
Kožní lymfomové onemocnění u geriatrického pacienta
:
T. Gregorová; K. Bielaková; J. Feit
:
Geriatrie a Gerontologie 2016, 5, č. 4: 223-228
:
Case Reports
Autoři v kazuistice prezentují případ kožního lymfomového onemocnění u 80letého pacienta. Poukazují na úskalí diagnostiky této nemoci především v jejích počátečních stadiích a zmiňují možnosti léčby. Autoři se také dotýkají problematiky antibiotické terapie u seniora s přítomnou pre-frailty, která u nemocného zřejmě vedla k rozvoji pseudomembranózní enterokolitidy vyvolané Clostridium difficile.
Klíčová slova:
primárně kožní T-lymfom – mycosis fungoides – Sézaryho syndrom – pseudomembranózní enterokolitida – Clostridium difficile
Maligní nonhodgkinské lymfomy představují v zastoupení výskytu maligních onemocnění pouze malé procento, i tak je na ně třeba v diferenciální diagnostice myslet. Lymfomy postihují primárně lymfatické uzliny, na druhém a třetím místě za nimi je postižení gastrointestinálního traktu a kůže. Dalšími oblastmi primární mimouzlinové manifestace lymfomů jsou kostní dřeň, paranazální dutiny, štítná žláza, varlata a centrální nervová soustava. Terapie a prognóza onemocnění závisí nejen na lokalizaci, ale také na histologickém typu lymfomu a rozsahu nemoci. Klasifikace REAL člení lymfomy do skupin podle průměrné míry agresivity na nízce, středně a vysoce agresivní formy(1).
O primárních kožních lymfomech mluvíme, pokud je v době diagnózy postižena pouze kůže. Zahrnují heterogenní skupinu vycházející jak z T-lymfocytů, tak z B-lymfocytů, lišící se fenotypem, histologickým obrazem a též stupněm biologické aktivity, agresivity, a tedy i prognózou. Na základě těchto kritérií sjednotila Světová zdravotnická organizace (WHO) a Evropská organizace pro výzkum a léčbu rakoviny (EORTC) praktickou klasifikaci kožních lymfomů(6).
Primární T-buněčné lymfomy představují pouhá 4 % z nehodgkinských lymfomů, jedná se tedy o onemocnění relativně vzácné. Mycosis fungoides (MF) je z nich potom nejčastější – tvoří téměř polovinu ze všech kožních T-buněčných lymfomů. Jeho incidence se uvádí 0,4/100 000 obyvatel ročně. Poprvé byl popsán francouzským lékařem Alibertem v roce 1806. MF je charakterizován jako nízce maligní T-lymfom s nízkým stupněm agresivity. Jeho progrese je pozvolná a mnoho let se infiltrace klonálními T-lymfocyty omezuje pouze na kůži – zachovává si svůj dermotropismus. Jeho výskyt je zhruba 2× častější u mužů než u žen, jeho vznik je možný v jakémkoliv věku, ale nejčastěji toto onemocnění začíná ve 4. až 6. dekádě života(2, 3). Vzhledem k nespecifickému klinickému i histologickému obrazu MF v počátečních stadiích je běžné, že od objevení se počátečních příznaků do stanovení diagnózy uběhne 6 i 7 let(2, 4, 5).
Kazuistika
80letý pacient byl původně hospitalizován na plicní klinice pro bilaterální bronchopneumonii. Byl léčen širokospektrými antibiotiky, amoxicilinem-klavulanátem, po dobu 14 dní. Vzhledem k rozvoji hlenovitých průjmů a pozitivitě testu na okultní krvácení byl senior přeložen na naše pracoviště k dalšímu došetření. V klinickém obraze dominuje slabost, zhoršené dýchání a svědivá vyrážka trvající déle než půl roku. Stolice je řídká, hlenovitá, asi 3× denně, bez makroskopických známek přítomnosti krve.
Rodinná anamnéza je nevýznamná. Z osobní anamnézy dominuje postižení kardiopulmonální – pacient je léčen pro chronickou obstrukční plicní nemoc, chronické srdeční selhávání se syndromem anginy pectoris, fibrilaci síní; je po infarktu myokardu v 65 letech, v 72 letech provedena angioplastika; podle ECHO srdce je dilatovaná levá komora s lokálními poruchami kinetiky, ejekční frakcí 30 %, významnou mitrální a trikuspidální regurgitací a známkami těžké plicní hypertenze. Pacient má implantovaný kardiostimulátor. Je sledován a léčen pro benigní hyperplazii prostaty, diabetes mellitus 2. typu a dyslipoproteinemii. Do 50 let byl kuřákem. Neguje lékové i jiné alergie. Medikace je četná a odpovídá výše uvedeným diagnózám. Chuť k jídlu je nezměněná, váha stabilní. Pacient je střední postavy s BMI 25, MMSE 23 bodů, ADL 90 bodů. Má sklon k hypotenzi (vstupní TK 100/40).
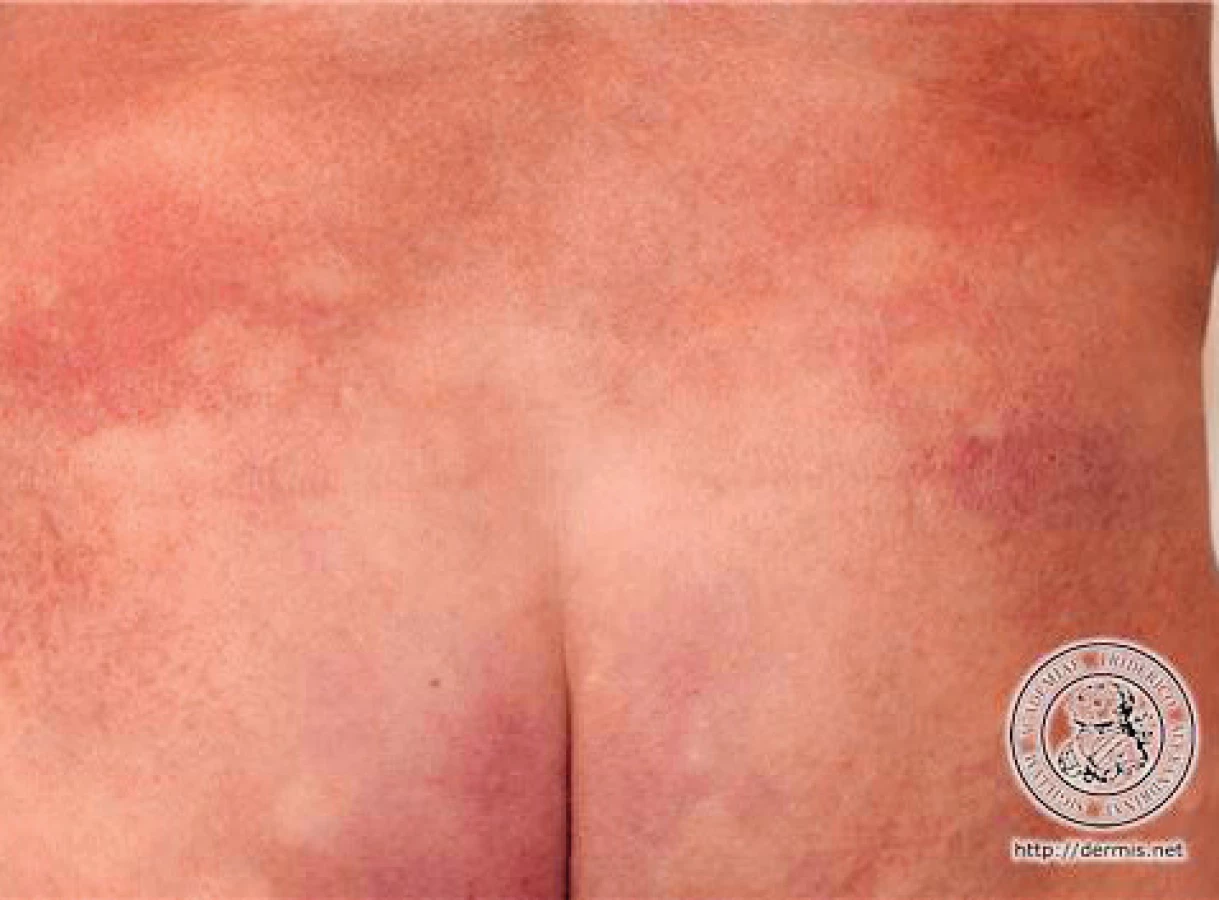
V klinickém obraze pacienta dominuje celotělový růžovočervený makulózní exantém, místy s drobným olupováním kůže, maximum postižení je na břiše, kde ložiska splývají. Bez lymfadenopatie. Na plicích přetrvává mírný poslechový nález chrůpek vpravo bazálně. Břicho je prohmatné, palpačně citlivější, ale nebolestivé, s přítomnou peristaltikou, bez hmatné rezistence. Na pravém kotníku je patrný defekt 0,5 × 1 cm s bělavou spodinou. Na EKG je eufrekvenční fibrilace síní.
V krevním obraze patrná mírná leukocytóza (11,05 × 109), v diferenciálním rozpočtu 61 % neutrofilů a 11 % lymfocytů, dále mírná mikrocytární anemie (hemoglobin 106 g/l, MCV 81 fl), přičemž z hodnot anemického souboru je jen mírně snížená hladina železa (7,7 μmol/l). Základní koagulační parametry jsou v normě. V biochemickém vyšetření bylo přítomno mírné zvýšení kreatininu (115 μmol/l) a snížení hladiny celkového vápníku v séru (2,04 mmol/l). Ostatní parametry včetně jaterních testů, amylázy i CRP byly v normě. Z onkomarkerů byla nadhraniční hladina TPS 102 μg/l, LD 3,96 μkat/l a beta-2 mikroglobulinu 5,29 mg/l.

V předchorobí byl pacient hospitalizován před 5 měsíci na dermatologii kvůli svědivému exantému, podle biopsie uzavřeno jako ekzém dermatitis. Před 2 měsíci byl potom léčen na chirurgické klinice a následně na infekčním oddělení pro bolesti břicha, podle CT břicha bylo patrné zesílení stěny terminálního ilea, léčen konzervativně, pro rozvoj průjmů podáván rifaximin a cloroxin s ústupem obtíží. Před přijetím na naši kliniku bylo provedeno sonografické vyšetření střev, kde bylo patrné přetrvávající mírné zesílení stěny terminálního ilea v úseku zhruba 10 cm. Ultrazvuk parenchymatózních orgánů nevykazoval pozoruhodnosti. Negativní bylo i vyšetření stolice na přítomnost klostridiového toxinu. Ve stěru z rekta byly masivně kvasinky, proto byl pacient přeléčen fluconazolem. To vedlo přechodně ke snížení četnosti stolic, i tak se nemocný nadále cítil sláb a intermitentně jej pobolívalo břicho. Bylo tedy provedeno kolonoskopické vyšetření, jež nebylo výtěžné, proto byla doporučena nová kolonoskopie v analgosedaci.
Mezitím ale došlo u seniora ke zhoršení průjmů, bolestem břicha a byla diagnostikována klostridiová enterokolitida, která bývá relativně častou komplikací u hospitalizovaných pacientů po intenzivní antibiotické terapii. Po zjištění infekce Clostridium difficile byl senior léčen metronidazolem, pozvolna tak došlo k odeznění průjmů a celkovému zlepšení stavu nemocného, nadále byla podávána probiotika. Kontrolní vyšetření stolice na klostridiový antigen byla opakovaně negativní. I přesto byly stolice nadále řídké a břicho palpačně citlivé.
Podle gastroskopického vyšetření byla patrná snížená kompetence kardie, hiátová hernie a chronická gastritida při duodenogastrickém refluxu. Kolonoskopie v analgosedaci prokázala jen vnitřní hemoroidy a klidné divertikly sigmoidea, sliznice terminálního ilea nejevila známky zánětu, biopsie byla odebrána z okraje Bauhinovy chlopně.
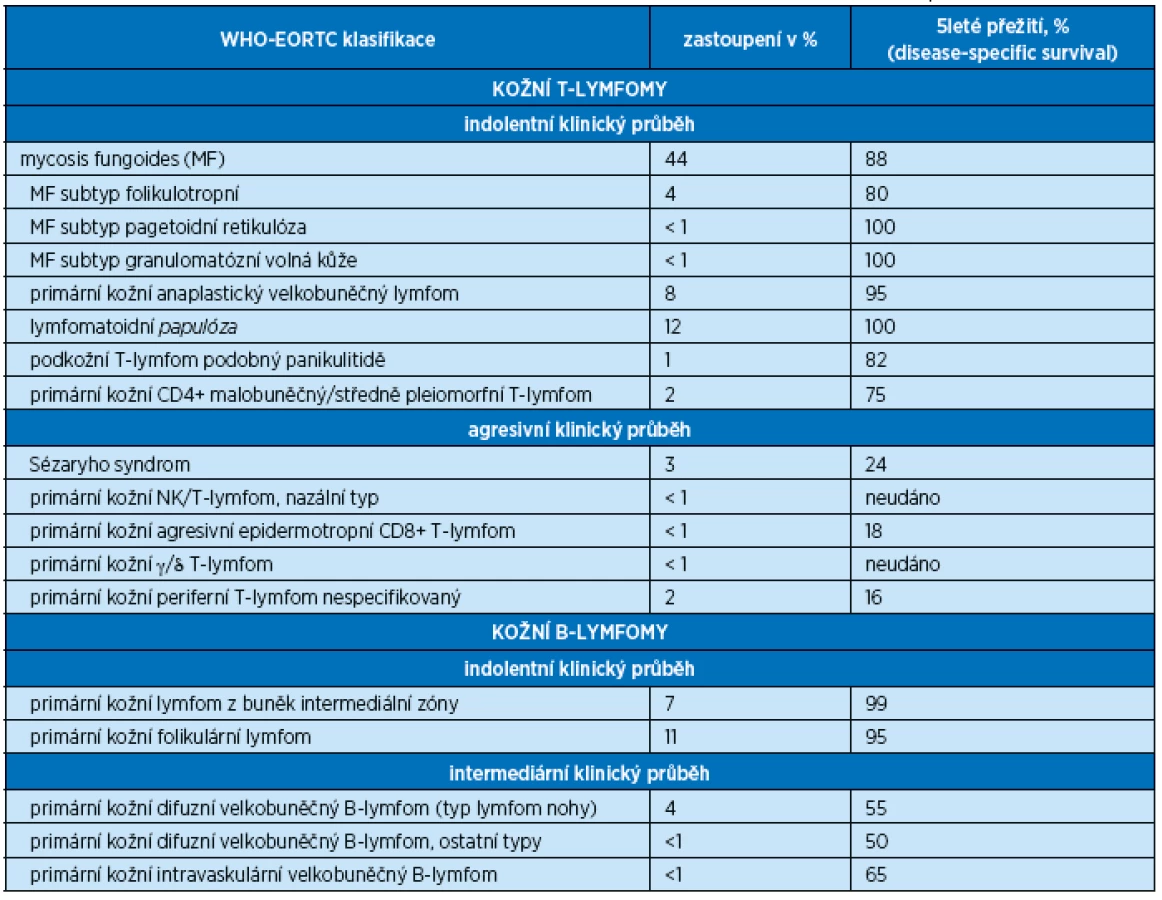
Pacienta i přes podávaná antihistaminika (bisulepin) nadále sužovalo svědění kůže, dermatolog zhodnotil nález jako možný mikrobiální ekzém, parapsoriázu či exantém paraneoplastický; lokálně doporučil kortikoidní masti a emoliencia. Na zavedené lokální terapii dochází k mírné regresi exantému, kůže je nadále suchá, lamelózně se olupuje. Vzhledem k ústupu kožních eflorescencí dermatolog považuje za nejpravděpodobnější diagnózu ekzém dermatitis a s léčbou je spokojen. Pacient ale nadále trpí nepříjemným svěděním kůže se zhoršenou kvalitou spánku, celkovou únavou a slabostí během dne. Zvažováno je i lymfoproliferatitvní onemocnění, provedená flowcytometrie periferní krve prokázala kromě relativní lymfopenie s granulocytózou výrazně zvýšený CD4/8 index ve prospěch CD4+ T-lymfocytů. Klonalita T-lymfocytů v periferní krvi nebyla prokázána. Lékaři naší kliniky tudíž trvali na provedení biopsie z kožních lézí. Probatorní excize byla provedena v oblasti sakra, podle histologie byl v horním koriu patrný lymfocytární infiltrát pronikající do epidermis, CD4-lymfocyty převažovaly nad CD8 a u T-lymfocytů v kůži byla prokázána T-klonalita. Histologické vyšetření prokázalo kožní T-lymfom mycosis fungoides.
Nyní, čekajíc na výsledek histologie z terminálního ilea, jsme byli spolu s hematoonkology na diagnostickém rozcestí a kladli jsme si otázku, zda se jedná o kožní T-lymfom mycosis fungoides se systémovou generalizací do GIT, primární T-lymfom střeva se sekundárním postižením kůže, anebo o koincidenci mycosis fungoides s jinou nemaligní ileitidou?
Protože se nám odběr vzorku z ilea nezdál být zcela reprezentativní, zvažovali jsme i provedení diagnostické laparoskopie, od kterého jsme museli ustoupit kvůli vysokému peroperačnímu riziku u polymorbidního pacienta s přítomnou frailty, navíc krátce po prodělané bronchopneumonii a klostridiové kolitidě. Vzhledem k příznivému kolonoskopickému nálezu, normálnímu nálezu z histologie terminálního ilea a pro nepřítomnost lymfadenopatie se hematoonkologové přiklonili k diagnóze primárního kožního T-lymfomu mycosis fungoides (zřejmě v koincidenci s jinou nemaligní ileitidou) a pacient byl předán do péče specializovaného dermatologického centra, kde bude dále sledován a léčen. V případě progrese onemocnění pak funguje úzká spolupráce s hematoonkology a topicky orientovaná léčba může být nahrazena systémovou onkologickou terapií.
Diskuse
Klinický a stejně tak histopatologický obraz MF je proměnlivý. V počátečních stadiích bývají projevy na kůži nespecifické. Nejčastěji nacházíme růžová až načervenalá ložiska dermatitidy, někdy splývající ve větší plochy. Typicky se vyskytují na částech těla chráněných před sluncem (hýždě, trup). Ložiska mohou i bez terapie přechodně vymizet nebo se zmenšovat. Kůže bývá suchá s olupováním, svědí a pro nespecifický klinický i histologický obraz jsou projevy často hodnoceny např. jako ekzém, psoriáza, parapsoriáza či erytém nejasného původu. Stejně nejednoznačně zněly diagnostické závěry i v případě našeho pacienta. Toto stadium se označuje jako premykotické.
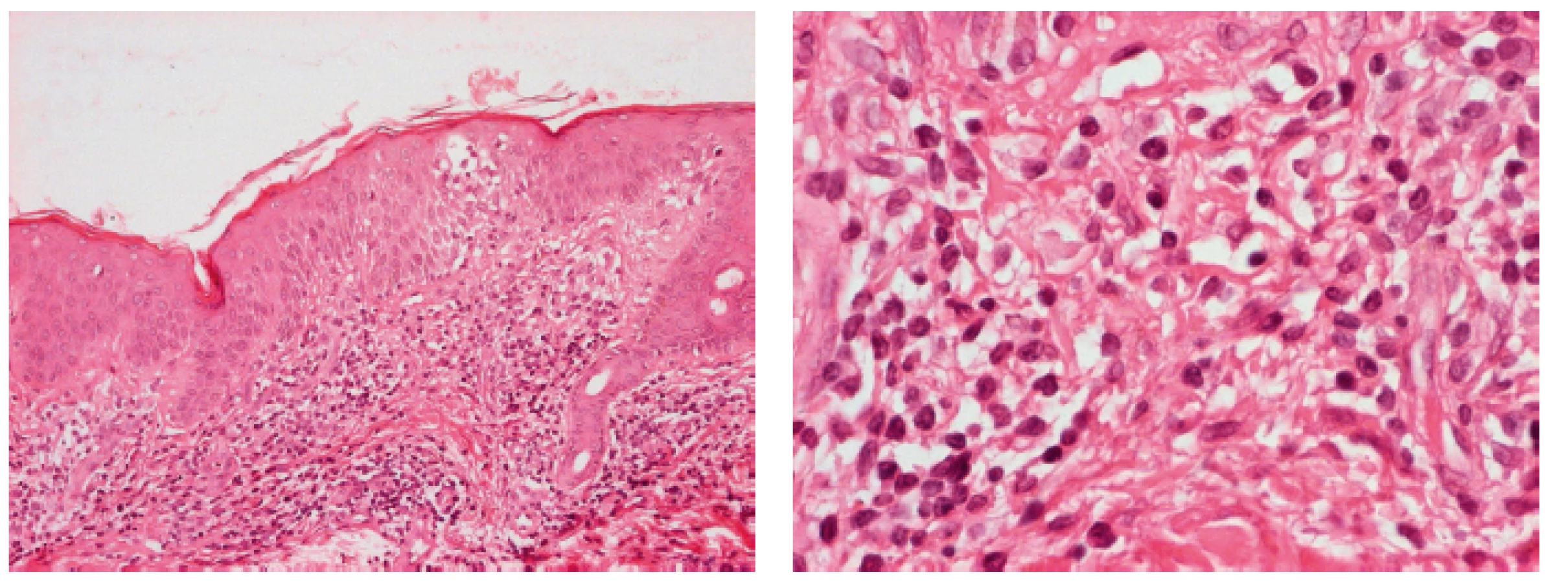
Dalším stadiem je stadium infiltrativní (stadium plaků), kdy dochází k infiltraci epidermis T-lymfocyty a ložiska mají vyvýšený charakter a plazivý, ostře ohraničený okraj (plaky). Pravidlem je nepříjemné svědění kůže. Onemocnění MF bylo u nemocného v této kazuistice odhaleno právě v tomto stadiu na základě kožní biopsie a vzniklého podezření na lymfoproliferativní chorobu (speciální barvení, stanovení imunofenotypizace a klonality lymfocytů v kůži jsou prováděna až při podezření na lymfoproliferaci).


Infiltráty v kůži se mohou postupně rozšiřovat a dávat vznik intraepidermálním kolekcím nádorových buněk – tzv. Pautrierovým mikroabscesům. Při vzniku velkých nakupení maligních buněk vznikají na kůži červenohnědé tumory, které mohou i exulcerovat nebo mohou být bolestivé a sekundárně se infikovat. Hovoříme o stadiu tumorózním(3, 4,7 ).
Maligní T-lymfocyty mohou postupně infiltrovat celou kůži a tak vzniká generalizovaná erytrodermie. Po mnoha letech izolovaného postižení kůže může dojít i k infiltraci regionálních lymfatických uzlin a vnitřních orgánů. V některých případech může dojít k transformaci v lymfom vysokého stupně malignity, který je typický přítomností CD30+ buněk. Jako leukemizující varianta MF je označován Sézaryho syndrom, jenž je typický generalizovanou erytrodermií i lymfadenopatií a přítomností patologických T-lymfocytů (tzv. Sézaryho buněk) v periferní krvi, uzlinách a kůži(6, 8).
Ve stanovení diagnózy MF se opíráme o klinické a histologické vyšetření kůže. Vzhledem k nespecifickému obrazu v počátečních stadiích bývá diagnostika často svízelná, důležité jsou opakované biopsie u podezřelých svědivých kožních eflorescencí. Význam má i stanovení krevního obrazu a imunofenotypizace lymfocytů, případně stanovení klonality T-lymfocytů. V rámci stagingu se uplatní sonografie lymfatických uzlin při klinicky suspektní lymfadenopatii, rentgen hrudníku a sonografie dutiny břišní k odhalení postižení viscerálních orgánů či uzlin. CT či PET/CT potom provádíme při diagnostických nejasnostech k vyloučení postižení viscerálních orgánů. U pacientů, u kterých je na základě klinického a zobrazovacích vyšetření podezření na postižení mimokožních lokalizací, je třeba toto verifikovat provedením biopsie (3, 5, 10, 11). V případě nemocného v naší kazuistice byla provedena biopsie z terminálního ilea, pokud by ale střevní potíže nadále přetrvávaly, zřejmě by bylo nutné přistoupit k opakování CT břicha či PET/CT. Určitý břišní diskomfort byl nyní u pacienta přisuzován proběhlému střevnímu mykotickému a bakteriálnímu zánětu a dysmikrobii po antibiotické terapii při preexistující divertikulóze kolon.
Mycosis fungoides se v naprosté většině případů vyvíjí roky až desetiletí a má indolentní průběh. Prognóza pacientů závisí na stadiu onemocnění, typu a rozsahu postižení kůže a případném rozšíření do mimokožních lokalizací. Podle studií bylo zjištěno, že u pacientů s ložisky či infiltrativními změnami kůže, které zasahují pod 10 % povrchu kůže, má šanci 10letého přežití 97–98 %; pokud tyto změny zasahují více než 10 % povrchu těla, je přežití asi 83 %. Ve stadiu tumorů potom 10leté přežití klesá na 42 % a u pacientů s histologicky ověřeným postižením lymfatických uzlin se toto procento snižuje na 20 %. U jedinců s postižením vnitřních orgánů nebo při maligní transformaci na velkobuněčný T-lymfom je průběh agresivní a tito pacienti často umírají během jednoho roku v důsledku orgánového postižení či infekčních komplikací(6, 11).
I přestože je mycosis fungoides jako T-buněčný lymfom lokalizován v kůži, probíhá pohyb T-lymfocytů mezi kůží, lymfou, krví a kostní dření, což limituje výsledky lokální terapie zaměřené pouze na kůži. Lokální terapie zahrnuje především místní aplikaci kortikosteroidů, fototerapii, fotochemoterapii, celotělové ozáření elektronovými paprsky, případně lokální radioterapii. Systémová terapie představuje celkové podání retinoidů, interferonu alfa, systémové chemoterapie a ve specifických případech podání alemtuzumabu nebo extrakorporální fotoferézu(2, 3, 5, 9, 12). Nedílnou součástí léčby mycosis fungoides je podpůrná léčba pruritu a řešení infekčních komplikací, které jsou u pacientů s tímto onemocněním velmi časté vzhledem k narušení celistvosti kožního krytu.
Závěr
Nejrůznější typy postižení kůže jsou v seniorském věku velmi časté a v jejich diagnostice by se nemělo zapomínat ani na lymfoproliferativní choroby. V případě mycosis fungoides, která ve svých počátečních stadiích často napodobuje jiné kožní choroby, je diagnostika velmi svízelná a trvá i několik let, než je odhalena. Při podezření na tuto nemoc jsou třeba opakované kožní biopsie. Samozřejmě je třeba myslet i na to, že se nemusí jednat o primární onemocnění kůže, ale o možný projev závažného systémového onemocnění.
MUDr. Tereza Gregorová1,
MUDr. Katarína Bielaková, Ph.D.1,
doc. MUDr. Josef Feit, CSc.2
1Klinika interní, geriatrie, ošetřovatelství a praktického lékařství FN Brno
2Ústav patologie, FN Brno
MUDr. Tereza Gregorová
e-mail: Gregorova.Tereza@fnbrno.cz

Vystudovala Lékařskou fakultu Masarykovy univerzity v Brně a od roku 2008 působí jako lékařka a asistentka na Klinice interní, geriatrie, ošetřovatelství a praktického lékařství ve FN Brno. Zpočátku pracovala na lůžkovém oddělení kliniky, nyní se po čtyřleté pauze vrátila od svých dětí k pacientům geriatrické ambulance FN Brno.
Sources
1. Adam Z, Krejčí M, Vorlíček J, et al. Speciální onkologie. Praha: Galén 2010.
2. Vašků V. Kožní T buněčné lymfomy – diagnostika a terapie. Postgraduální medicína 2007; 9(5): 561–568.
3. Adam Z, Krejčí M, Vorlíček J, et al. Hematologie. Praha: Grada Publishing 2008.
4. Cetkovská P. Primární kožní T lymfomy: mycosis fungoides a Sézaryho syndrom. Onkologie 2010; 4(4): 233–236.
5. Weller R, Hunter J, Savin J, et al. Clinical Dermatology. 4. vyd. Malden, USA: Blackwell Publishing 2008.
6. Willemze R, Jaffe ES, Burg G, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005; 105(10): 3768–3785.
7. Štork J, Arenberger P, Pizinger K. Dermatovenerologie. Praha: Galén 2008.
8. Olsen E, Vonderheid E, Pimpinelli N, et al. Revisions to the staging and classification of mycosis fungoides and Sezary syndrome. Blood 2007; 110(6): 1713–1722.
9. Trautinger F, Knobler R, Willemze R, et al. EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome. Eur J. Cancer 2006; 42: 1014–1030.
10. Vlašín Z, Jedličková H, Feit J, et al. Praktická dermatologie v obrazech a schématech. Brno: Vladerma 2001.
11. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T cell lymphoma (mycosis fungoides and Sézary syndrome): part I. Diagnosis: clinical and histopathologic features and new molecular and biologic markers. J Am Acad Dermatol 2014; 70(2): 221–222.
12. Jawed SI, Myskowski PL, Horwitz S, et al. Primary cutaneous T cell lymphoma (mycosis fungoides and Sézary syndrome): part II. Prognosis,management, and future directions. J Am Acad Dermatol 2014; 70(2); 240–242.
13. Dermatology information system – čerpána fotografická dokumentace: obr. 2, 3 a 4. Dostupné na: http://www.dermis.net/dermisroot/en/home/index.htm
Labels
Geriatrics General practitioner for adults Orthopaedic prostheticsArticle was published in
Geriatrics and Gerontology

2016 Issue 4
Most read in this issue
- Obstipation in the elderly
- Diabetes and dementia – what is known about their relationship?
- Acute and late onset complications of diabetes – do they pose a significant problem in the elderly?
- A cutaneous lymphomatic disease at geriatric patient