikována vývojová vada mozku – susp. semilobární holoprosencefalie. K přesné identifikaci vady a event. přidružených malformací byla doplněna ve 3,5 měsících věku MRI mozku s potvrzením sonografického nálezu (obr. 1).
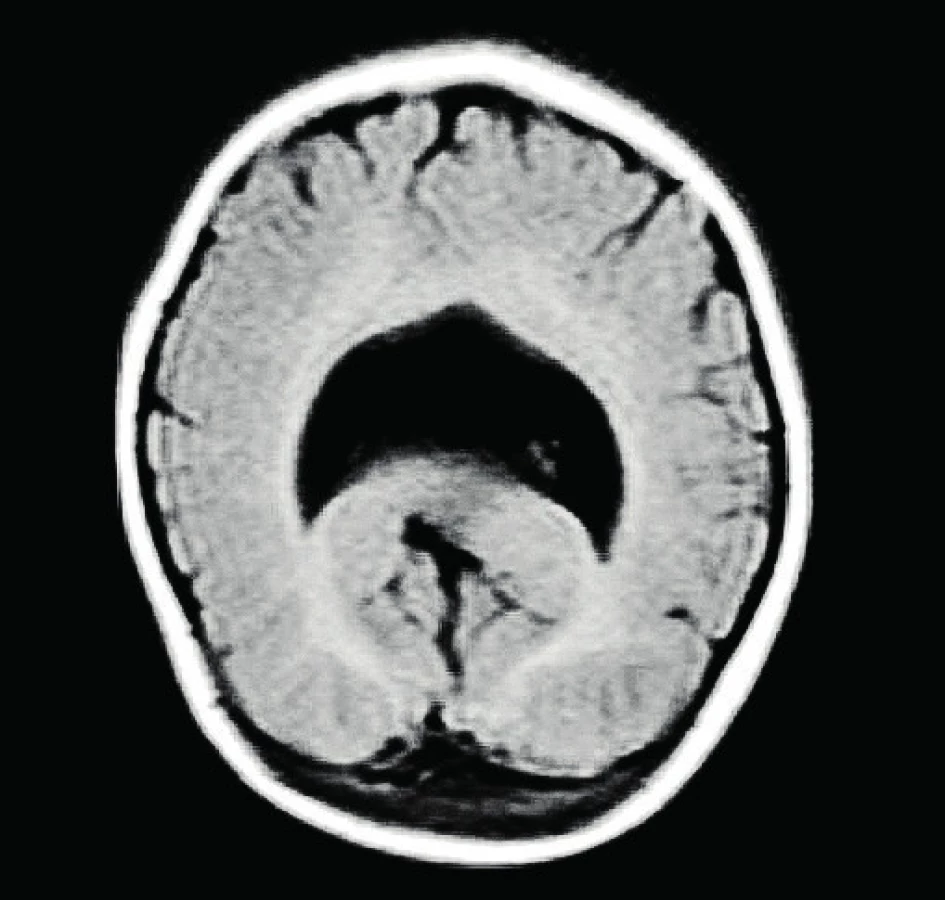
Podle genetického vyšetření – karyotyp 46,XX. Molekulárně genetické vyšetření nachází mutaci c.748C>T p. (Gln250Ter) v exonu 1 genu ZIC2 v heterozygotním stavu, vyšetření genů SHH, SIX3 a TGIF bylo negativní. Zjištěná mutace vede ke vzniku předčasného terminačního kodonu a ztrátě exprese funkčního proteinu na mutované alele. Daná mutace podle současných poznatků je patogenní a je molekulární příčinou HPE u probandky. Jedná se o mutaci de novo vzniklou, oba rodiče byli dovyšetřeni s negativními nálezy. Výsledky dalších paraklinických vyšetření – OAE nevýbavné, vyšetření BERA oboustranně bez odpovědi i v nejvyšších stimulačních hladinách – potvrzuje oboustranně těžkou sluchovou vadu. Podle kardiologického vyšetření hemodynamicky nevýznamné FOA. Sonografie břicha bez patologie. EEG je opakovaně bez specifické abnormity, s opožděním základní elektrogenezy. Z důvodu základní diagnózy středočárové abnormity CNS byla dívka časně zařazena i do péče endokrinologa.
Zpočátku byly odběry s nálezy v mezích normy. V 8 měsících věku při rutinních biochemických testech byla zjištěna iontová dysbalance – hypernatremie (s max. hodnotou 170 μmol/l), hyperchloremie (s max. hodnotou 125 μmol/l), hyperosmolalita až 346 mmol/kg. Kalium v mezích, glykemie opakovaně v normě. Z výsledků hormonálního screeningu: nízká hodnota IGF-1 – 16 μg/l (norma 50–170), kortizol 234 nmol/l (norma 50–650), ACTH 220 ng/l (norma 8–40). Prolaktin v normě. Osmolalita moče v rozmezí 135–250 mmol/kg (norma 50–1160). Odpad Na+ močí/24 hodin byl 14,5 mmol (norma 20–60). Opakovaně byla vyšetřena bilance tekutin: příjem tekutin za 24 hodin činil 550–700 ml (90–120 ml/kg/den), výdej 300–440 ml (hodinová diuréza 2–2,9 ml/kg/hod).
Z terapie nasazen Minirin (desmopressinum – syntetický analog vazopresinu), pro vysoké hodnoty ACTH byla léčba doplněna o hydrocortison. Na medikaci pozvolna dochází k úpravě vnitřního prostředí, hodnoty však výrazně kolísají podle aktuálního stavu hydratace dítěte a v průběhu akutních respiračních infektů. V kompenzovaném stavu jsou hodnoty sérové koncentrace Na+ v rozmezí 142–152 mmol/l, Cl- 107–117 mmol/l. Nedošlo k zásadnímu ovlivnění osmolality moče, zůstává na hodnotách dolní hranice normy (maxim. zaznamenaná hodnota osmolality moče byla 343 mmol/kg). Odpad Na+ močí/24 hodin na medikaci je 16,1 mmol, koncentrace Na+ v moči 29 mmol/l.
V 10 měsících vývojově dívka odpovídá přelomu I.–II. trimenonu, je výrazně hypertonická. Celkově je neklidná, dráždivá, špatně usíná. OH je 39 cm (výrazně pod 3. percentilem). Má okohybnou poruchu charakteru konvergentního strabismu. Sluchová vada je řešena naslouchadly. V dosavadním průběhu sledování nedošlo k rozvoji epileptických záchvatů. Dívka neprospívá. Váží 6050 g, měří 66,5 cm. Poměr výška/věk je na 3. percentilu, váha/výška na 25. percentilu (obr. 2a, 2b). Prozatím živena per os.


DISKUSE
HPE je vadou, která vzniká v období ventrální indukce. V tomto údobí mohou vzniknout také další typy vad, např. septooptická dysplazie, Dandyho-Walkerova malformace atd. Podstatou HPE je porucha indukční funkce prechordálního mezodermu. Jde o anomálii střední čáry, kdy není dokončena separace prosencefala na dva telencefalické váčky. Za fyziologických okolností z telencefalických váčků vznikají hemisféry s postranními komorami a diencefalon, z něhož pak vycházejí oční váčky, thalamy a hypothalamus. Telencefalické váčky po rozdělení mediálně rotují a tím v sobě uzavírají postranní komory. V místě jejich kontaktu vzniká interhemisferální rýha [3]. HPE je vadou vzácnou, vzniká velmi časně, fyziologicky se jednotlivé hemisféry začínají oddělovat zhruba 33. den gestace.
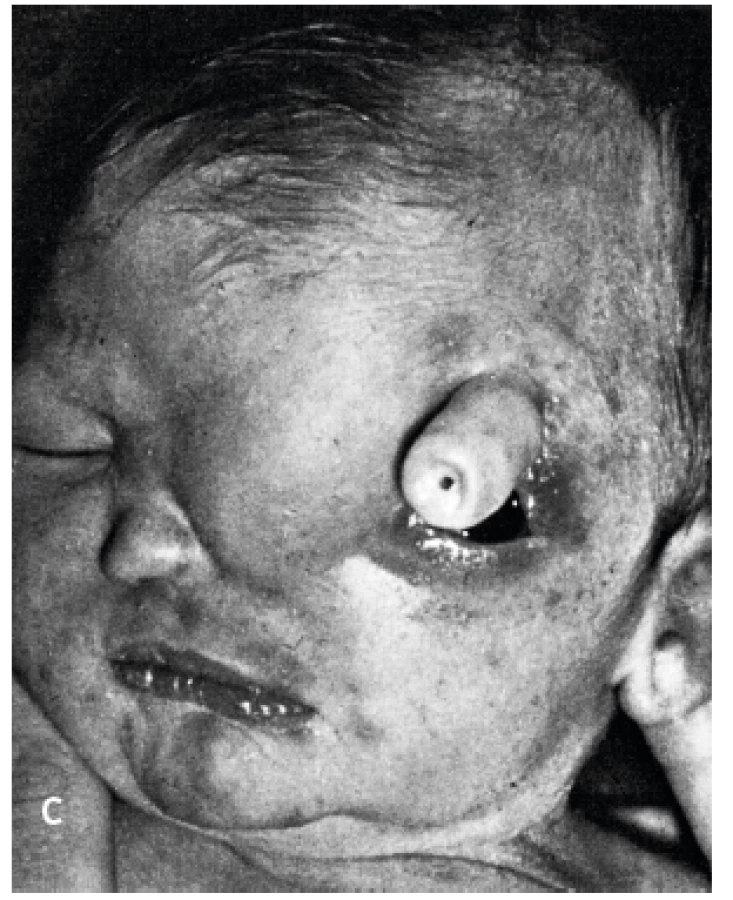
HPE má několik typů (obr. 4). Nejtěžší vadou je alobární HPE, která tvoří více než 60 % všech HPE [4]. Komorový systém splývá do jediného monoventriklu, kolem kterého je tenký lem mozkové tkáně, zcela chybí středočárové struktury, není patrný falx ani interhemisferická rýha [1–3]. Mozek je zásoben jen jednou arteria cerebri anterior, aa. cerebri mediae jsou ale párové [2]. Postižení jedinci mívají často těžké vady obličeje (proboskis1, etmocefalii2, cebocefalii3, premaxilární agenezi4, kyklopismus5, hypoplazii nosu, rozštěpové vady (obr. 3a, 3b, 3c ), rigiditu, poruchy dýchání ( apnoické pauzy ), poruchy termoregulace [2, 5].
1proboskis – chobotovitý výběžek
2etmocefalie – okulární hypotelorismus s/bez proboskisem
3cebocefalie – okulární hypotelorismus s 1 nosním otvorem
4premaxilární ageneze – okulární hypotelorismus, plochý nos, střední rozštěp rtu
5kyklopie – ve střední čáře uložená 1 orbita s očním bulbem a nad ní prominence rudimentárního nosu
(Zdroj: Wiedemann HR, Kunze KJ. Atlas klinických syndromů pro kliniku a praxi. 1. české vyd. Martin: Osveta, 1996: 48–49. ISBN 80-217-0517-5.)


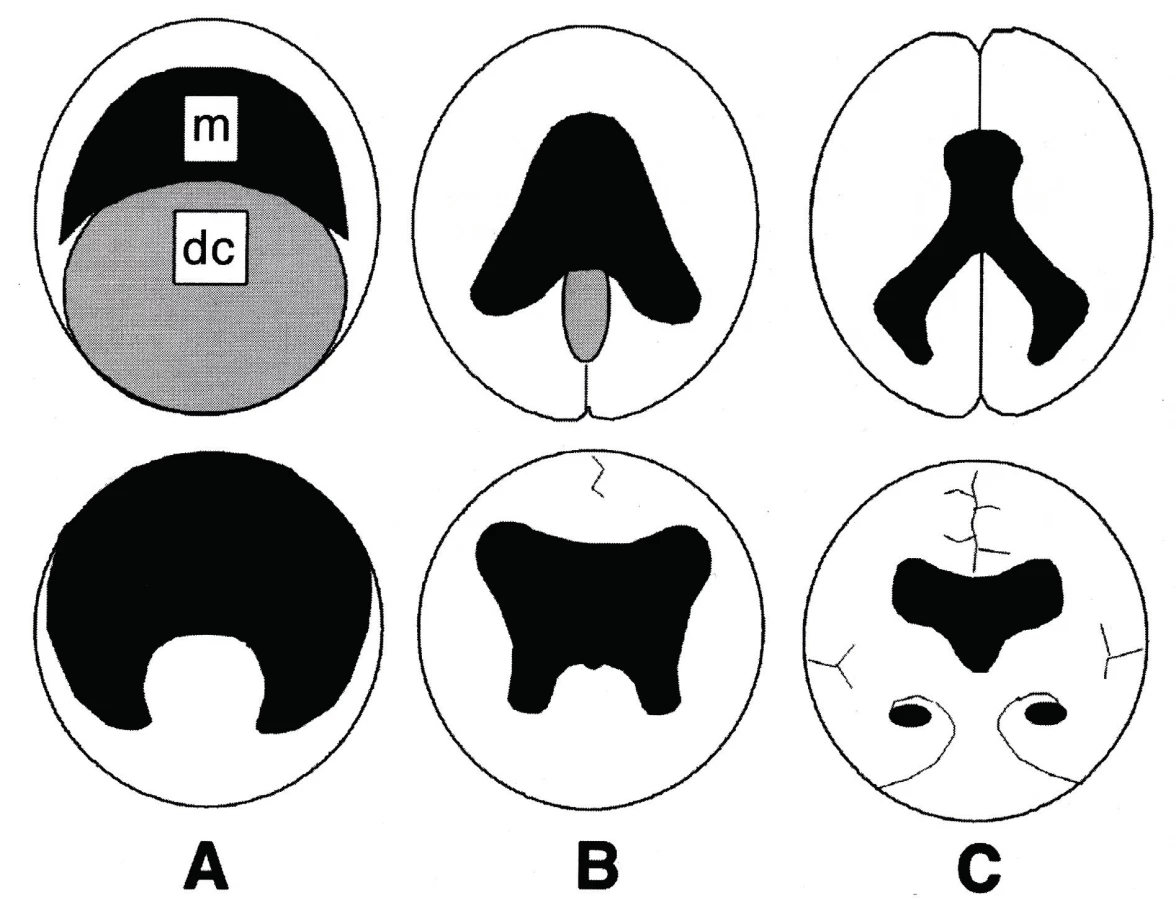
Semilobární HPE je nejčastější vadou u přeživších novorozenců a tvoří zhruba 25 % všech HPE [4]. Parciálně jsou vytvořeny a odděleny okcipitální rohy, hemisféry mají silnější plášť, okcipitálně je naznačená interhemisferická rýha. Je přítomna částečná separace bazálních ganglií a thalamů [1]. U formy lobární jsou nedokonale separovány oblasti předních a dolních oblastí frontálních laloků, chybí septum pellucidum. U těchto lehčích forem může být přítomna jen minimální faciální stigmatizace, např. hypotelorismus, ploché čelo, široké filtrum, větší uši atd. Děti bývají mikrocefalické [1–2].
Incidence HPE je odhadována na 1:10 000–1:16 000 živě narozených dětí, prevalence HPE v raném embryonálním údobí je však daleko vyšší (1:250) [4]. Riziko opakování vady v další graviditě – pokud nejde o chromozomální aberaci – je cca 6% [2–3]. Častěji se vyskytuje u dívek.
Rizikovými faktory pro vznik HPE jsou maternální diabetes, syfilis, infekce CMV , toxoplazmóza (komplex TORCH). Z exogenních nox např. alkohol, nikotinismus a toxiny cigaretového kouře, drogová závislost, z léků kyselina acetylsalicylová, lithium, retinoidy, antikonvulziva.
HPE se zhruba ve 25–50 % pojí se strukturálními či numerickými aberacemi chromosomů (1q+, 11q+, trisomie 13, 18...) [3, 6]. Jsou popsány formy syndromické autosomálně dominantní (Pallisterův-Hallův syndrom, Rubinsteinův-Taybiho syndrom, Kallmannův syndrom atd.). Mezi formy syndromické s autosomálně recesivní dědičností patří syndrom Smithův-Lemliho-Opitzův, Meckelův syndrom atd. [6]. Formy non-syndromické jsou podmíněny mutacemi mnoha genů, které se uplatňují v řízení ventrodorzálního gradientu vývoje mozku. Prvním genem asociovaným s HPE, který byl identifikován, jsou mutace genu SHH (Sonic Hedgehog), který kóduje jeden z transkripčních faktorů nutných pro časný vývoj CNS. Mutace genu ZIC2, který je lokalizován na chromosomu 13q32, je druhou nejčastější příčinou non-syndromické HPE [6, 7]. Mezi další příčiny patří mutace ventralizačních genů – SIX3, TGIF, receptoru pro SHH označovaného jako PTCH atd. [2, 6]. Existuje určitá korelace mezi genotypem a fenotypem jedince.
HPE často doprovází i další vady CNS – cysty zadní jámy [3], defekty neurální trubice, hydrocefalus, syndrom kaudální dysgeneze. Postižení jedinci často trpí epileptickými záchvaty, mají abnormní spánkový cyklus. Mohou být přítomny abnormity uropoetického traktu, kardiovaskulárního systému, omfalokéla, stenóza anu, poly/syndaktylie, hypoplazie až chybění palců ruky [5]. Postižení jedinci trpí závažnou oromotorickou dysfunkcí, častý je gastroezofageální reflux. Z toho plyne i zvýšené riziko aspiračních komplikací. HPE mohou provázet projevy endokrinní dysfunkce s poruchou vodní a elektrolytové rovnováhy. Hypernatrémie vzniká na podkladě centrální adipsie v kombinaci s poruchou sekrece vazopresinu (adipsický diabetes insipidus). Vazopresin (AVP) vylučují peptigerní neurony hypothalamu. Produkce AVP je regulovaná hodnotou osmolality plazmy, na kterou reagují centrální osmoreceptory uložené na anterolaterální stěně III. postranní komory. Na neosmotické regulaci sekrece AVP se podílí baroreceptory centrálních žil a srdce. Podle lokalizace defektu mozku existují různé druhy abnormit osmoregulačního systému. Postižení jedinci nemají pocit žízně, je vyšší osmotický práh sekrece vazopresinu (tzv. „reset osmostat“). Parciální centrální diabetes insipidus je definován sníženou sekrecí vazopresinu. Třetí možností je defekt v uvolňování vazopresinu na osmotické podněty při zachované baroregulaci sekrece hormonu [8, 9]. Hypernatrémie bývá akcentována během akutních infektů a při nedostatečném přívodu tekutin. Častý je deficit růstového hormonu a z toho plynoucí růstová retardace [10].
Děti s klinicky těžkými formami HPE přežívají jen několik měsíců po narození.
Terapie je převážně symptomatická – hormonální substituce, antikonvulzivní terapie, prevence aspiračních přívod, pomůcky ke krmení včetně zavedení perkutánní gastrostomie. Korektivní a plastické operace rozštěpových vad, shuntové operace hydrocefalu. Rehabilitační a ošetřovatelská péče.
Diagnostika je možná prenatálně. Uplatňují se strukturální zobrazovací metody – sonografie, MRI. Zásadní je vyšetření genetikem.
ZÁVĚR
Vývojové vady centrální nervové soustavy stojí zhruba za 75 % úmrtí ve fetálním období. V postnatálním období pak často zodpovídají za postižení dítěte, jsou příčinou rozvoje farmakorezistentní epilepsie atd. Diagnostika těchto vad je možná prenatálně, nicméně i přes mohutný rozvoj strukturálních zobrazovacích metod mohou uniknout diagnostice, tak jak tomu bylo právě v našem případě. Multioborová péče je základem péče o tyto děti.
Zkratky:
ACTH – adrenokortikotropní hormon
AVP – vazopresin
BERA – Brainstem Evoked Responses Audiometry – sluchové kmenové evokované potenciály
CNS – centrální nervový systém
EEG – elektroencefalografie
FOA – foramen ovale apertum
HPE – holoprosencefalie
IGF-1 – insulin-like growth factor 1
MRI – magnetic resonance imaging – magnetická rezonance
OAE – otoakustické emise
OH – obvod hlavy
Došlo: 23. 3. 2017
Přijato: 17. 5. 2017
MUDr. Alice Nogolová
Oddělení dětského lékařství
Městská nemocnice Ostrava, p.o.
Nemocniční 20
728 80 Ostrava 2
e-mail: alice.nogolova@mnof.cz
Sources
1. Seidl Z, Vaněčková M. Diagnostická radiologie – neuroradiologie. 1. vyd. Praha: Grada, 2007: 57–59, 66–68. ISBN 978-80-247-1106-5.
2. Menkes JH, Sarnat HB, Maria BL. Dětská neurologie, 1. díl. 7. vyd. Praha: Triton, 2011: 414–417, 455–461. ISBN 978-80-7387-341-7.
3. Hadač J. Ultrazvukové vyšetření mozku přes velkou fontanelu. 1. vyd. Praha: Triton 2000: 45–50. ISBN 80-7254-110-2.
4. Paulussen A, Schrander-Stumpel CT, Tserpelis DCJ, et al. The unfolding clinical spectrum of holoprosencephaly due to mutations in SHH, ZIC2, SIX3 and TGIF genes. Europ J Hum Genet 2010; 18: 999–1005.
5. Wiedemann HR, Kunze KJ. Atlas klinických syndromů pro kliniku a praxi. 1. české vyd. Martin: Osveta, 1996: 48–49. ISBN 80-217-0517-5.
6. Savastano CP, El-JAick KB, Costa-Lima MM, et al. Molecular analysis of holoprosencephaly in South America. Genet Mol Biol 2014 Mar; 37 (1 Suppl): 250–262.
7. Solomon BD, Lacbawan F, Mercier S, et al. Mutations in ZIC2 in human prosencephaly: description of a novel ZIC2 specific phenotype and comprehensive analysis of 157 individuals. J Med Genet 2010; 47 (8): 513–524.
8. Lebl J, Al Taji E, Koloušková S, a kol. Malý atlas dětské endokrinologie. 1. vyd. Praha: Galén, 2013: 9–11. ISBN 978-80-7492-065-3.
9. Kovács L, Jankó V, Nagyová G, Dallos T. Adipsický diabetes insipidus u pacienta s dysgenézou corpus callosum. Čes-slov Pediat 2014; 69 (1): 12–20.
10. Lebl J, Dušátková P, Malíková J, Obermannová B. Má genetické vyšetření u dětí s nedostatkem růstového hormonu klinický význam? Pediatr praxi 2013; 14 (6): 376–378.
Labels
Neonatology Paediatrics General practitioner for children and adolescentsArticle was published in
Czech-Slovak Pediatrics

2017 Issue 6
Most read in this issue
- Holoprosencefalie
- Odporúčania pre pohybovú aktivitu detí a mládeže na Slovensku (6–18 rokov)
- Nehojící se atopický ekzém
- Specifika péče o děti narozené po asistované reprodukci