
Familial hemophagocytic lymphohistiocytosis: from autopsy to prenatal diagnosis. Report of a case
Authors:
Marta Ježová 1; Renata Gaillyová 2
Authors‘ workplace:
Department of Pathology, University Hospital Brno and Medical College of Masaryk University, Czech Republic
1; Department of Clinical Genetics, University Hospital Brno and Medical College of Masaryk University, Czech Republic
2
Published in:
Čes.-slov. Patol., 53, 2017, No. 1, p. 29-34
Category:
Original Article
Overview
Hemophagocytic lymphohistiocytosis is a rare immunologic disorder affecting small children. It is characterized by an excessive and injurious immune response which turns rapidly fatal unless promptly and effectively treated. The main clinical signs are prolonged fever, hepatosplenomegaly, bleeding and laboratory findings of pancytopenia, increased serum transaminases, hypertriglyceridemia and hypofibrinogenemia. Four genes responsible for familiar hemophagocytic lymphohistiocytosis, which is inherited in autosomal recessive manner, have been identified so far. This case report describes a fatal case of familiar hemophagocytic lymphohistiocytosis caused by compound heterozygote mutation for perforin. A previously healthy neonate, first child of noncosanguineous young healthy parents, presented with hypothermia and fulminant hepatic failure at 28 days of life and succumbed short after. The diagnosis was made at autopsy and confirmed by genetic testing postmortem. Five months later prenatal testing confirmed carrier status in the sibling to be born. This is to our knowledge only the second case of familiar hemophagocytic lymphohistiocytosis caused by perforin deficit in a Czech patient.
Keywords:
familial hemophagocytic lymphohistiocytosis – liver failure – hypothermia – neonate – prenatal diagnosis
The condition now termed hemophagocytic lymphohistiocytosis (HLH) was first described as familial hemophagocytic reticulosis in two affected siblings in 1952 (1). HLH is classified into primary (hereditary) and secondary (acquired). Primary HLH is further divided into familial hemophagocytic lymphohistiocytosis (FHLH) and closely related immunodeficiencies which manifest by partial albinism, bleeding, ceroidosis and high susceptibility to HLH (Chediak-Higashi, Heřmanský-Pudlák, Griscelli syndrome). They are transmitted in an autosomal recessive manner. X-linked lymphoproliferative syndrome is another cause of primary HLH in men. Secondary HLH is a reactive condition associated with certain infections, rheumatologic disorders and malignancies. Epstein-Barr virus is the most common cause of infection-associated HLH (formerly virus-associated hemophagocytic syndrome). Viral infection can trigger an attack in genetically predisposed individuals too (2).
The peak incidence of FHLH is between one and six months. 70 – 80 % of patients are below one year of age, however older age groups may be affected including adults (late-onset FHLH). Approximately 10 % manifest in the neonatal period, some immediately after birth or in utero (3). The estimated incidence varies because of uneven geographic distribution. The annual incidence rate of 0.12 – 0.15 per 100 000 children which equals to 1.8 – 2.2 per 100 000 live births was calculated in Sweden (4).
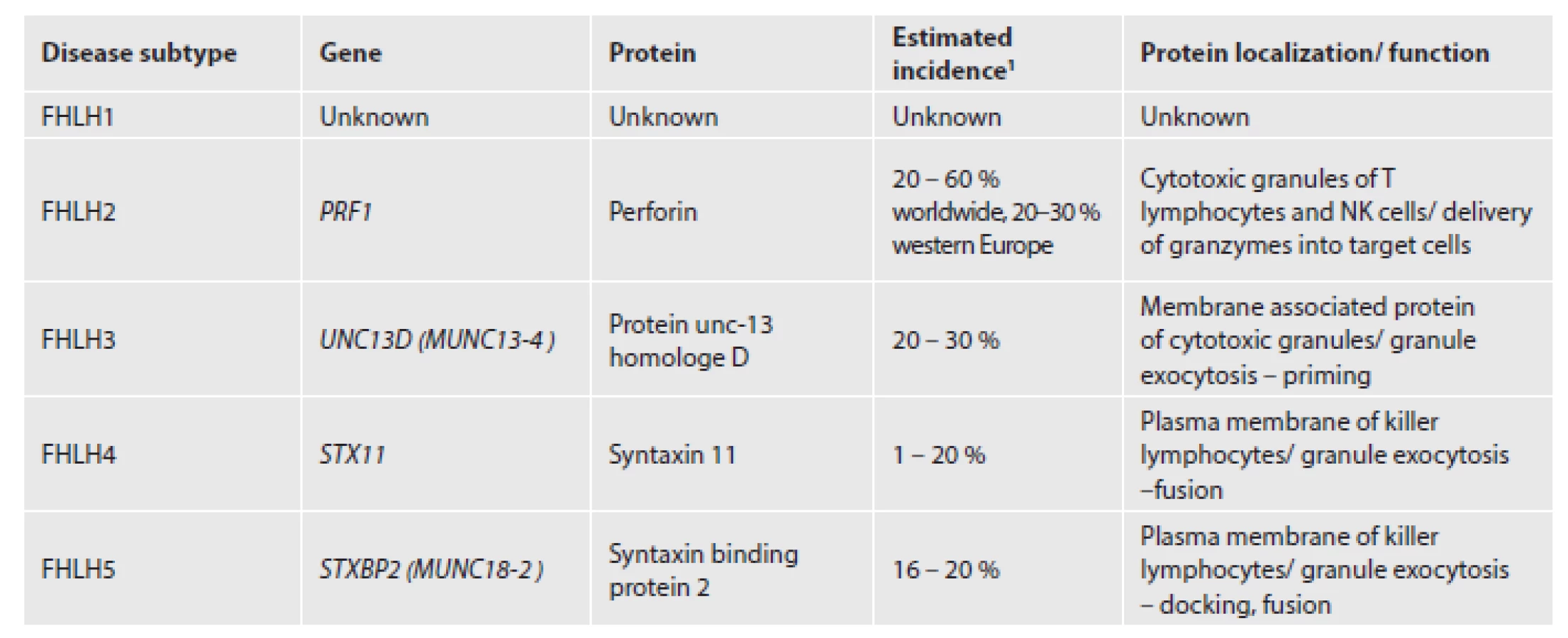
The genetic background is heterogeneous and five disease subtypes have been described (Tab. 1). Approximately one third of FHLH cases are caused by mutations in the perforin gene which maps to 10q22. Since discovery of perforin mutation in 1999, three other causative genes have been identified (5). FHLH type 1 has been linked to chromosome 9 with unknown genetic defect (6). Still 20 – 50 % of involved genes are yet to be explored. Perforin is a pore-forming protein stored in secretory granules of cytotoxic lymphocytes and natural killer (NK) cells. It has an absolutely essential role in granzyme mediated apoptosis of virus-infected and malignant cells. Perforin provides delivery of granzymes into the cytosol of target cells where they activate caspases responsible for the execution phase of programmed cell death (7). Protein encoded by UNC13D and two partner proteins syntaxin 11 and syntaxin binding protein 2 are employed in secretory granules trafficking (8,9). Cytotoxic lymphocytes and NK cells from these patients carry normal perforin but fail to degranulate.
The diagnosis can be established if molecular genetic testing for FHLH is positive or at least five of eight diagnostic criteria are fulfilled (Tab. 2). The cardinal symptoms are fever, splenomegaly with or without hepatomegaly and cytopenias. Hepatosplenomegaly is observed from the beginning and worsens with time. Normocytic anemia and thrombocytopenia are early signs, neutropenia evolves later. Without treatment severe pancytopenia is the rule (3). Additional findings in less than 50 % of patients are lymphadenopathy and polymorphous skin rash (10,11). Children with respiratory distress, hypotension, severe bleeding and multiorgan dysfunction syndrome need intensive care (12). Severe meningoencephalitis may emerge at initial clinical presentation or later with progression of the disease (13).

The pathogenesis involves uncontrolled activation and proliferation of non-malignant CD8+ T lymphocytes and macrophages, infiltration of these cells into host tissues and hypersecretion of proinflammatory cytokines. Characteristic laboratory values include high levels of ferritin, hypofibrinogenemia, hyperlipidemia with increased triglycerides and normal cholesterol, hypoalbuminemia and hyperbilirubinemia (3,14). Hepatic dysfunction occurs early and diagnosing HLH in the absence of at least increased transaminases is discouraged (14).
The median survival without treatment is less than two months (10). Most patients die of hemorrhage or infection. Timely diagnosis is crucial and saves the lives. Current therapeutic protocol recommends initial combination of chemotherapy and immunosuppressive therapy. Subsequent allogenic hematopoietic stem cell transplantation (HSCT) is the only curative option for all primary HLH (14,15). Modern therapeutic regimens reached an overall 5-year survival of 54 % with a probability of survival of 66 % after HSCT. No patient with familial HLH survived without transplantation. The prognosis is considerably worse in infants below 6 months of age (16). The survival in a small cohort of neonates was only 40 % (17).
CLINICAL HISTORY
The proband, a full term baby girl, was born by normal vaginal delivery with birth weight of 2940 g. Birth history was unremarkable except for ABO incompatibility. Clinical examination at 2 weeks reported healthy newborn with satisfactory weight gain on formula. The baby became sick and was referred to pediatric intensive care unit by the local pediatrician at 4 weeks of age because of sudden onset of jaundice and black stool. On physical examination the baby presented with icterus, sunken fontanelle, protuberant abdomen with liver 3 cm below the costal margin, week peristalsis, and bloody stool on the nappy. She was tachypnoic, somnolent and hypothermic (34.5
oC). Capillary refilling time was sluggish, nasogastric tube drained milk with fresh blood. The condition rapidly deteriorated with enlarging abdomen, bradycardia and significant hemorrhage from the oral cavity. Astrup acid-base balance (capillary blood) showed severe acidosis (pH 7.02), hypocapnia and low oxygen saturation. Initial blood count revealed anemia. Ultrasonography suspected bilateral hydrothorax and free gas in the abdominal cavity, but paracentesis drained only a small amount of serous fluid. Despite aggressive treatment including blood cell transfusion, fresh frozen plasma, volume resuscitation and vasopressors to restore the circulation, the baby died 2 hours after admission. The last laboratory test showed high transaminases, hyperbilirubinemia, low serum albumin, thrombocytopenia and elevated CRP (20.7 mg/l). Coagulation profile test was critically abnormal with INR > 5 and low fibrinogen. The death was attributed to fulminant liver failure of unknown etiology.
MATERIALS AND METHODS
Autopsy was performed 24 hours after death. Tissue specimens were fixed in formalin and embedded in paraffin. Small specimens of liver and muscle were deeply frozen. Workup for viral infections using the panel of PCR assays (EBV, CMV, HSV 1, 2) was negative.
Histopathological slides were prepared and stained with hematoxylin-eosin. Special stains Gram, Giemsa and Groccot were carried out to detect microorganisms in the lungs, and Pearls reaction was used to evaluate iron deposits in the liver. Immunohistochemical stains for CD1a (Leica Biosystems, RTU – ready to use, clone MTB1), CD3 (Leica, dilution 1:100, clone PS1), CD4 (Leica, RTU, clone 1F6), CD56 (Leica, dilution 1:100, clone 1B6), perforin (Leica, dilution 1:50, clone 5B10), CD8 (Dako, prediluted, clone C8/144B), CD45/LCA (Dako, dilution 1:500, clone 2B11+ PD7/26), CD68 (Dako, dilution 1:200, clone KP1) and granzyme B (Zytomed Systems, RTU, polyclonal) were used. Oil red was used to detect fat deposits in frozen material.
Proband´s DNA was isolated from both muscle and liver tissue. The coding region as well as the adjacent intronic regions of the gene PRF1 (NM_005041.4, NP_005032.2) were amplified by PCR and subsequently analysed by direct sequencing. Large deletions/duplications were excluded using MLPA (Kit: P028, MRC, Holland). Mutations were confirmed in the parents.
RESULTS
Autopsy findings
The body showed pale icterus and fresh blood in both nostrils. Signs of hemorrhagic diathesis included pleural, epicardial, thymic and renal capsular petechias and ecchymosis in superficial fascia of the scalp. Both lungs were heavy and deep red. Small effusions in left pleural, abdominal and pericardial cavity were detected. The liver was soft and enlarged (198 g), a cut section displayed prominent congestion. The spleen was firm and enlarged three times its normal weight (30 g). Lymph nodes were unremarkable except for the enlargement of some pelvic and hepatic nodes. The gallbladder contained white bile.
Histopathological findings
Histopathological examination revealed diffuse hemorhagic and necrotizing pneumonia with hyaline membranes, special stains demonstrated mixed bacterial flora. The inflammatory response was sparse. The thymus showed profound stress involution.
Hepatic portal tracts were expanded by dense lymphohistiocytic infiltrate (Fig. 1). The lymphocytes damaged portal bile ducts and permeated portal vein branches being attached to their endothelial surface. This kind of phlebitis was also seen in some central veins. Intraepithelial lymphocytosis in large bile ducts was distinctive (Fig. 2). Portal and sinusoidal infiltrates were composed of CD3+ lymphocytes with predominance of CD8+ subset (Fig. 3A, 3B) and coexpression of granzyme B and perforin. No CD56+ NK/T cells were found and CD1a was negative. The sinusoids were plugged by numerous hypertrophic Kuppfer cells with engulfed erythrocytes, lymphocytes and nuclear fragments (Fig. 4A). Immunohistochemistry for CD68 confirmed their histiocytic nature (Fig. 4B). The hepatocytes were damaged with microvesicular steatosis and multifocal centrilobular necrosis. Hemosiderin was not increased.




It was hard to identify erythrophagocytosis in the spleen. Splenic red pulp was expanded and Malpighi bodies were atrophic. Enlarged lymph nodes showed paradoxical lymphocytic depletion with karyorrhectic debris. The sinuses were ample and contained large, partially foamy histiocytes with obvious erythrophagocytosis. Mild lymphohistiocytic infiltrate was also found in the gallbladder and renal cortex. The bone marrow was not available for examination.
Genetic findings
The analysis in the proband revealed two heterozygous mutations: c.214A>C, p.T72P (paternal) and c.185_195delACACACAAAGG, p.D62Vfs*12 (maternal). Both variants have not been described in the literature yet, but predictive software tools (MutationTaster, Polyphen) considered both mutations disease-causing. The clinical diagnosis of FHLH type 2 was confirmed on the genetic level with high probability. The disease is transmitted in an autosomal recessive manner. We offered genetic counselling and prenatal analysis in the next pregnancy due to 25 % risk of FHLH2. Genetic testing of the second sibling used fetal DNA obtained by chorionic villus sampling. The crucial regions of the PRF1 gene were explored using the same testing method. The sequences were scanned for the familiar mutations. The fetus inherited the paternal mutation, but the maternal mutation was not passed on to it.
DISCUSSION
Hemophagocytic lymphohistiocytosis is a rare disorder which is difficult to recognize for both pediatricians and pathologists nowadays, just as it was 25 years ago (18). HLH is a generalized illness that can manifest in atypical clinical forms such as a fever of unknown origin, acute liver failure, meningoencephalitis or Kawasaki disease-like. Diagnostic criteria are helpful but some of them occur late in the disease process (3). The correct diagnosis is frequently delayed or completely missed. Secondary bacterial and fungal infections prevail in autoptic findings, candidiasis is found in more than a half of autoptic cases (18). Hemophagocytosis is difficult to detect if there is pronounced autolysis and after previous treatment with steroids and/or chemotherapy (19).
Our patient presented with acute liver failure at the end of the neonatal period. The course was extremely aggressive with nonspecific symptoms lasting no more than two days. Icterus and hemorrhagic diathesis appeared only a few hours before death. Acute liver failure in neonates and infants is very rare and the causes differ from those in adults. These are birth asphyxia, congenital infections, neonatal hemochromatosis, metabolic defects and hematologic disorders (HLH, congenital leukemia) (20). HLH should be suspected if a combination of hepatomegaly, splenomegaly, fever, petechiae, pleural effusions, thrombocytopenia, anemia, elevated CRP ≥ 5 mg/dl and hypalbuminemia ≤ 2.5 g/dl occurs (21). These clinical and laboratory parameters are routinely checked in sick children which is not true for triglycerides and ferritin, as was also evident in our case (Tab. 2). The spleen in HLH is always significantly enlarged form 3 to 20 times the normal weight (18). The liver is also enlarged in contrast to fulminant hepatitis, neonatal hemochromatosis, and massive necrosis after toxic exposure which cause the liver to shrink.
The histological hallmark of HLH is phagocytosis of intact erythrocytes, nucleated hematopoietic elements and their fragments. Bone marrow aspiration or biopsy is the method of choice for the detection of hemophagocytosis, but its absence does not rule out HLH. Unfortunately, the bone marrow examined at initial clinical presentation is often normal or shows only nonspecific abnormalities. Serial marrow aspirates with the progressive disease yield better results (3). The sensitivity of the initial bone marrow hemophagocytosis in the diagnosis of HLH was only 60 %. Moreover, the number of phagocyting cells tended to be low (single cells) (22). Lymph node, spleen or liver biopsies may be useful if the bone marrow investigation is inconclusive. Cerebrospinal fluid or ascites should be screened for hemophagocytic cells if available. In any case, bone marrow hemophagocytosis, respectively erythrophagocytosis, is not specific for HLH and can be seen in a variety of conditions with activated macrophages (blood transfusion, hemolytic anemia, sepsis, graft versus host disease). Even high amounts of hemophagocytosis correlate poorly with disease probability without confirmatory clinical and laboratory findings (23).
The liver probably shows hemophagocytosis to a lesser extent than other reticuloendotelial organs. Hemophagocytic activity was observed in more than 70 % of lymph nodes and spleens, one third of bone marrow specimens, but only 11 % of autoptic livers. Lymphohistiocytic infiltration of portal tracts resembling chronic hepatitis was found in the majority of patients (19). In 25 liver specimens from patients with severe hepatic dysfunction morphological changes suggestive of HLH were recognized. There was portal and sinusoidal lymphohistiocytic infiltrate of CD3+, predominantly CD8+, granzyme B+ lymphocytes admixed with CD68+ histiocytes exhibiting hemophagocytosis. The degree of lymphohistiocytic infiltrate ranged from mild to extensive and correlated with clinical activity. Lymphocytic cholangitis with little epithelial injury and phlebitis of portal and/or central veins were invariably seen. Giant cell transformation of hepatocytes was observed in some biopsies. Four histopathologic patterns of liver injury in HLH were characterized: chronic hepatitis-like, leukemia-like, histiocytic storage disease-like and giant cell hepatitis-like (24).
Our case showed most of the morphological signs described above with a leukemia-like pattern. Lymphocytic cholangitis and lymphocytic cholecystitis were evident. Large histiocytes in sinusoids were difficult to distinguish from hepatocytes due to their abundant, granular eosinophilic or foamy cytoplasm. Hemophagocytosis was conspicuous if looked for. We found CD68 helpful to visualise the hemophagocyting cells despite substantial background staining.
Fever, a key sign of HLH, is induced by high levels of proinflammatory cytokines IL-1, IL-6 and TNFα (3). The incidence of fever is reported close to 100 %, but it seldom occurs in neonates, especially premature ones (16). HLH is a kind of systemic inflammatory response syndrome (SIRS) with excessive levels of circulating cytokines (cytokine storm). Fever and alternatively hypothermia are considered one diagnostic criterion for SIRS. Actually, hypothermic patients with sepsis have higher mortality than febrile patients. Our patient fulfilled 4 of 8 diagnostic criteria (Tab. 2). Fever was missing, but we think that hypothermia can be taken as an equivalent of fever for diagnostic purposes. We can also speculate that our patient would have developed fever later in the course of the disease if she had survived. Supportive laboratory criteria, liver histology, and the overall clinical picture were in agreement with hemophagocytic lymphohistiocytosis.
The familial type of HLH was probable because of a very young age (below 1 year) and absence of infections which are commonly associated with secondary HLH. Molecular genetic testing is recommended even in HLH which is supposed to be acquired. Sequencing in our patient revealed two heterozygous mutations in the PRF1 gene. Neither cytotoxic cell activity studies nor perforin analysis by flow cytometry were performed, but lymphocytes immunoreactive for perforin were detected in the liver. More than 120 unique DNA variants have been reported throughout the entire coding region of PRF1. Pathogenic variants include missense and nonsense mutations, small deletions and small insertions (24). This is only the second case of FHLH with perforin mutation (26) as well as the first case of successful prenatal testing for FHLH2 reported in the Czech Republic. Antenatal diagnosis of FHLH5 in one patient was previously mentioned by Czech authors (2). All siblings who inherit two mutations will eventually develop HLH and need hematopoietic stem cell transplantation. Prenatal testing in the next offspring confirmed an asymptomatic carrier status.
CONCLUSION
HLH is a life threatening illness and both pediatricians and pathologists should have more awareness of this rare disorder. The disease may masquerade as acute liver failure of unknown etiology. The incidence of hypothermia in HLH is not known, but analogous to SIRS, hypothermia could be equated to fever in diagnostic criteria of HLH. In a family with a suspicion of HLH genetic counselling is advised.
ACKNOWLEDGEMENTS
The authors thank Martin Gencik, MD (Praxis für Humangenetik, Wien) for molecular genetic testing; Eliška Tvrdíková, MD for autoptic examination of the proband; Michaela Richtrová, MD for providing clinical data and Associate Professor Josef Feit, MD, PhD for English language correction.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest regarding the publication of this paper.
Correspondence address:
MUDr. Marta Ježová, Ph.D.
Department of Pathology, University Hospital Brno
Jihlavská 20
625 00 Brno,
Czech Republic
tel.: +0042 532233083
e-mail: mjezova@fnbrno.cz
Sources
1. Farquahar JW, Claireaux AF. Familial hemophagocytic reticulosis. Arch Dis Child 1952; 27(136): 519–525.
2. Suková M, Mejstříková E, Vodičková E, et al. Hemofagocytující lymfohistiocytóza. Vnitř Lék 2010; 56(Suppl2): 157–169.
3. Janka GE. Familial and aquired hemophagocytic lymphohistiocytosis. Eur J Pediatr 2007; 166(2): 95–109.
4. Meeths M, Horne A, Sabel M, et al. Incidence and clinical presentation of primary hemophagocytic lymphohistiocytosis in Sweden. Pediatr Blood Cancer 2015; 62(2): 346-352.
5. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science 1999; 286(5446): 1957–1959.
6. Ohadi M, Lalloz MR, Sham P, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet 1999; 64(1): 165–171.
7. Trapani JA, Smyth MJ. Functional significance of the perforin/granzyme cell death pathway. Nat Rev Immunol 2002; 2(10): 735–747.
8. Lieberman J. Cell-mediated cytotoxicity. In: Paul WE. Fundamental Immunology (7th ed). Philadelphia, PA: Lippincott Williams& Wilkins, a Walters Kluwer business; 2013: 891–910.
9. Voskoboinik I, Dunstone MA, Baran K, Whisstock JC, Trapani JA. Perforin: structure, function, and role in human immunopathology. Immunol Rev 2010; 235(1): 35–54.
10. Janka GE. Familial erythrophagocytic lymphohistiocytosis. Eur J Pediatr 1983; 140(3): 221–230.
11. Ishii E, Ogha S, Tanimura M, et al. Clinical and epidemiological studies of familial hemophagocytic lymphohistiocytosis in Japan. Med Pediatr Oncol 1998; 30(5): 276–283.
12. Karapinar B, Yilmaz D, Balkan C, Akin M, Ay Y, Kvakii K. An unusual case of multiple organ dysfunction syndrome in the pediatric unit intensive care: hemophagocytic lymphohistiocytosis. Pediatr Critic Care Med 2009; 10(3): 285–290.
13. Henter JI, Nennesmo I. Neuropathological findings and neurological symptoms in twenty-three children with hemophagocytic lymhohistiocytosis. J Pediatr 1997; 130(3): 358–365.
14. Weitzman S. Approach to hemophagocytic syndromes. Hematology Am Soc Hematol Educ Program 2011; 2011: 178–183.
15. Janka GE, Lehmberg K. Hemophagocytic lymphohistiocytosis: pathogenesis and treatment. Hematology Am Soc Hematol Educ Program 2013; 2013: 605–611.
16. Trottestam H, Horne A, Aricó M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long term results of the HLH-94 treatment protocol. Blood 2011; 118(17): 4577–4584.
17. Suzuki N, Morimoto A, Ohga S, et al. Characteristics of hemophagocytic lymphohistiocytosis in neonates: a nationwide survey in Japan. J Pediatr 2009; 155(2): 235–238.
18. Stejskal J, Hrodek O, Elleder M. Familiární hemofagocytující lymfohistiocytóza. Cesk Patol 1990; 26(1): 14–24.
19. Öst A, Nilson-Ardnor S, Henter J. Autopsy findings in 27 children with hemophagocytic lymphohistiocytosis. Histopathology 1998; 32(4): 310–316.
20. Dhawan A, Mieli-Vergani G. Acute liver failure in neonates. Early Hum Dev 2005; 81(12): 1005–1010.
21. Ryu J, Kim KM, Oh SH, et al. Differential clinical characteristics of acute liver failure caused by hemophagocytic lymphohistiocytosis in children. Pediatr Int 2013; 55(6): 748–752.
22. Gupta A, Tyrrell P, Valani R, Benseler S, Weitzman S, Abdelhaleem M. The role of initial bone marrow aspirate in the diagnosis of hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2008; 51(3): 402–404.
23. Ho C, Yao X, Tian L, Li FY, Podoltsev N, Xu ML. Marrow assessment for hemophagocytic lymphohistiocytosis demonstrates poor correlation with disease probability. Am J Clin Patol 2014; 141(1): 62–71.
24. Chen JH, Fleming MD, Pinkus GS, et al. Pathology of the liver in familial hemophagocytic lymphohistiocytosis. Am J Surg Patol 2010; 34(6): 852–867.
25. Sieni E, Cetica V, Hackmann Y, et al. Familial hemophagocytic lymphohistiocytosis: when rare disease shed light on immune system functioning. Front Immunol 2014; 5(167): 1–19.
26. Špíšek R, Mejstříková E, Formánková R, et al. Familiární hemofagocytující lymfohistiocytóza na podkladě deficitu perforinu úspěšně léčená transplantací hematopoetických buněk - první diagnostikovaný případ v České republice. Cas Lek Cesk 2006; 145(1): 50–54.
27. Henter JI, Horne A, Aricó M, et al. HLH 2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007; 48(2): 124–131.
28. Zhang K, Filipovich AH, Johnson J, Marsh RA, Villanueva J. Hemophagocytic lymphohistiocytosis, familial. In: Pagon RA, Adam MP, Ardinger HH, eds. Gene Reviews (Internet). Seattle (WA): University of Washington; 2006 (updated 2013): 1993–2015 (cit. May 2015).
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2017 Issue 1
Most read in this issue
-
Update on the 2016 WHO classification of tumors of the central nervous system
– Part 1: Diffusely infiltrating gliomas -
Update on the 2016 WHO classification of tumors of the central nervous system.
Part 2: Embryonal tumors and other tumor groups (except for diffuse gliomas) - Unusual histopathological picture of acute lung injury in different stages of resorption with predominance of organizing pneumonia in a young man with influenza A (H1N1)
- WHO´s next?