
Mladá žena se synkopami
Authors:
T. Mikušová; P. Bothová; J. Meluzín
Authors‘ workplace:
I. interní kardioangiologická klinika FN u sv. Anny Brno a LF MU Brno
Published in:
Kardiol Rev Int Med 2011, 13(4): 258-259
Category:
Case reports- competitive
Overview
Nekompaktní kardiomyopatie je vzácné onemocnění srdečního svalu, pro jehož léčbu nemáme guidelines. V kazuistice uvádíme případ mladé ženy se synkopami, u které bylo zjištěno toto onemocnění. Vzhledem k výskytu náhlého úmrtí v rodině, opakovaným synkopám a neischemické kardiomyopatii s dobrou ejekční frakcí byla indikována k zavedení implantabilního kardioverteru-defibrilátoru (ICD).
Klíčová slova:
nekompaktní kardiomyopatie – srdeční selhání – synkopa
Kazuistika
30letá žena, matka dvou dětí sledovaná s autoimunitní tyreoiditidou, jinak zdravá, byla po recidivě synkopy hospitalizována v okresní nemocnici. Pacientka prodělala krátkodobou ztrátu vědomí, které předcházelo bušení srdce a pocení. EKG křivka byla při přijetí do nemocnice normální.
Za hospitalizace byla resuscitována pro 10sekundovou asystolii. Po zavedení dočasné kardiostimulace si pacientka začala stěžovat na torakalgie, nově byl zjištěn pansystolický šelest.
Pro podezření na perforaci myokardu elektrodou dočasné kardiostimulace byla přeložena k dořešení na naše pracoviště. CT srdce a transtorakální i transezofageální echokardiografické vyšetření bylo bez jasných známek perforace, na echokardiografii bylo vysloveno podezření na intramurální hematom ve stěně pravé komory a zároveň byl popsán obraz odpovídající diagnóze nekompaktní kardiomyopatie. EKG monitorace byla bez záchytu závažné arytmie. Pod echokardiografickou kontrolou byla vytažena elektroda dočasné kardiostimulace, o perforaci myokardu nešlo. Po výkonu vymizely torakalgie i pansystolický šelest. Pacientka byla nadále stabilní, trval sinusový rytmus, bez arytmií.
Provedení magnetické rezonance potvrdilo diagnózu nekompaktní kardiomyopatie. Jiná závažná patologie nebyla popsána, systolická funkce levé komory byla normální, ejekční frakce 60 %, nebyly známky patologického enhancementu po podání gadolinia.
Po doplnění rodinné anamnézy jsme zjistili, že děd z otcovy strany zemřel náhle ve 33 letech, otec byl vyšetřován pro arytmie, strýc a sestřenice prodělali opakovaně kolapsové stavy. Anamnéza z matčiny strany byla vzhledem k výskytu nekompaktní kardiomyopatie negativní.
K došetření arytmií a arytmogenního potenciálu pacientky bylo provedeno elektrofyziologické vyšetření, které neprokázalo AVNRT či akcesorní dráhu, taktéž programovaná stimulace komor byla negativní, Wenckebachův bod byl 150/min. Test na nakloněné rovině pacientka opakovaně absolvovala v minulosti s normálním nálezem.
Koronarografie vzhledem k nízké pravděpodobnosti výskytu ischemické choroby srdeční u mladé ženy bez rizikových faktorů rozvoje aterosklerózy nebyla provedena.
Přestože pacientka měla dobrou systolickou funkci levé komory srdeční a negativní elektrofyziologické vyšetření, bylo pro diagnózu nekompaktní kardiomyopatie, výskyt recidivujících synkop u pacientky a náhlou smrt v rodině indikováno zavedení implantabilního kardioverteru-defibrilátoru (ICD). Implantace byla komplikována iatrogenním pneumotoraxem vlevo, který byl řešen zavedením hrudní drenáže s postupnou úpravou stavu. Antikoagulační terapie nebyla nasazena pro dobrou ejekční frakci levé komory. Byl zaveden ramipril v dávce 1,25 mg denně. Pacientka byla propuštěna do ambulantní péče bez subjektivních potíží, kardiopulmonálně kompenzovaná. Bylo zahájeno genetické vyšetření, výsledky však ještě v těchto dnech nejsou k dispozici. Doporučili jsme kardiologické došetření ostatních členů rodiny.
Diskuze
Nekompaktní kardiomyopatie levé komory (left ventricular non-compaction – LVNC) je onemocnění, které si získalo pozornost především v posledních 25 letech. Poprvé bylo popsáno Grantem v roce 1926, ale jako klinickou jednotku jej poprvé uvedli Dušek et al v roce 1975 [1]. Nekompaktní kardiomyopatie může být spojena s dalšími vrozenými anomáliemi. V případě, že je pouze samotné srdce postiženo výraznými trabekulami a hlubokými recesy, se jedná o tzv. izolovanou nekompaktní kardiomyopatii, kterou se budeme dále zabývat.
Nekompaktní kardiomyopatie nejspíš vzniká při poruše embryonálního vývoje kompaktní vrstvy myokardu, kdy ve fetálním a finálně vyvinutém srdci nadále přetrvává síť trabekul. Onemocnění je geneticky heterogenní s výskytem sporadických i familiárních forem. Zdá se, že autozomálně dominantní dědičnost převažuje nad X-vázanou. Bylo zaznamenáno i několik autozomálně recesivních případů [2].
Prvotní diagnóza je především echokardiografická. V literatuře je zmíněno několik klasifikací, podle kterých lze nález hodnotit. V praxi jsou nejčastěji užívaná kritéria dle Jenniho et al [3]:
- ztluštělý myokard s dvouvrstvou strukturou složenou z tenké kompaktní epikardiální vrstvy (C) a daleko silnější nekompaktní endokardiální vrstvy (N) tvořenou sítí trabekul s hlubokými endomyokardiálními prostory; poměr N : C je > 2 (obr. 1),
- predominantní lokalizace je mid-laterální, mid-inferiorní a apikální (obr. 2),
- typické je barevné dopplerovské zobrazení hlubokých intertrabekulárních výběžků vyplněných krví z levé komory,
- absence dalších srdečních abnormalit (v případě izolované nekompaktní kardiomyopatie),
- typ zobrazení k hodnocení: krátká osa, měření poměru N/C na konci systoly.
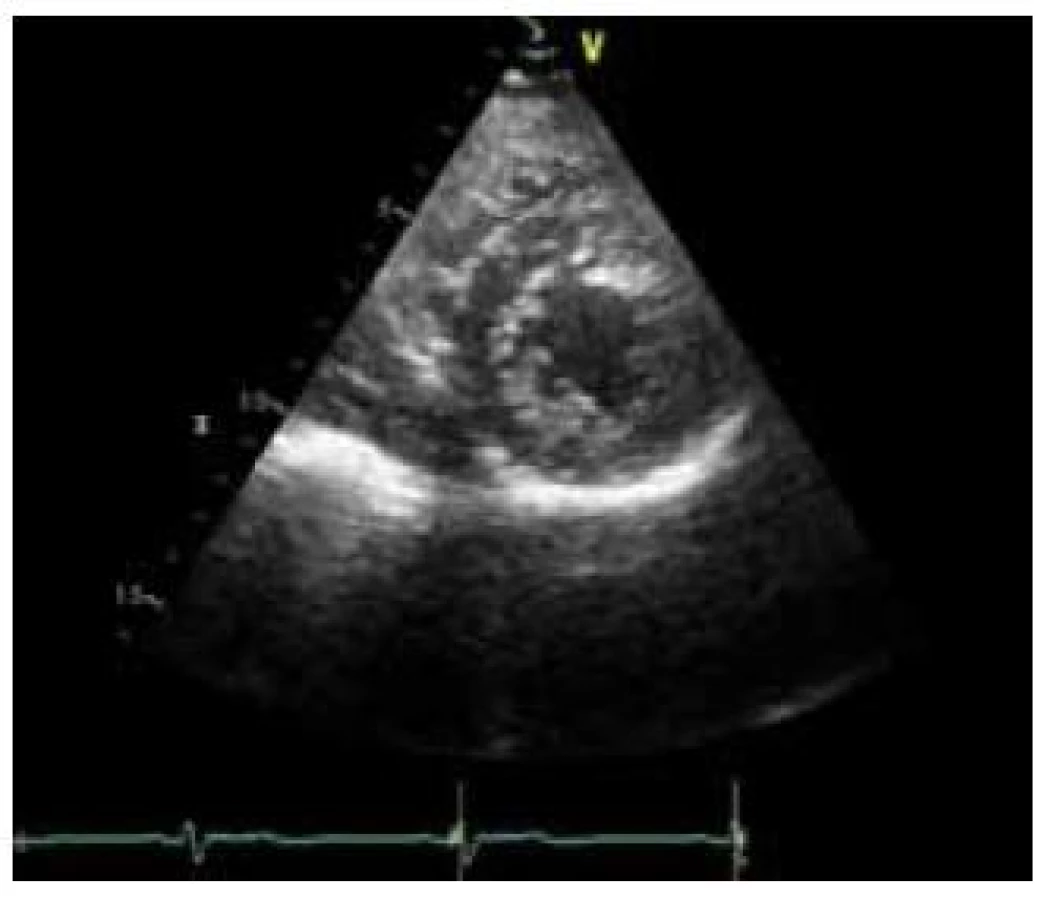
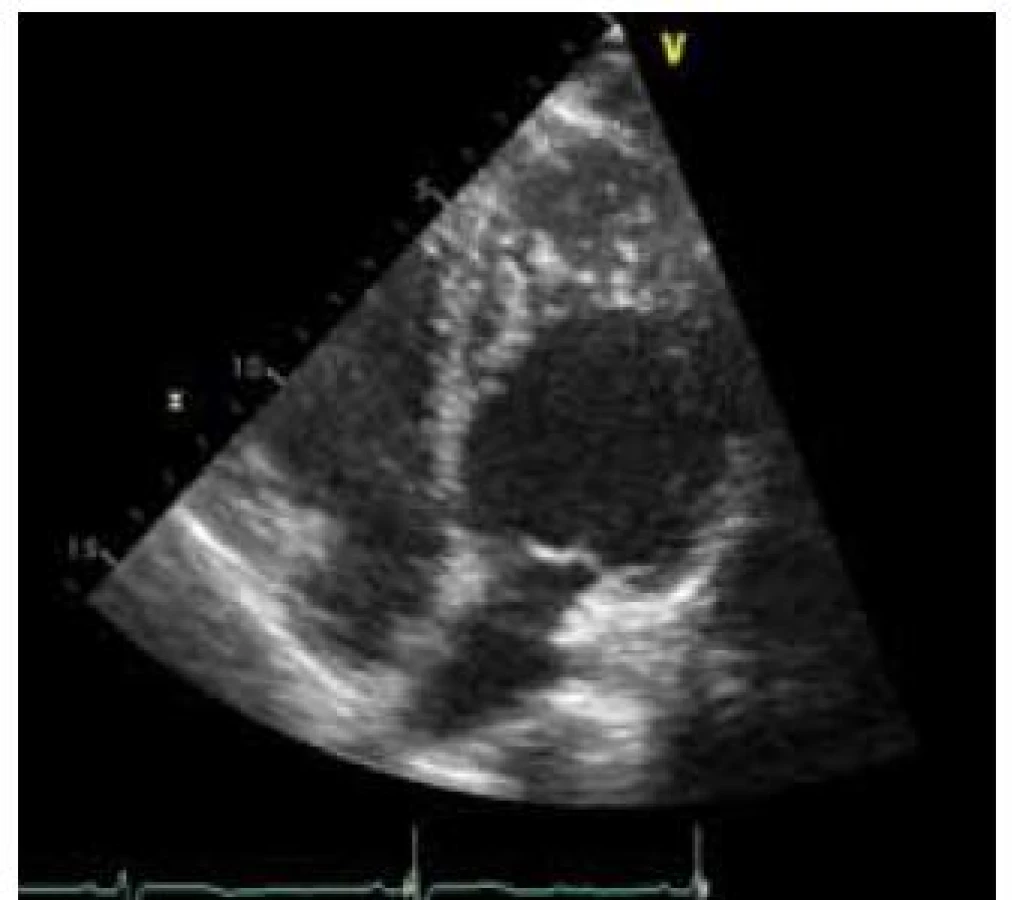
Při zobrazení magnetickou rezonancí je také klasifikací více. Nejpoužívanější je kritérium dle Petersena et al [4], kdy poměr mezi nekompaktní a kompaktní vrstvou je > 2,3. Měření je provedeno na konci diastoly (obr. 3). Známky opožděného enhancementu detekují fibrózu myokardu, která je spojena s horší prognózou.
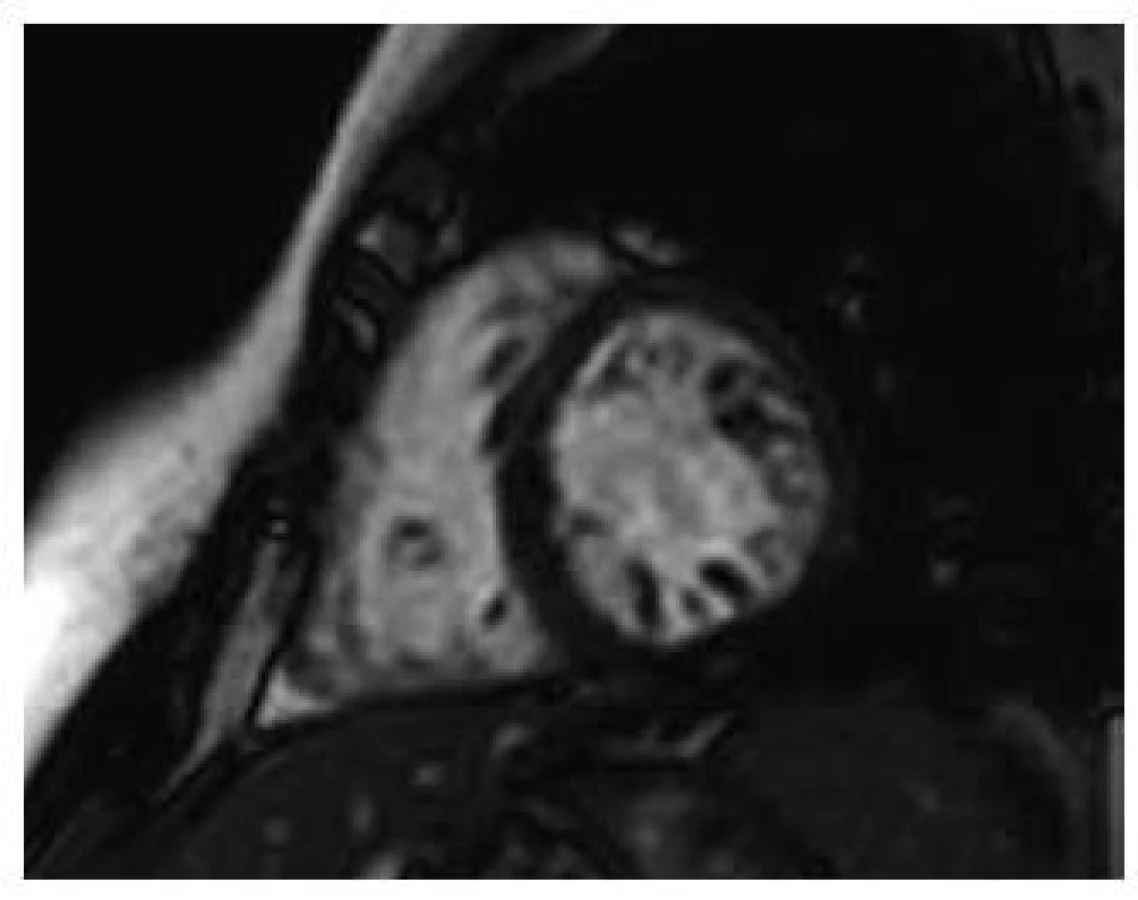
K dořešení symptomatických arytmií a synkopálních stavů je třeba provedení elektrofyziologického vyšetření. Opomenuto by nemělo být genetické vyšetření a rodinný screening u příbuzných prvního stupně. Při podezření na myopatii (skeletální/mitochondriální) by měl být pacient vyšetřen neurologem.
Počátečními symptomy bývají atypické torakalgie, dušnost nebo palpitace. EKG známky jsou nespecifické – bývá přítomna blokáda levého Tawarova raménka, známky hypertrofie levé komory srdeční, abnormality v repolarizační fázi, ale křivka může být především u mladších lidí i zcela normální.
Typickými komplikacemi pokročilých stadií této choroby je klasická triáda, do které náleží srdeční selhání, komorové arytmie a systémová embolizace [5].
Život ohrožující komorová tachykardie byla zaznamenána u více než 20 % pacientů s nekompaktní kardiomyopatií. Nebezpečím náhlé smrti jsou ohroženi především pacienti s pokročilým onemocněním, u kterých dochází pravděpodobně v důsledku poruchy mikrocirkulace k fibróze myokardu, jež tvoří arytmogenní substrát [6–8].
Indikace primárně preventivní implantace ICD u pacienta s nekompaktní kardiomyopatií a dobrou funkcí levé komory může být sporná. V literatuře je doporučeno přistupovat k takovým případům podobně jako k pacientům s neischemickou kardiomyopatií.
O indikaci sekundárně preventivní implantace ICD či zavedení resynchronizační terapie u osob s pokročilým srdečním selháním, EF LK pod 35 % v případě diagnózy nekompaktní kardiomyopatie není třeba pochybovat [9].
Asymptomatičtí pacienti s dobrou funkcí levé komory by měly být sledováni minimálně každé dva roky s provedením echokardiografie a 24hodinového sledování EKG. Léčba srdečního selhání by měla být vedena dle guidelines. Antikoagulační terapie je v literatuře doporučena u pacientů s poklesem ejekční frakce levé komory pod 40 % [5].
Závěr
Nekompaktní kardiomyopatie je vzácné onemocnění, které i vzhledem k absenci guidelines vyžaduje ve stanovení vyšetřovacího a léčebného postupu individuální přístup, který by měl být v souladu s nejnovějšími poznatky medicíny.
V souvislosti se zdokonalováním zobrazovacích metod je možno očekávat častější záchyt tohoto onemocnění.
Práce byla vypracována v rámci Výzkumného záměru MŠMT – MSM0021622402.
Doručeno do redakce 30. 9. 2011
Přijato po recenzi 17. 10. 2011
MUDr. Tereza Mikušová
MUDr. Pavla Bothová, Ph.D.
prof. MUDr. Jaroslav Meluzín, CSc.
I. interní kardioangiologická klinika FN u sv. Anny Brno a LF MU Brno
tereza.mikusova@fnusa.cz
Sources
1. Dusek J, Ostadal B, Duskova M. Postnatal persistence of spongy myocardium with embryonic blood supply. Arch Pathol 1975; 99: 312–317.
2. Lorsheyd A, Cramer MJ, Velthuis BK et al. Familial occurence of isolated non-compaction cardiomyopathy. Eur J Heart Fail 2006; 8: 826–831.
3. Jenni R, Oechslin E, Schneider J et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 2001; 86: 666–671.
4. Petersen SE, Selvanayagam JB, Wiesmann F et al. Left ventricular non-compaction: insights from cardiovascular magnetic resonance imaging. J Am Coll Cardiol 2005; 46: 101–105.
5. Oechslin E, Jenni R. Left ventricular non-compaction revisited: a distinct phenotype with genetic heterogeneity? Eur Heart J 2011; 32: 1446–1456.
6. Burke A, Mont E, Kutys R et al. Left ventricular noncompaction: a pathological study of 14 cases. Hum Pathol 2005; 36: 403–411.
7. Oechslin EN, Attenhofer Jost CH, Rojas JR et al. Long-term follow-up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol 2000; 36: 493–500.
8. Jenni R, Wyss CA, Oechslin EN et al. Isolated ventricular noncompaction is associated with coronary microcirculatory dysfunction. J Am Coll Cardiol 2002; 39: 450–454.
9. Táborský M, Kautzner J, Bytešník J et al. Zásady pro implantace kardiostimulátorů, implantabilních kardioverterů-defibrilátorů a systémů pro srdeční resynchronizační léčbu 2009. Cor Vasa 2009; 51: 602–618.
Labels
Paediatric cardiology Internal medicine Cardiac surgery CardiologyArticle was published in
Cardiology Review

2011 Issue 4
Most read in this issue
- Není hypertrofie jako hypertrofie aneb Nezapomínejme na amyloidózu
- Kalcifikace perikardu
- Peripartální kardiomyopatie
- Nesarkomerické formy hypertrofické kardiomyopatie v dospělosti