
Neobvyklý pľúcny nález masívneho vyplnenia alveolov penovitými makrofágmi pri kongenitálnej epidermolysis bullosa po aspirácii súčastí plodovej vody u novorodenca prežívajúceho 15 dní bez akýchkoľvek príznakov poškodenia dýchacích funkcií
:
Daniel Farkaš 1; Marián Švajdler ml. 2; Lucia Fröhlichová 2; Jana Šprláková 3; Silvia Farkašová Iannaccone 4; Zoltán Szép 5; Oľga Nyitrayová 5
:
Bioptická laboratoř s. r. o., Plzeň, Česká Republika
a Oddelenie patológie, Univerzitná nemocnica Louisa Pasteura, Košice, Slovenská Republika
; Úrad pre dohľad nad zdravotnou starostlivosťou, SLaPA pracovisko, Košice
1; Šiklův ústav patologie, Univerzita Karlova Praha, Lékařská fakulta Plzeň, Česká Republika
2; Detská fakultná nemocnica, Košice
3; Ústav súdneho lekárstva UPJŠ LF, Košice
4; Cytopathos s. r. o., Bratislava
5
:
Čes.-slov. Patol., 51, 2015, No. 2, p. 89-93
:
Original Article
Epidermolysis bullosa predstavuje skupinu mechanobulóznych ochorení, ktoré sú najčastejšie dedične podmienené. V článku opisujeme prípad 15-dňového novorodenca ženského pohlavia s kongenitálnou epidermolysis bullosa postihujúcou približne 1/3 povrchu kože. Dieťa zomrelo v dôsledku nezvládnuteľnej sepsy s multiorgánovým zlyhaním. Predmetom nášho príspevku je popis neobvyklého pľúcneho nálezu s masívnym vyplnením alveolov penovitými makrofágmi po aspirácii súčastí plodovej vody obsahujúcej nadmerné množstvo odlúčených epitélií pokožky. Prezentovaný prípad poukazuje na výraznú diskrepanciu negatívneho klinického a závažného histopatologického nálezu.
Kľúčové slová:
epidermolysis bullosa – alveolárne penovité makrofágy – aspirácia plodovej vody
Epidermolysis bullosa (EB) zahŕňa viaceré klinickopatologické varianty mechanobulóznych ochorení, ktoré sú najčastejšie podmienené dedičnými mutáciami génov kódujúcich buď štrukturálne proteíny bazálnych keratinocytov (simplexné formy EB), súčastí bazálnej membrány (junkčný typ EB) alebo proteíny zóny sublamina densa papilárnej dermis (dystrofický typ EB) (1). V súčasnom období je známych minimálne 14 génových mutácii, ktoré vedú k tomuto ochoreniu (2). Patofyziologicky sú zmeny indukované minimálnou traumou, ktorá vedie k poškodeniu mimoriadne fragilnej kože. Predmetom nášho príspevku je opis neobvyklého pľúcneho nálezu u 15-dňového novorodenca s kongenitálnou formou epidermolysis bullosa zo skupiny EB simplex, u ktorého negatívny klinický nález a anamnestické údaje nezodpovedali závažnému pľúcnemu nálezu v zmysle masívneho vyplnenia alveolov penovitými makrofágmi.
OPIS PRÍPADU
Jednalo sa o ľahko hypotrofické dieťa (2630 g, 47 cm) z prvej gravidity, ženského pohlavia, bielej rasy, narodené v 40. týždni gestácie cisárskym rezom pre nepostupujúci pôrod. Už pri narodení boli približne na 1/3 povrchu kože prítomné pľuzgiere (buly) a plošné defekty kože v zmysle jej odlučovania, ktoré výrazne pripomínali odlučovanie kože pri intrauterinnom odumretí plodu (macerácia). Vak pôrodných blán bol zachovaný, plodová voda bola skalená odlúčenými areálmi kože, ale nejavila znaky imbibície mekóniom. Apgarovej skóre bolo 10 v prvej aj piatej minúte života, pričom neboli zistené žiadne klinické známky aspirácie plodovej vody, alebo smolky (mekónia). Pri preklade na špecializované oddelenie nebola nutná ventilačná podpora, dýchanie bolo eupnoické, auskultačne čisté, nebola nutnosť podávať surfaktant. Okamžite bolo započaté ošetrovanie kožných defektov, ordinovaná infúzna terapia a profylaktická antibiotická terapia. Počas medikamentóznej terapie nebola zistená žiadna vedľajšia reakcia na podávané lieky. Na 8. deň života bol novorodencovi zavedený centrálny žilový katéter a následne vykonané RTG vyšetrenie hrudníka, pri ktorom bola zistená primeraná transparencia pľúc a neboli zistené žiadne závažnejšie odchýlky bronchiálneho stromu. Vzhľadom na chýbanie akýchkoľvek prejavov poškodenia dýchacieho systému (napr. sťažené dýchanie, kašeľ) nebolo počas celej hospitalizácie indikované ďalšie RTG alebo iné zobrazovacie vyšetrenie pľúc (napr. CT). Izolované akrálne postihnutie kože, postihnutie skvamóznych slizničných plôch orofaryngu, anusu a ezofágu, poškodenie nechtov, kĺbov, pseudosyndaktýlia, respektíve striktúry čreva neboli diagnostikované.
Aj napriek minimálnemu mechanickému kontaktu dochádzalo k tvorbe nových pľuzgierov, pričom došlo k pridruženiu infekčných komplikácii s rozvojom sepsy s multiorgánovým zlyhaním, ktoré na 15. deň života viedlo k smrti. Počas celej hospitalizácie dieťa nemalo suchý kašeľ, neboli prítomné prejavy dušnosti (dyspnoe), známky respiračnej insuficiencie, nebola nutná ventilácia pľúc, dokonca ani nutnosť podávania kyslíka. Počas liečby mu neboli podávané žiadne olejovité preparáty vo forme kvapiek. V rodine dieťaťa sa nevyskytla kožná choroba v zmysle epidermolysis bullosa, ale otec dieťaťa sa liečil na psoriázu. Z dostupnej dokumentácie vyplýva, že u matky dieťaťa počas tehotenstva neboli zistené žiadne závažnejšie poruchy zdravia (napr. systémové ochorenia, nežiadúce reakcie na lieky, otrava ťažkými kovmi).
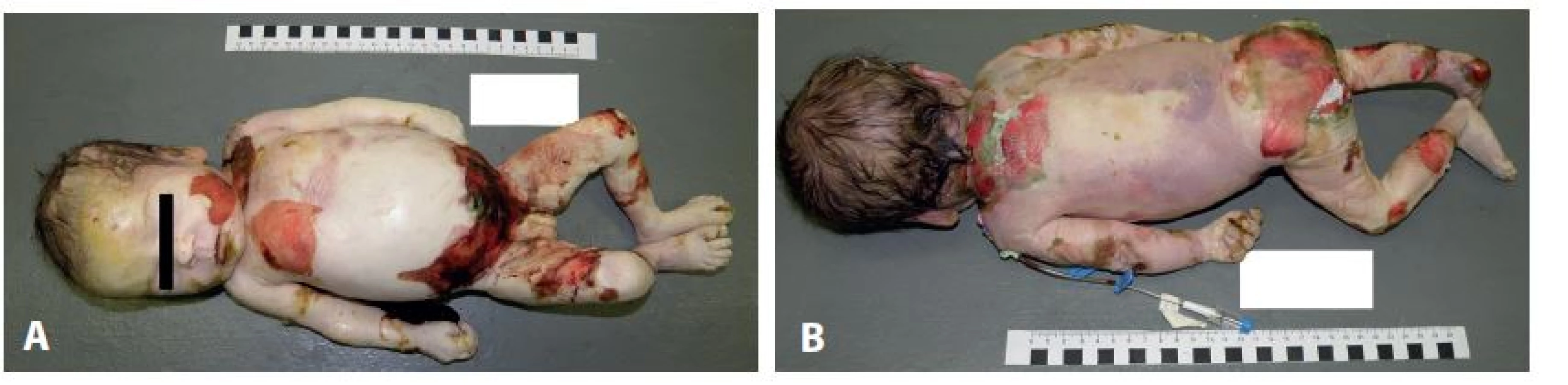
Pitva bola vykonaná 17 hodín po smrti. Tkanivá boli fixované vo formole, nie v glutaraldehyde. Pri vonkajšej obhliadke boli zistené na všetkých častiach tela (hlava, trup a končatiny) rozsiahle mapovité defekty kože v zmysle jej čerstvého odlučovania ako aj hojace sa oblasti, celkovo postihujúce približne tretinu povrchu kože (obr. 1). Poškodenia kože makroskopicky miestami pripomínali hojace sa popáleniny alebo poleptania.
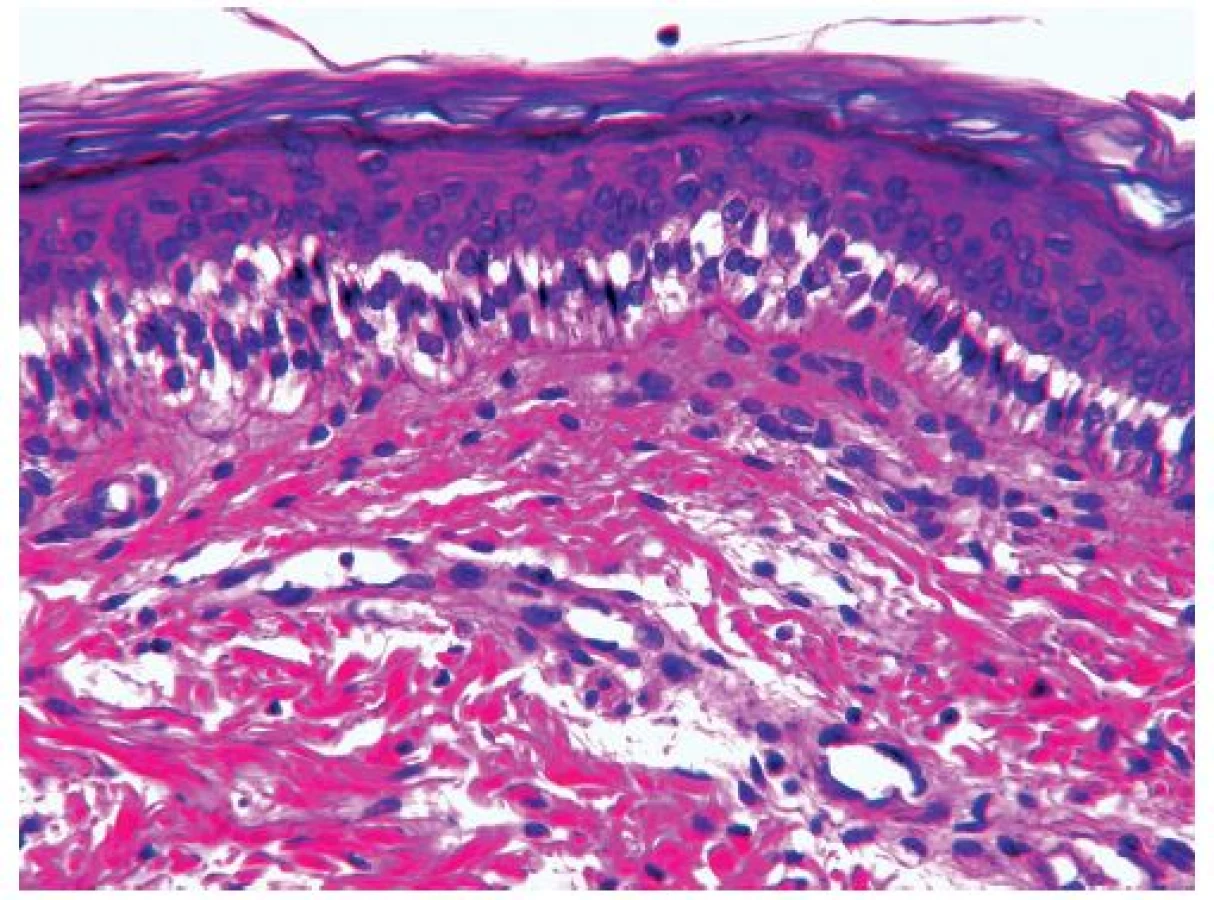
Mikroskopickým vyšetrením kože sme zistili nápadnú separáciu epidermis pod vrstvou bazálnych buniek (ložiskovo bolo možné pozorovať začiatok tvorby buly v spodných častiach bazálnych keratinocytov) (obr. 2), areálovite nápadné kompletné odlúčenie epidermy s leukocytárnou reakciou. V oblastiach staršieho poškodenia kože boli fokálne prítomné prejavy reepitelizácie a v miestach čerstvej kompletnej denudácie epidermy ložisková kandidóza. Na okraji incipientných vezikúl a búl bola zachytená charakteristická cytolýza bazálnych keratinocytov (bledé vakuolizované keratinocyty vretenitého tvaru – tzv. „eraser effect“) (obr. 3). V dermis bol prítomný iba diskrétny zápalový infiltrát tvorený lymfocytmi, neutrofilmi a eozinofilmi. Excízie kože pôvodne fixované vo formaldehyde a zaliate v parafíne boli dodatočne vyšetrené pomocou transmisného elektrónového mikroskopu. Vyšetrenie preukázalo charakteristické štiepenie v dolnej tretine bazálnych keratinocytov, v zóne pod bunkovým jadrom (obr. 4).


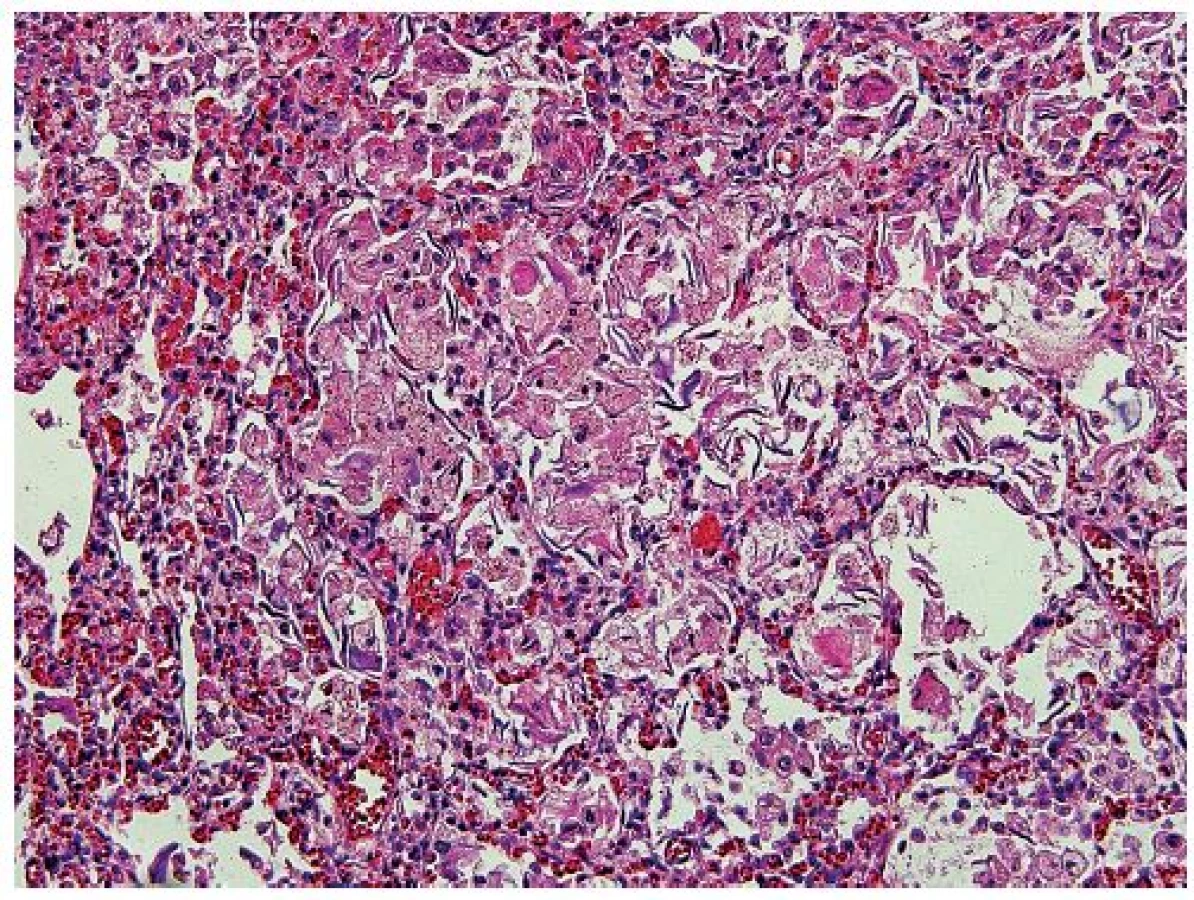
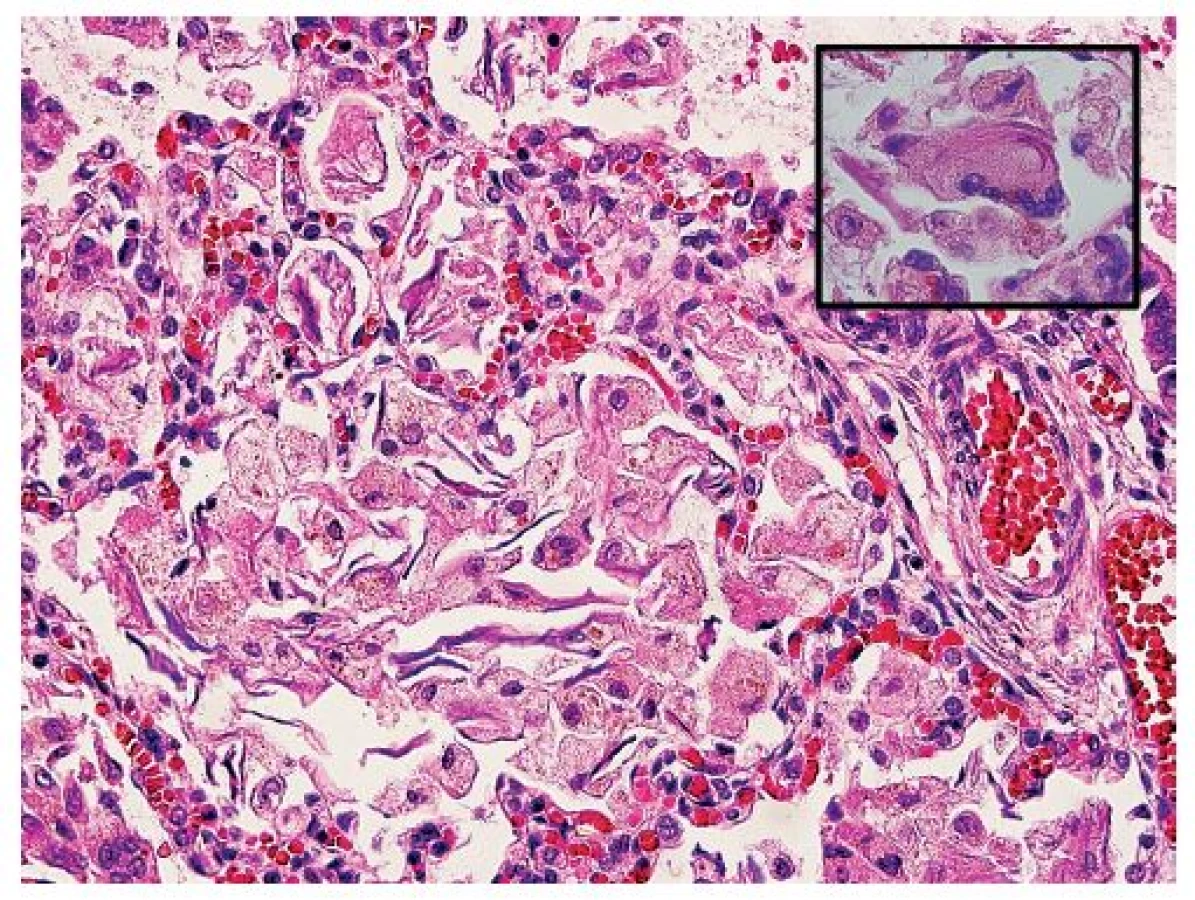
Vo všetkých lalokoch pľúc bol okrem intraalveolárneho edému prítomný masívny nález nahromadenia penovitých makrofágov, ktoré takmer kompletne vypĺňali väčšinu alveolov (obr. 5). Početné penovité makrofágy, a miestami aj mnohojadrové obrovské bunky obsahovali intracytoplazmatické inklúzie zlatohnedého a svetloružového sfarbenia (obr. 6), ktoré boli PAS pozitívne, avšak nevykazovali pozitivitu pri dôkaze na železo (Perlsova reakcia), ani na žlčové farbivá (Fouchet). Ďalším nápadným a neprehliadnuteľným nálezom v pľúcach bola prítomnosť plochých epiteliálnych (skvamóznych) buniek, ktoré miestami mali zachované jadrá (obr. 5). Prítomnosť mekónia a lanuga v alveoloch nebola zistená. Imunohistochemickým vyšetrením bola dokázaná silná cytoplazmatická pozitivita penovitých makrofágov s protilátkami proti CD68 (obr. 7) a CD163. Množstvo a objem makrofágov niekoľkonásobne prevyšovali počty makrofágov zistených v kontrolnom tkanive pľúc od novorodenca takmer identického veku (obr. 8). Skvamózne bunky lokalizované v alveoloch boli pozitívne pri dôkaze CK5/6 (obr. 9). Pri porovnaní s kontrolným prípadom boli makrofágy výrazne zvýšené nielen v alveolárnych priestoroch, ale aj v interstíciu. V interstíciu pľúc bol len minimálne zvýšený počet lymfocytov a plazmatických buniek, alveolárna architektúra bola zachovaná, fibrotizácia vo farbení podľa van Giesona v pľúcach nebola zistená. Špeciálnym farbením podľa Grama, Ziehl-Neelsena nebola zistená prítomnosť žiadnych mikroorganizmov, farbením pomocou PAS nebola zistená prítomnosť plesní, čím bol vylúčený infekčný pôvod pľúcneho nálezu. Neboli zistené žiadne mikroskopické zmeny vyskytujúce sa pri difúznom alveolárnom poškodení ako napr. hyalínne membrány, exsudácia fibrínu alebo krvácania.

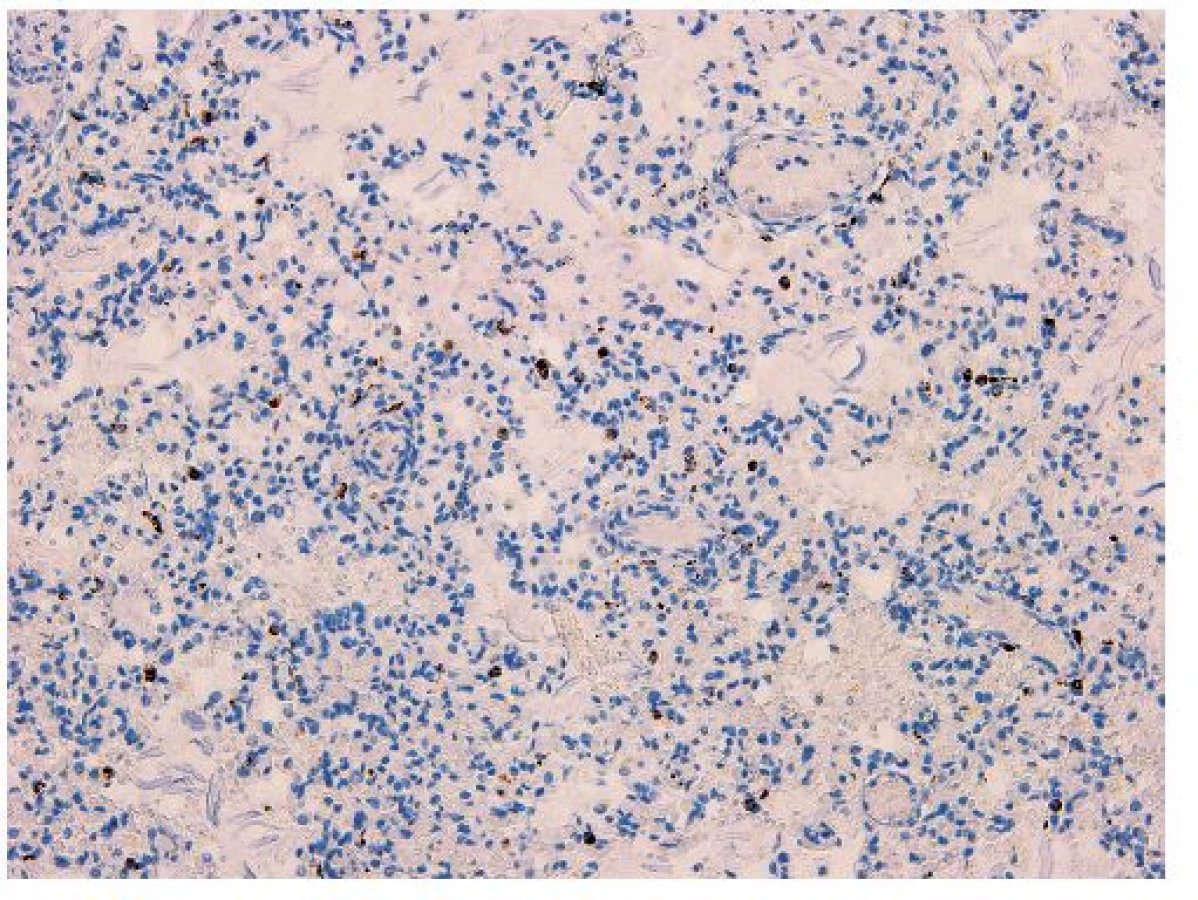
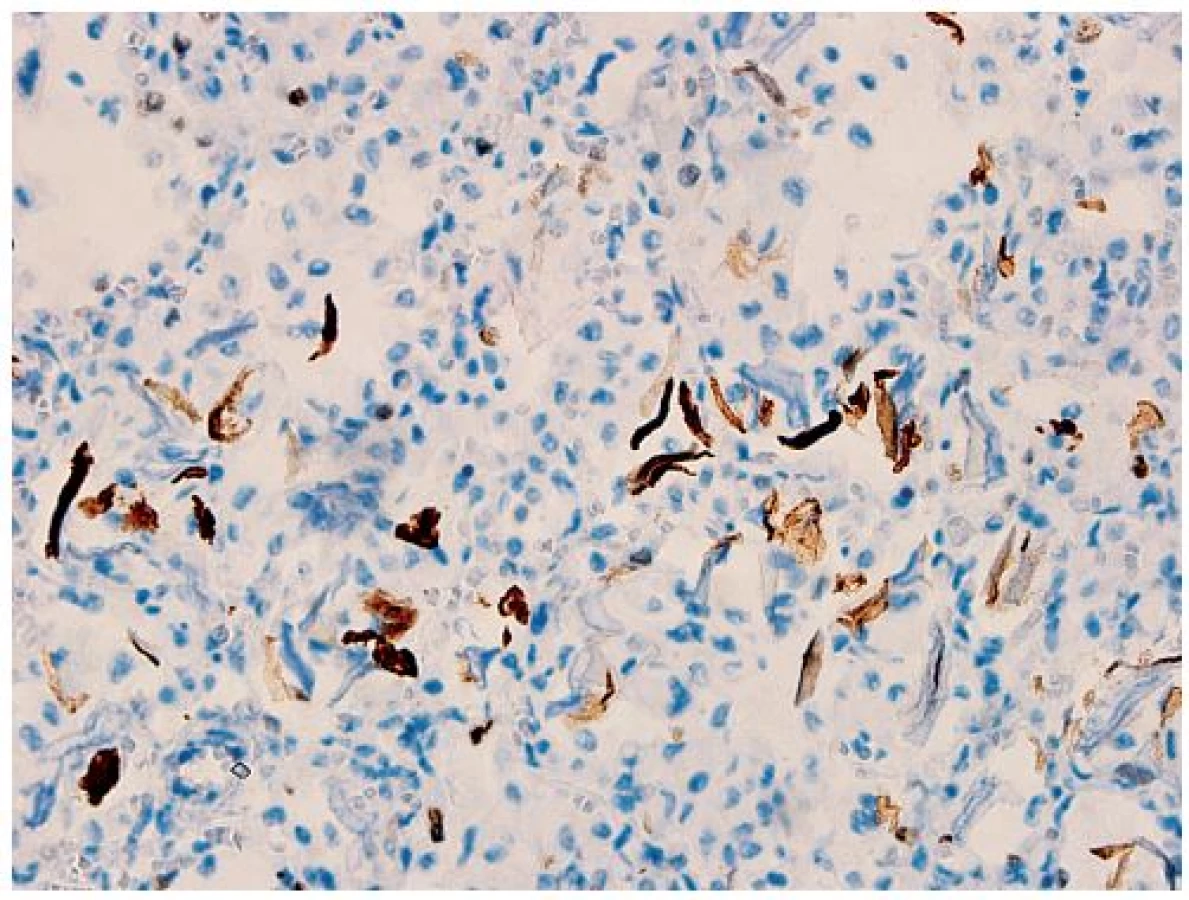
Na základe klinického obrazu a priebehu ochorenia, celkovej distribúcie kožných lézií, ako aj na podklade mikroskopických a elektrónovomikroskopických zmien kože bol prípad uzavretý ako kongenitálna epidermolysis bullosa zo skupiny EB simplex. Presnejšie určenie konkrétneho podtypu v rámci tejto skupiny by mohlo priniesť až molekulovo-genetické vyšetrenie. Bezprostrednou príčinou smrti novorodenca bola sepsa s multiorgánovým zlyhaním. Kultivačným vyšetrením krvi bol ako infekčný agens zistený staphylococcus haemolyticus a staphylococcus epidermidis. Pri histologickom vyšetrení sme naviac zistili ložiskovú kandidózu kože a pažeráka. V nami popísanom prípade sme nezistili žiadne zjavné vývinové poruchy (napr. bronchiálneho stromu, zmeny nechtov, atréziu duodena, hydronefrózu).
DISKUSIA
Podľa národného registra epidermolysis bullosa (EB) v USA je prevalencia EB 4,6/106, incidencia 10,75/106 (živonarodených detí) a celková prevalencia vrodenej EB je 8,22/106 (3). Ide o geneticky heterogénnu skupinu ochorení, ktoré sa dedia až na ojedinelé výnimky autozomálne dominantne, patofyziologicky sú zmeny indukované malou traumou, ktorá v prípade EB simplex vedie k cytolýze bazálnych alebo suprabazálnych keratinocytov s tendenciou hojiť sa bez jazvovatenia (pokiaľ nedôjde k sekundárnym zmenám erózií, napr. infekciou). Existujú viaceré klinické fenotypy ochorenia (napr. generalizovaný Köbnerov variant a Dowlingov-Mearov variant alebo lokalizovaný Weberov-Cockayneov variant), ktoré sa líšia vekom v čase prezentácie, distribúciou pľuzgierov, asociovanými abnormalitami a mutovaným génom.” (4). Histomorfologicky v prípade všetkých variant epidermolysis bullosa simplex dochádza k tvorbe vezikúl cytolýzou v bazálnej vrstve alebo v dolných vrstvách stratum spinosum (5).
V našom prípade, berúc do úvahy klinický nález, považujeme histologický nález v pľúcach za úplne neočakávaný a prekvapivý. Vo všetkých lalokoch obidvoch pľúcnych krídiel bola u 15-dňového novorodenca zistená prítomnosť jednak plochých, epiteliálnych buniek ako aj penovitých makrofágov. Prítomnosť mekónia a lanuga nebola zistená. Imunohistochemickým vyšetrením boli ploché, epiteliálne bunky identifikované ako keratinizujúce bunky epidermy, ktoré sa do pľúc dostali ešte počas intrauterinného vývinu a perzistovali tam až do úmrtia. Samotná prítomnosť buniek pochádzajúcich z aspirácie v 15. dni života nie je v rozpore s publikovanými pozorovaniami a údajmi (6). Zvyšky aspirácie plodovej vody boli zistené u detí (v kontrolnej skupine, ako aj v skupine detí, ktoré zomreli na syndróm náhleho úmrtia dojčaťa) až do 4. mesiaca života (7). Za najprekvapujúcejší histologický nález v tomto prípade považujeme masívnu akumuláciu penovitých makrofágov. Mikroskopický obraz do istej miery pripomínal obraz pri deskvamatívnej intersticiálnej pneumónii (DIP) (8). DIP sa vzácne môže vyskytovať aj u detí, ale na rozdiel od dospelých v etiológii nehrá úlohu fajčenie. DIP bola popísaná napr. pri niektorých variantach deficitu surfaktantu B alebo Gaucherovej chorobe. V dostupnej literatúre sme zistili len jeden prípad epidermolysis bullosa a súčasného intersticiálneho pľúcneho postihnutia u novorodenca, s intersticiálnymi a peribronchiálnymi lymfocytovými infiltrátmi a lymfoidnými folikulmi a miernou intraalveolárnou deskvamáciou a hypertrofiou médie artérií, ktorý však klinicky bol sprevádzaný pľúcnou hypertenziou, zrýchleným dýchaním a cyanózou (9). So zreteľom na to, že u dieťaťa v našom prípade neboli prítomné žiadne prejavy aspirácie plodovej vody, alebo mekónia, neboli prítomné žiadne akútne, alebo subakútne klinické prejavy pľúcneho postihnutia, ako ani žiadne patologické zmeny zistiteľné RTG vyšetrením pľúc, histologický nález nespadá do kategórie detských intersticiálnych ochorení (10, + osobná komunikácia). Je nutné podotknúť, že aj niektoré infekčné ochorenia pľúc (napr. pneumocystis Jiroveci, atypická mykobakterióza a pod.) môžu pripomínať obraz podobný DIP, čo sme v našom prípade vylúčili.
Domnievame sa, že masívna akumulácia penovitých makrofágov bola najpravdepodobnejšie dôsledkom aspirácie plodovej vody s nadmerným množstvom intrauterinne deskvamovaných buniek epidermy pri kongenitálnej EB a následnou aktiváciou pľúcnych makrofágov pri ich odstraňovaní. Na tomto mieste je nutné podotknúť, že aj minimálna aspirácia mekónia by viedla k poruchám dýchania s nutnosťou odsávania tracheobronchiálneho obsahu a umelej pľúcnej ventilácii, čo sa v danom prípade nestalo.
Aj keď prípady s kongenitálnou EB sú publikované len zriedkavo (11) a jedná sa o vzácne dedičné ochorenie, je nutné myslieť na túto jednotku v prípadoch pitiev intrauterinného odumretia plodu tak zo známych, ako aj neznámych príčin. S touto chorobnou jednotkou je nutné počítať nielen pri mŕtvorodených plodoch s pôrodom v nemocnici, ale aj mimo nej. V súdnolekárskej praxi je nevyhnutné v prípadoch intrauterinnej smrti plodu myslieť na mechanobulózne ochorenia najmä vtedy ak sa zdá, že je rozpor medzi klinickými údajmi o čase posledných prejavov života a makroskopickým nálezom v zmysle výraznejšej deskvamácie kože (macerácie), ktorý by mohol svedčiť pre dlhšie trvajúce intrauterinne odumretie plodu. Nesprávna interpretácia môže mať právne následky pre klinických lekárov. Na základe korelácie klinických a pitevných nálezov (12,13) je možné v niektorých prípadoch upresniť čas vnútromaternicového odumretia plodu. Pri overených klinických údajoch a nekorešpondujúcich kožných zmenách v zmysle nadmerného a predčasného odlučovania kože zistených pri pitve je vhodné uvažovať o kongenitálnom bulóznom ochorení kože, histologicky kožu adekvátne vyšetriť (vrátane elektrónovomikroskopického vyšetrenia) a prípadne navrhnúť rodičom genetické vyšetrenie.
ZÁVER
V kazuistike sme prezentovali neočakávaný a prekvapivý pľúcny nález u donoseného 15-dňového novorodenca s kongenitálnou EB simplex, ktoré zomrelo na sepsu s multiorgánovým zlyhaním. Vzhľadom na to, že sa v praxi sporadicky stretávame s nesúladom medzi údajmi o posledných prejavoch intrauterinného života a nekorelujúcimi posmrtnými zmenami zistenými pri pitve (pokročilejšia macerácia) je v takýchto prípadoch vhodné myslieť aj na kongenitálne mechanobulózne ochorenie, zabezpečiť mikroskopické vyšetrenie kože a interpretáciu nálezov na kompetentných pracoviskách.
Najpozoruhodnejším pozorovaním v tomto prípade bol masívny nález vyplnenia alveolov penovitými makrofágmi, ktorý do istej miery pripomínal obraz deskvamatívnej intersticiálnej pneumónie, ktorý však paradoxne nebol sprevádzaný žiadnou pľúcnou klinickou symptomatológiou a bol teda iba vedľajším nálezom. Po vylúčení iných možných príčin sa domnievame, že tento nález bol podmienený aspiráciou plodovej vody s nadmerným množstvom intrauterinne deskvamovaných buniek epidermy. Toto pozorovanie si však vyžaduje overenie v ďalších štúdiách.
POĎAKOVANIE
Za cenné pripomienky, postrehy a poznámky ďakujeme Megan Dishop, M.D., Department of Pathology and Laboratory Medicine, Children´s Hospital in Colorado a doc. MUDr. Radoslavovi Matějovi, Ph.D., Oddělení patologie a molekulární medicíny, Thomayerova nemocnice Praha.
PREHLASENIE
Autor práce prehlasuje, že v súvislosti s témou, vznikom a publikácií tohto článku nie v konflikte záujmov a vznik ani publikácia článku neboli podporené žiadnou farmaceutickou firmou. Toto prehlasenie sa týka i všetkých spoluautorov.
Adresa pre korešpondenciu:
MUDr. Daniel Farkaš
Úrad pre dohľad nad zdravotnou starostlivosťou
SLaPA pracovisko Košice
Letná 47, 040 01 Košice
tel.: +421552852660, fax: +421552852655
e-mail: farkas.dany@gmail.com
Sources
1. Barnhill RL, Crowson AN, Magro CM, Piepkorn MW. Dermatopathology (3rd edn). The McGraw-Hill: New York; 2010: 324-327.
2. Fine JD. Inherited epidermolysis bullosa: recent basic and clinical advances. Curr Opin Pediatr 2010; 22(4): 453-458.
3. Fine JD, Hintner H. Life with Epidermolysis Bullosa (EB): Etiology, Diagnosis, Multidisciplinary Care and Therapy, Springer: Wien New York; 2009: 27.
4. Horn HM, Tidman MJ. The clinical spectrum of epidermolysis bullosa simplex. Br J Dermatol 2000; 142: 468-472.
5. Smith LT. Ultrastructural findings in epidermolysis bullosa. Arch Dermatol 1993; 129: 1578-1584.
6. Dettmeyer RB. Forensic Histopathology. Springer: Berlin Heidelberg; 2011: 218-219.
7. Fracasso T, Karger B, Vennemann M, Bajanowski T, Golla-Schindler UM, Pfeiffer H. Amniotic fluid aspiration in cases of SIDS. Int J Legal Med 2010; 124 (2): 113-117.
8. Leslie KO. My approach to interstitial lung disease using clinical, radiological and histopathological patterns. J Clin Pathol 2009; 62(5): 387-401.
9. Doeker B, Hussein A, Trowitzsch E. Interstitial lung diseases with pulmonary hypertension associated with epidermolysis bullosa in an infant. Klin Pädiatr 1998; 210(5): 340-344.
10. Dishop MK. Diagnostic pathology of diffuse lung disease in children. Pediatr Allergy Immunol Pulmonol 2010; 23(1): 69-85.
11. Smith LT, Miller AW, Kirz DA, Elias S, Brumbaugh S, Holbrook KA. Separation of noncutaneous epithelia in a fetus diagnosed in utero with junctional epidermolysis bullosa. Pediatr Res 1992; 31(6): 561-566.
12. Genest DR, Williams MA, Greene MF. Estimating the time of death in stillborn fetuses: I. Histologic evaluation of fetal organs; and autopsy study of 150 stillborns. Obstet Gynecol 1992; 80: 575-584.
13. Genest DR, Singer DB. Estimating the time of death in stillborn fetuses: III. External fetal examination; a study of 86 stillborns. Obstet Gynecol 1992; 80: 593-600.
Labels
Anatomical pathology Forensic medical examiner ToxicologyArticle was published in
Czecho-Slovak Pathology

2015 Issue 2
Most read in this issue
- Practical comments on examination of placenta in the second and third trimester of gravidity
- Molecular pathology of endometrial carcinoma – a review
- Unusual lung finding of massive alveolar filling with foamy macrophages in congenital epidermolysis bullosa after amnion fluid aspiration in 15-day-old newborn without any clinical signs of respiratory impairment
- EGFR in triple negative breast carcinoma: significance of protein expression and high gene copy number