
Získaná neuromyotonie s nevelkými centrálními příznaky s průkazem protilátek proti napěťově řízeným kaliovým kanálům – kazuistika
Authors:
J. Latta 1; E. Ehler 1; J. Zámečník 2
Authors‘ workplace:
Neurologická klinika, Pardubická krajská nemocnice, a. s., 2Ústav patologie a molekulární medicíny, UK 2. LF a FN Motol, Praha
1
Published in:
Cesk Slov Neurol N 2009; 72/105(4): 373-377
Category:
Case Report
Overview
U 52letého, dosud zdravého muže se v průběhu několika týdnů rozvinul myalgický syndrom s únavností, slabostí, brněním a záškuby ve svalech, a to zejména na dolních končetinách (DK). Pro tyto potíže pak už nebyl schopen samostatné chůze. V klinickém nálezu při přijetí byla slabost a tuhost svalů s bolestivo u palpací, vegetativní příznaky s pocením, intermitentní tachykardi í, zácpo u, dále osobnostní a behavi orální změny s nespavostí a noční zmateností. V elektromyografii (EMG) byla patrna klidová trvalá aktivita s výboji, které byly provokovány volní aktivito u a zejména stimulací motorických vláken. Nemocného jsme léčili karbamazepinem a metylprednizolonem. Došlo k ústupu hypertoni e, bolestí svalů i centrálních příznaků. V té fázi došel silně pozitivní nález a utoprotilátek proti kali ovým kanálům. Paci ent však náhle zemřel na maligní arytmii.
Klíčová slova:
neuromyotonie – Morvanův syndrom – centrální příznaky Morvanova syndromu – protilátky proti kaliovým kanálům – maligní arytmie – moykymie – fascikulace
Úvod
„Stiff-muscles“ (tuhé, neohebné svaly) jsou podmíněny trvalou svalovou aktivitou. Nejedná se o přímé onemocnění svalu a tento stav je nutno odlišit od rigidity či spasticity. Stiff-muscles se nejčastěji vyskytují u syndromu kontinuální svalové aktivity (neuromyotonie), encefalomyelitidy se svalovou hypertonií, stiff-person syndromu a stiff-leg syndromu [1]. Neuromyotonie se vyznačuje pseudomyotonickou poruchou svalové relaxace v kombinaci s myokymiemi a fascikulacemi. V klinickém nálezu se nachází tuhost svalů a krampy [2].
V roce 1890 Augustin Marie Morvan popsal pacienta s myokymií, bolestmi svalstva, nadměrným pocením a poruchou spánku [3]. Morvanův syndrom je vzácné autoimunitní onemocnění charakterizované nepravidelnými kontrakcemi svalstva, krampy, slabostí, hyperhidrózou, nespavostí a zmateností [4]. V anglicky psané literatuře bylo doposud popsáno jen asi 14 případů Morvanova syndromu a jen několik případů s kompletním spektrem centrálních příznaků [5]. Předkládáme případ dospělého muže, u kterého se vyvinuly typické projevy získané neuromyotonie s centrálními příznaky Morvanova syndromu.
Kazuistika
52letý muž byl přijat pro tři týdny progredující bolesti a brnění chodidel a lýtek s postupným šířením proximálně až do bederní oblasti a třísel. Bolesti byly doprovázeny výraznými fascikulacemi a slabostí při chůzi. Byl schopen jen několika krůčků s pomocí dvou podpažních berlí. Pracoval jako řidič, byl sledován pro syndrom karpálního tunelu profesionálního původu a před čtyřmi lety podstoupil operaci vlevo s výrazným efektem. Pět let se léčil pro hypertenzi (isradipin, perindopril), jinak byl vždy zdráv a křeče či záškuby ve svalech nemíval.
V klinickém nálezu při přijetí dominovala slabost a tuhost svalů, ve svalech byly spontánní záškuby charakteru myokymií i rychlé izolované nerytmické záškuby. Při poklepu na svaly – více na dolních končetinách, ale i horních končetinách a jazyku – se akcentovaly myokymie i náhlé záškuby (nejspíše fascikulace). Idiomuskulární dráždivost byla také zvýšena a vibrační čití bylo zkráceno na 5/8. Pohyby byly pomalé, mimika ztuhlá, až maskovitá, patrno bylo i „myotonické“ zpoždění víček při pohledu dolů a mydriáza s omezeným rozsahem fotoreakce. Dále hyperhidróza na končetinách, trupu i obličeji a zácpa. Během hospitalizace se postupně vyvinuly i psychické změny. V popředí dominovala noční agitovanost nasedající na krátké stavy zmatenosti při častém buzení ze spánku, naopak během dne stěží udržel pozornost a pospával. Byly přítomny i alterované reakce na některé nepříjemné vyšetřovací a léčebné metody (per rectum vyšetření, zavádění čípků, klyzma).
V laboratorních odběrech jsme zjistili zvýšenou hodnotu kreatinkinázy (CK): 12,82 ukat/l (0,58–3,87) a její izoenzym CK-MB: 0,42 ukat/l (0,00–0,10). Volný myoglobin v séru byl výrazně vyšší: 196,4 ug/l (25–72). Jaterní enzymy byly zvýšeny jen lehce. Z dalších vyšetření byla vyšší hladina imunoglobulinů třídy G: 22,09 g/l (6,81–16,4), gamaglobulinů v séru: 0,236 (0,10–0,19) a C‑reaktivní protein: 33,8 mg/l (0–10). Nádorové markery (PSA, AFP, CEA, C19.9, C12.5, C15.3) v séru byly negativní.
Následně byla provedena lumbální punkce s normálním cytologickým nálezem s proteiny 0,59 g/l (0,20–0,40). Při vyšetření proteinových frakcí byl prokázán zvýšený orosomukoid 17,8 mg/l (1,5–4,5) a při izoelektrické fokuzaci nebyly nalezeny žádné oligoklonální pásy.
Při motorické neurografii byla nalezena jen delší distální latence pro n. medianus vpravo (8,70 ms) a repetitivní následné výboje po stimulaci motorických vláken. Pro tyto následné a bezprostředně nastupující výboje nebylo možno vyšetřit F-vlny (obr. 1) a ani H-reflex (obr. 2). Senzitivní nervové akční potenciály byly nižší amplitudy, rychlost vedení senzitivním nervem byla lehce snížena. Při vyšetření svalů koncentrickou jehlovou elektrodou byly v popředí spontánní a často repetitivní až rytmické výboje charakteru dupletů, tripletů i multipletů a někdy i vysokofrekvenčního krátce trvajícího výboje, fascikulace a myokymie (obr. 3, 4). Patologická aktivita (multiplety, výboje) se provokovala jak pohybem jehly, poklepem na sval, tak v menší míře i volní kontrakcí. Při analýze potenciálů motorických jednotek byl jen zvýšený podíl polyfázických potenciálů, jinak trvání, amplituda i frekvence pálení motoneuronů byly v mezích normy.
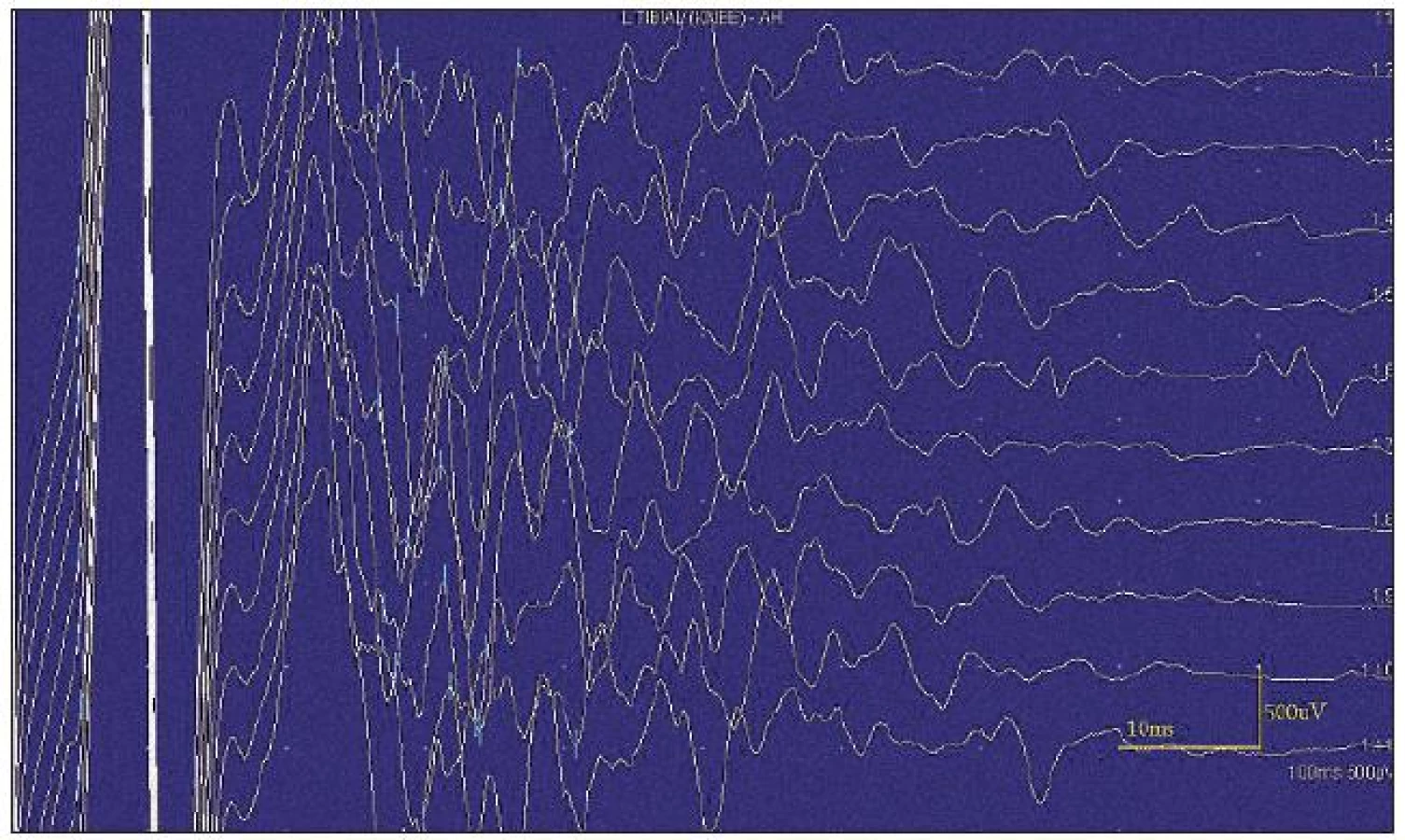
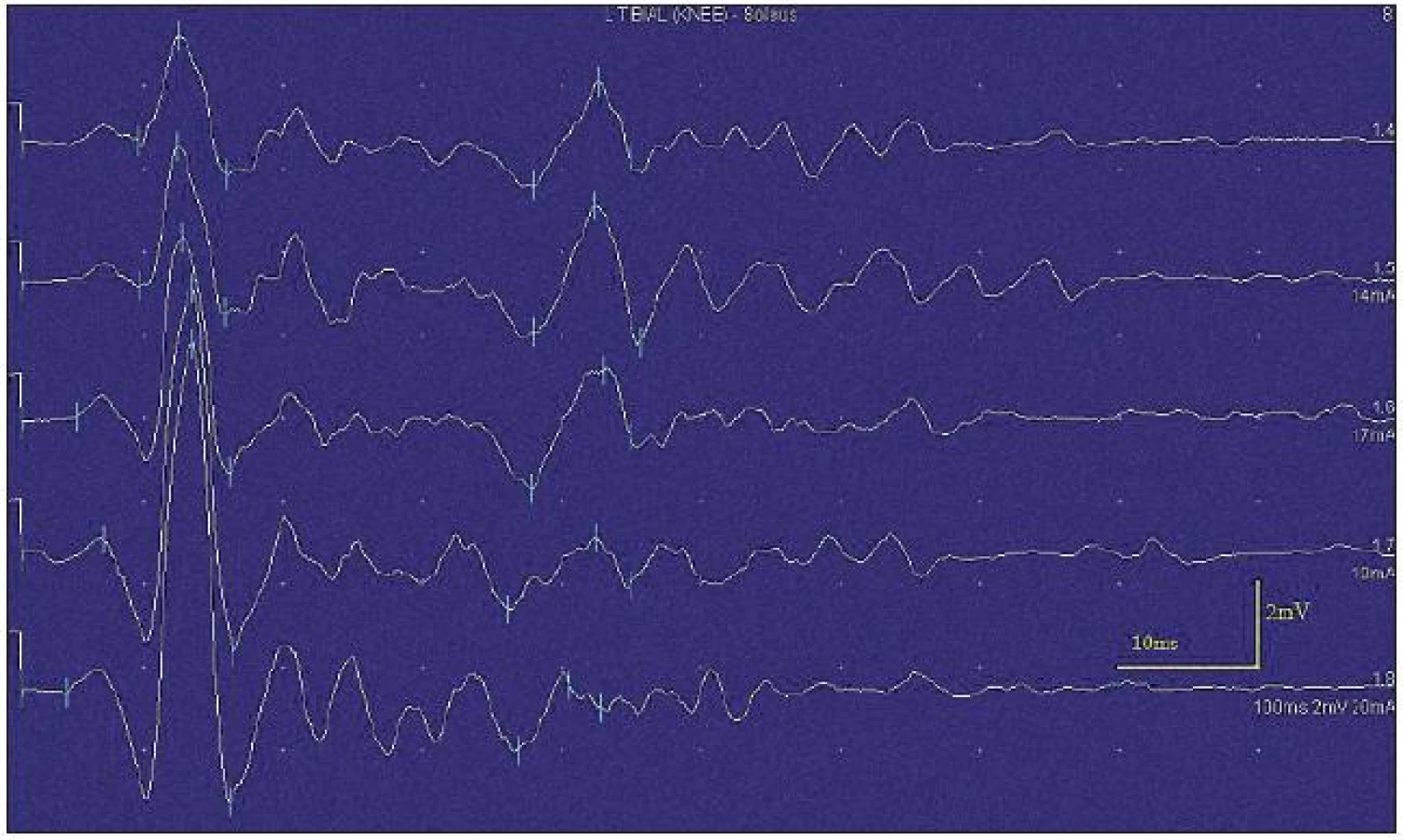
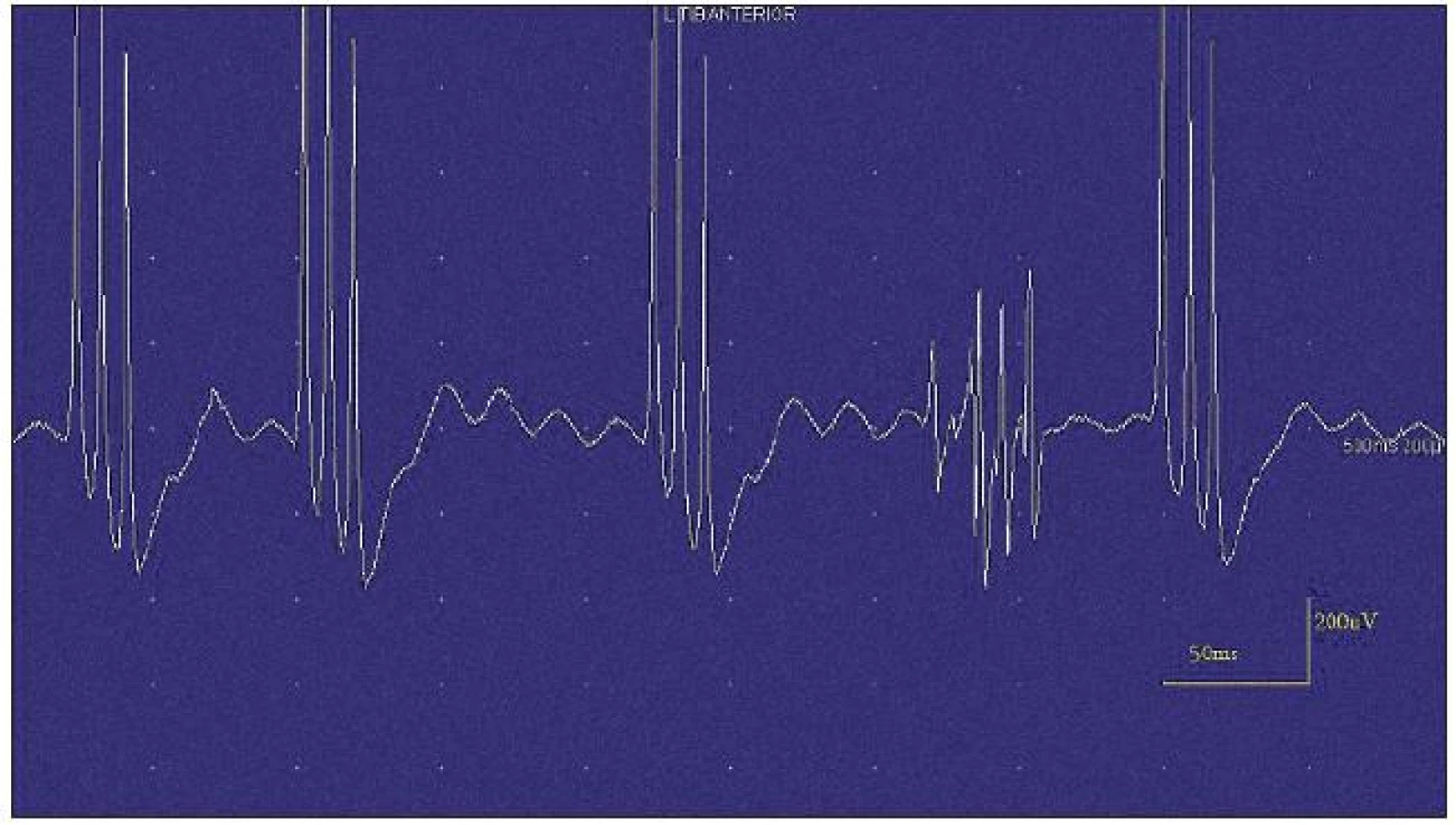

EEG neprokázala patologickou aktivitu a brain mapping zobrazil dominanci alfa aktivity 10 Hz nad zadními kvadranty. EKG ukázalo klidovou sinusovou tachykardii, QTc (Bazettův index) byl v normě. Bez průkazu změn QT úseku (do 400 ms).
Pro vyloučení paraneoplastické etiologie jsme provedli vyšetření paraneoplastických protilátek (anti‑Hu, anti‑Ri, anti‑Yo, anti‑myelin), UZ břicha, CT hrudníku, biopsii uzliny v levé nadklíčkové jamce a kožní biopsii s negativním nálezem.
Biopsie levého m. vastus medialis neprokázala žádné myopatické ani neurogenní změny. Standardní enzymově histochemické vyšetření i imunohistologie s průkazem sarkolemálních proteinů byly v normě. Patologické změny nebyly pozorovány ani při elektronmikroskopickém vyšetření.
Vzhledem k možnému autoimunitnímu charakteru postižení jsme nechali vyšetřit protilátky proti gangliosidům a anti‑GAD (protilátky proti glutamátdekarboxyláze), které byly rovněž s negativním výsledkem.
EMG nález svědčil pro neuromyotonii a byl v souladu s klinickým obrazem i laboratorním nálezem. Nemocného jsme začali léčit karbamazepinem (až 2 × 400 mg) v kombinaci se slabými opioidy (tramadol a pak dihydrokodein 2 × 90 mg) a zahájili jsme imunosupresivní léčbu metylprednizolonem i. v. (250 mg denně). Následně jsme obdrželi výsledek vyšetření protilátek proti napěťově řízením kaliovým kanálům (anti‑VGKC), které bylo silně pozitivní: 1 439 pM/l (Oxford Radcliff Hospital – radio immunoassay, norma 0–100). Nemocný se postupně zlepšoval a byl schopen již samostatné chůze, bylo menší pocení, zácpa i tuhost svalstva. Přechodně ustoupily i fascikulace a myokymie. Z centrálních příznaků přetrvávala již jen porucha usínání.
Přes zlepšení klinického nálezu i subjektivních potíží však pacient po 30 dnech hospitalizace náhle umírá. Kromě známek akutního srdečního selhání při maligní arytmii nebyly pitevním vyšetřením prokázány žádné další změny na vnitřních orgánech, včetně mozku a míchy.
Diskuze
Existuje celá řada poruch periferního neurogenního původu, která může produkovat kontinuální svalovou aktivitu a je pak podkladem svalové tuhosti a křečí. (tab. 1). Tato porucha byla popsána jako kontinuální aktivita svalového vlákna či Isaacsův syndrom (Isaacs 1961). Pro svalovou hypertonii s poruchou relaxace připomínající myotonii ji Mertens a Zschocke v roce 1965 pojmenovali neuromyotonií. Další názvy vždy obsahovaly popis jak hypertonie, tak i záškubů svalových snopců – myokymie s poruchou svalové relaxace či pseudomyotonie s myokymií (tab. 2) [1,3].
![Charakteristiky neuromyotonie [1,9].](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image/332e33df6aae6bab91ccc73ab8ba50f9.jpg)
![Klasifikace neuromyotonie [16].](https://www.prolekare.cz/media/cache/resolve/media_object_image_large/media/image/b3da5a0b9df87cb6589bde587a63c019.jpg)
Nemoc se vyvíjí postupně a je klinicky charakterizována nárůstem svalové tuhosti v klidu, trvalými záškuby ve svalech – twitching (fascikulace) a kontrakcemi svalů šířícími se ve vlnách – rippling (myokymie), často sval připomíná pohybující se červy („sack of worms“) [6]. Křeče, mnohdy bolestivé, navazují na volní svalovou kontrakci a jsou důsledkem opožděné relaxace svalu (pseudomyotonie). Spontánní bolesti jsou vzácné, ale téměř pravidelně jsou přítomny bolesti svalů při křečích a je palpační bolestivost svalů. Nejprve bývají postiženy distální svaly dolních končetin (DK) a postupně se nemoc šíří proximálně, na horní končetiny (HK), trup i bulbární svaly. Touto distribucí se neuromyotonie odlišuje od stiff-person syndromu (nejprve bývá postiženo svalstvo trupu a proximálních segmentů končetin) a naopak se blíží až obrazu tetanických křečí (karpo-pedální) [1,7]. U našeho nemocného se objevila tuhost svalů a křeče zpočátku na akrech DK a postupně se šířily na HK i proximálně na trup. Ihned od počátku byly provázeny akroparesteziemi. Fascikulace i myokymie se objevily asi s týdenním zpožděním po ztuhlosti svalů.
Sérové hladiny svalových proteinů (volný myoglobin, CK, aldoláza, laktát dehydrogenáza) mohou být u neuromyotonie zvýšeny. Je to pravděpodobně známka svalového poškození při nadměrné svalové zátěži [8].
Výboje při myokymiích jsou krátké pršky jednotlivých motorických jednotek, které pálí frekvencí 5–150 Hz. Tyto výboje se projevují ve formě dupletů, tripletů či multipletů a jsou následovány krátkou periodou elektrického ticha. Mohou být pravidelné i nepravidelné a jejich frekvence často závisí na délce myokymických výbojů – delší pršky se objevují méně často. Generátor myokymických výbojů může být umístěn na periferním motoneuronu velmi proximálně (např. u roztroušené sklerózy či gliomu pontu) i velmi distálně (např. u akutní polyradikuloneuritidy) [1,7]. Výboje se nejčastěji objevují spontánně. Méně často jsou indukovány volní aktivitou, kdy volně spuštěný akční potenciál prochází přes generátor – tedy oblast axonu, ve které je iniciováno repetitivní pálení potenciálu motorické jednotky. Dysfunkce VGKC v axonální membráně vede k výraznému zvýšení supernormality a ke spontánním repetitivním výbojům na podkladě hyperexcitabilní axolemy. Role efaptické transmise je v místě generátoru nejistá [9].
Neuromyotonické výboje mají stejnou patofyziologii i lokalizaci generátoru, avšak mají vyšší frekvence (150–300 Hz). Pršky výbojů jsou prodloužené, mají náhlý začátek i náhlý konec a dochází ke snižování amplitudy jednotlivých potenciálů v průběhu výboje. Výboje bývají spontánní i spouštěné volní aktivitou, pohyby jehly, hypoxií či poklepem na sval. Od myokymií se liší pouze delším trváním a klesající amplitudou [9,10].
Neuromyotonické i myokymické výboje přetrvávají i po proximálních blokádách nervu lokálními anestetiky a mizí u blokády nervosvalové ploténky (kurare, botulotoxin). Jsou přítomny při celkové anestezii i ve spánku [9,11]. U autoimunitně podmíněných neuromytonických syndromů se obvykle nachází generátor výbojů distálně od terminálního větvení axonu, a přitom však nepostihuje presynaptickou část nervosvalové ploténky (rozdíly mezi spontánními a volně spouštěnými motorickými akčními potenciály při metodě stanovení velikosti motorických jednotek) [12]. My jsme klinicky i v EMG nacházeli myokymie i neuromyotonické fenomény, také však fascikulace, ojediněle i rytmické fibrilace. Tyto fenomény byly přítomny spontánně, jejich četnost se mírně zvýšila při volní aktivitě (s delší latencí), výrazně se zvýšila po poklepu na sval (s rychlým nástupem) i při mechanické iritaci EMG jehlou. Rovněž při motorické neurografii a zejména při vyšetření F-vln vždy bezprostředně následovala série následných výbojů. Na rozdíl od literárních údajů jsme latenci repetitivních výbojů neprokázali [2,11]. Docházelo však ke snižování amplitudy motorických odpovědí.
Neuromyotonické syndromy jsou poruchy periferních nervů. Jsou to léze lokalizované – fokální či spíše multifokální, porucha je v axonální membráně, a to na podkladě dysfunkce (upregulace) pomalých kaliových kanálů v nodální oblasti [1,9,13]. Se získanou neuromyotonií a Morvanovým syndromem jsou vázané „Shaker‑type“ kaliové kanály (Kv1) [14]. Nejsou však postiženy pouze motorické axony, ale také vegetativní nervy, někdy i převodní systém v srdci, rovněž i senzitivní vlákna. Kromě motorických projevů jsme prokázali i výrazné poruchy autonomních nervů – mydriázu s omezeným rozsahem fotoreakce, výrazné pocení, slzení, tachykardii a zácpu. Již od počátku se objevilo brnění na akrech HK i DK, což bylo podkladem pro úvahu o akutní polyradikuloneuritidě. Po zahájení léčby karbamazepinem se zmírnila nejen tuhost svalstva a omezil se i výskyt fascikulací a myokymií, ale ustoupilo i pocení, mydriáza a zčásti tachykardie i úporná zácpa. Ustoupila nespavost a noční zmatenost.
Potřeba spánku je u Morvanova syndromu výrazně redukována až na 2–4 hodiny za den. Klinické projevy spojené s insomnií jsou denní ospalost, narušená struktura REM spánku a porucha usínání spojená s halucinatorními projevy [5]. Obdobné příznaky jsme pozorovali i u našeho nemocného.
V roce 1890 popsal Morvan myokymie s bolestmi svalů, excesivním pocením a poruchou spánku. Tento stav nazval „fibrilární choreou“. Jeho nemocný zemřel pět týdnů po vzniku potíží. Jako Morvanův syndrom se označuje neuromyotonie jak s periferními projevy, tak i projevy centrálními – nespavost, deprese, psychické změny, halucinace. U tohoto syndromu bývají obzvláště výrazné změny autonomních funkcí a zejména srdeční arytmie. Popisují se frekventní supraventrikulární extrasystoly až „syndrom pomalého Q-T“. Mozkové a kardiální změny jsou v souvislosti s poruchou kaliových kanálů, a tedy s výskytem protilátek proti VGKC [3,15]. Mírní se s podáním imunosupresivní léčby či zejména po plazmaferéze [15]. Náš nemocný se vyznačoval výraznými vegetativními příznaky i výraznými psychickými problémy (nespavost, fixace k jednotlivým příznakům – i vegetativním, noční agitace, behaviorální a osobností změny). Po karbamazepinu a po zahájení imunosupresivní léčby se zmírnila hypertonie svalů, psychické projevy i vegetativní příznaky (pocení, zácpa, méně arytmií). Pátrali jsme po převodní poruše i po změnách v repolarizační fázi komor (ekg, Bazettův index), avšak žádnou z těchto kardiálních poruch jsme neprokázali. Nemocný však zemřel náhle, na maligní arytmii. Bylo to sedm týdnů od začátku onemocnění. Klinický obraz a průběh nemoci našeho pacienta do značné míry odpovídal Morvanovu syndromu.
Závěr
Neuromyotonické syndromy se vyznačují zvýšeným svalovým napětím, pseudomyotonickými fenomény, přítomností myokymií a fascikulací ve svalech, počátečním postižením akrálních svalů a postupným šířením na trup a HK, akroparesteziemi, poruchami autonomního systému, charakteristickým neurofyziologickým nálezem a dysfunkcí axonální membrány periferních nervů s poruchou kaliových kanálů. I když se může projevit dosti různorodým obrazem, přesto jsou základní neurofyziologické charakteristiky jasně přítomny a nemoc má autoimunitní charakter, s průkazem protilátek a je dobře ovlivnitelná léčbou.
MUDr. Jan Latta
Neurologická klinika
Pardubická krajská nemocnice, a.s.
Kyjevská 44
532 03 Pardubice
e-mail: janci82@gmail.com
Sources
1. Fahn S, Jankovic J. Principles and practice of movement disorders. Philadelphi a: Churchill Livingstone Elsevi er 2007.
2. Kadaňka Z, Bednařík J. Ne uromyotoni e – nová kanalopati e. Cesk Slov Ne urol N 2000; 63/ 96(3): 128– 133.
3. Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P et al. Morvan’s syndrome: peripheral and central nervo us system and cardi ac involvement with antibodi es to voltage- gated potassi um channels. Brain 2001; 124(12): 2417– 2426.
4. Lee EK, Maselli RA, Ellis WG, Agi us MA. Morvan’s fibrillary chore a: a parane oplastic manifestati on of thymoma. J Ne urol Ne urosurg Psychi atry 1998; 65(6): 857– 862.
5. Bajaj BK, Shrestha S. An interesting case report of Morvan’s syndrome from the Indi an subcontinent. Ne urol Indi a 2007; 55(1): 67– 69.
6. Oh SJ. Principles of clinical electromyography. Case studi es. Baltimore: Willi ams & Wilkins 1993.
7. Valls- Solé J, Montero J. Role of EMG evalu ati on in muscle hyperactivity syndromes. J Ne urol 2004; 251(3): 251– 260.
8. Han IK, Newsom- Davis J. Ne uromyotoni a (Isaacs‘ Syndrome). In: Lane RJM (ed). Handbo ok of Muscle Dise ase. 1st ed. New York: Informa He althcare 1996: 355– 363.
9. Gutmann L, Gutmann L. Myokymi a and ne uromyotoni a. J Ne urol 2004; 251(2): 138– 142.
10. Maddison P. Ne uromyotoni a. Clin Ne urophysi ol 2006; 117(10): 2118– 2127.
11. Bednarík J, Kadanka Z. Voliti onal and stimulati on induced ne uromyotonic discharges: unusu al electrophysi ological pattern in acquired ne uromyotoni a. J Ne urol Ne urosurg Psychi atry 2001; 70(3): 406– 407.
12. Arimura K, Arimura Y, Ng A, Uehara A, Nakae M, Osame M et al. The origin of spontane o us discharges in acquired ne uromyotoni a. A Macro EMG study. Clin Ne urophysi ol 2005; 116(8): 1835– 1839.
13. Nodera H, Kaji R. Nerve excitability testing and its clinical applicati on to ne uromuscular dise ases. Clin Ne urophysi ol 2006; 117(9): 1902– 1916.
14. Kle opa KA, Elman LB, Lang B, Vincent A, Scherer SS. Ne uromyotoni a and limbic encephalitis sera target mature Shaker‑type K+ channels: subunit specificity correlates with clinical manifestati ons. Brain 2006; 129(6): 1570– 1584.
15. Irani S, Lang B. Auto antibody- medi ated disorders of the central nervo us system. Auto immunity 2008; 41(1): 55– 65.
16. Hart I, Vincent A, Willison H. Ne uromyotoni a. In: Engel A. Myastheni a Gravis and Myasthenic Disorders. Oxford: Oxford University Press US 1999: 230.
Labels
Paediatric neurology Neurosurgery NeurologyArticle was published in
Czech and Slovak Neurology and Neurosurgery

2009 Issue 4
Most read in this issue
- Nádory tretej mozgovej komory
- Získaná neuromyotonie s nevelkými centrálními příznaky s průkazem protilátek proti napěťově řízeným kaliovým kanálům – kazuistika
- Botulotoxin v léčbě spasticity
- Paroxysmální kinezigenní dyskineze: případ mladé ženy s alternující hemidystoni í – kazuistika